
Abstract
Chibiraga is a mall East Asian genus in the family Limacodidae (slug-moths). The latter includes many agricultural pests. Mitochondrial genome analysis is an important tool for studying insect molecular identification and phylogenetics. However, there are very few mitogenome sequences available for Limacodidae species, and none for the genus Chibiraga at all. To explore the mitogenome features of Chibiraga and verify its phylogenetic position, the complete mitogenome of Chibiraga houshuaii was sequenced and annotated. The complete 15,487 bp genome encoded 37 mitochondrial genes, including 13 protein-coding genes (PCGs), 22 transfer RNA (tRNA) genes, two ribosomal RNA (rRNA) genes, and a control region (CR). Most of the PCGs had typical ATN start codons and terminated with TAA or a single T residue. UUA (Leu2), AUU (Ile), UUU (Phe), AUA (Met) and AAU (Asn) were the five most frequently used codons. All tRNAs were folded into cloverleaf secondary structure, except for trnS1, which lacked the DHU arm. Phylogenetic analyses within the superfamily Zygaenoidea were performed based on multiple datasets from mitochondrial genes. The results showed that the families Phaudidae, Limacodidae and Zygaenidae were respectively recovered as monophyly; C. houshuaii was clustered in a clade with nettle type larvae in Limacodidae.
Similar content being viewed by others


Introduction
Limacodidae, commonly known as slug-moths, is a large family of Lepidopteran insects in the superfamily Zygaenoidea1,2. Limacodidae currently includes more than 310 genera and about 1800 species distributed worldwide, and is especially species-rich in the tropical and subtropical areas1,2,3. Unlike common caterpillars, the larvae of Limacodids usually have extremely reduced legs, and instead they developed suckers, so while advancing, their ventral surfaces are in direct contact with the substrate and make slug-like peristaltic movements1,2. The larvae of most Limacodid species have bright conspicuous warning colors and bear diverse cuticular processes of various types on the dorsum. The processes can be in the forms of spiny scoli (nettle), hairy tubercules, verrucae, or gelatinous warts among different species3,4. In particular, the nettle ones can produce urticating toxins, such as histamine and histidine, that can cause painful stings and skin inflammations when touched inadvertently5,6,7,8. Many of the Limacodidae are notorious pests, as they damage all kinds of tropical economic crops, such as banana, tea, sugarcane, cocoa and palm trees4,9,10,11.
Nowadays, Limacodidae has been widely accepted as a valid family by most modern taxonomists, although there remain some minor debates about its boundary delimitation within Zygaenoidea1,12,13,14,15. In Limacodidae, no tribal level taxon has ever been formally proposed, other than several morphology-based generic complexes raised decades ago by Holloway16 and Epstein1 respectively. Furthermore, the validation of these complexes has never been strictly tested with molecular methods since their establishments. As Limacodidae expanded in species number, these complexes have become obsolete, with many newly discovered species not fitting in. Therefore, the tribal level phylogenetic analysis for Limacodidae has been urgently demanded. Unfortunately, only two molecular phylogenetic studies investigating into the evolution of larval traits within Limacodidae have been published in recent years12,17. In these studies, the resolution of six ‘lineages’ that partially agreed with the old generic complex system in Limacodidae was generated, but no high-level classification has been carried out. To date, the problems of the overall classification within Limacodidae remain unsettled.
The insect mitochondrial genome (mitogenome) is a compact double-stranded circular DNA molecule, in the size typically varying between 15 and 18 kb, and usually harbors a set of 37 genes (13 protein-coding genes, 22 transfer RNA genes and two ribosomal RNA genes), which is conserved across bilaterian metazoans18,19. In addition to the genes, a mitogenome usually at least includes one A + T-rich noncoding control region of various lengths, which is responsible for the regulation in transcription and replication19,20. The small size and high cellular abundance of the mitogenome make it relatively much easier to obtain than nuclear genome19,21. In fact, the sequence data of insect mitogenomes are being accumulated rapidly in recent years, benefiting from the development of the next generation sequencing (NGS) technology, and meanwhile, used extensively in evolutionary studies, especially in phylogenetics19,22,23.
To date, there are only 21 complete or near complete mitochondrial genome sequences published for Limacodidae, covering mere 16 species in 13 genera (up to 31 December 2023). That is too small a proportion for such a large family as Limacodidae that consists of hundreds of genera and thousands of species. Therefore, in this study, we reported the first complete mitochondrial genome of the genus Chibiraga. Chibiraga is a mall East Asian Limacodid genus that includes three known species (C. banghaasi, C. houshuaii and C. yukei). All the adults of the genus are middle-sized plain-looking brown moths with darker markings on the forewings. The type species C. banghaasi is widely distributed in China, Korea and Russia. Its larvae are of the nettle-type, yellow in color, with blue-black markings and a bright-colored eye spot on dorsum, and they feed on Burseraceae and Cupressaceae plants24. C. houshuaii is distributed in Yunnan and Henan Provinces, China. C. yukei is in Sichuan Province, China. The larvae of the latter two species are yet unknown. The mitogenome was sequenced from C. houshuaii. We described and analyzed the mitogenome in details. we also further analyzed the phylogenetic relationships of three families in Zygaenoidea and explored the phylogenetic location of C. houshuaii. The establishment of this genome resource will provide insights for future relevant taxonomic and phylogenetic researches.
Results
Mitogenome structure and organization.
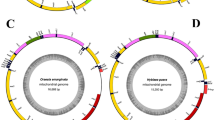
The complete mitogenome of C. houshuaii was a double-stranded circular DNA molecule of 15,487 bp in length (GenBank: OR619569). It shared the typical composition of 37 genes (13 PCGs, 22 tRNA genes and two rRNA genes) and an A + T-rich control region with other metazoan animals25 (Fig. 1). The gene arrangement pattern of this mitogenome followed that of the other Ditrysian moths26,27, with nine PCGs and 14 tRNAs encoded on the majority (J) strand, while the remaining four PCGs, eight tRNAs and the two rRNAs on the minority (N) strand (Table 1).

The circular structure of the mitogenome of C. houshuaii. Genes shown on the inner circle are transcribed in a clockwise direction, whereas those on the outer circle are transcribed counterclockwise. Different gene types are shown as filled boxes in different colors. ND1-6 are indicated in yellow; COX1-3 in light purple; ATP6 and 8 in green; CYTB in purple; tRNA genes in blue; rRNA genes in red and the control region in pink.
The mitogenome of C. houshuaii exhibited a distinct overall composition bias towards A + T nucleotides (79.8%). This predominance of A + T content varied slightly among different types of genes. It was most significant in the control region (90.1%), followed by rRNAs (84.4%), tRNAs (82.0%), and least in PCGs (78.0%) (Table 2). The AT and GC-skew values were used to describe the compositional bias between the two DNA strands28. In this study, no great AT-skew had been detected either over the entire sequence or in the different genic regions (− 0.02 to 0.04) (Table 2). Nonetheless, the GC-skew was significant among the whole genome. The GC-skew values were mostly negative (− 0.19 to − 0.13), and positive only in the rRNA gene regiones (0.36) (Table 2).
In the mitogenome of C. houshuaii, there were 17 spacer regions of 1–52 bp between adjacent genes, in addition to five gene overlaps of 1–8 bp (Table 1). The longest spacer appeared between trnQ-ND2 genes, which was conservative among all lepidopterans. The largest overlap was located between trnW and trnC genes.
PCGs and codon usage.
The total length of the 13 PCGs in the mitogenome of C. houshuaii was 11,182 bp, accounting for 72.2% of the entire sequence. Most PCGs of C. houshuaii used typical start codon ATN at initiation (six with ATG, three with ATT, two with ATA, and one with ATC), only COX1 started with an unorthodox putative codon CGA (Table 1). In respect to stop codons, COX1, COX2 and ND4 used a single T residue to stop transcription, all other ten genes terminated with the regular stop codon TAA (Table 1).
The calculation of the relative synonymous codon usage (RSCU) of C. houshuaii indicated that the five most frequently used codons were UUA (Leu2), AUU (Ile), UUU (Phe), AUA (Met) and AAU (Asn), having the counts of 438, 384, 360, 261 and 238, while the least commonly used codons were CCG (Pro), UCG (Ser2), CUG (Leu1), each having only one count, and CGC (Arg), UGC (Cys), CAG (Gln), each having two counts. In conclusion, the synonymous codons ending with A or T were distinctly more preferably used than those ending with C or G, when coding the same amino acid (Fig. 2).

Relative synonymous codon usage (RSCU) of the PCGs in the C. houshuaii mitogenome.
In addition, the synonymous substitutions (Ks), non-synonymous substitutions (Ka), and the Ka/Ks ratios of the 13 PCGs from Limacodidae were calculated (Fig. 3). The Ka/Ks values were used to estimate the natural selection patterns for the PCGs among a population29. In our study, the Ka/Ks values of all 13 PCGs were less than 1 (0.05–0.6), indicating the presence of purifying selection in these Limacodidae species.

The Ka, Ks, and Ka/Ks values of the 13 mitochondrial PCGs in Limacodidae.
tRNA genes, rRNA genes, and the control region.
The total length of the 22 transfer RNA genes of C. houshuaii was 1465 bp, accounting for 9.5% of the entire mitogenome. Their respective lengths ranged from the shortest trnR with 63 bp to the longest trnL1 with 73 bp (Table 1). The predicted secondary structures were shown in Supplementary Fig. S1. All tRNAs could be folded into the iconic cloverleaf secondary structure, except for trnS1, which did not have the typical DHU arm, but a single-stranded loop instead. A total of 19 wobbled G-U pairs (six on acceptor arms, six on DHU arms, six on anticodon arms, and one on TΨC arms), that formed weak bonds were found in 13 tRNAs. In addition, the atypical U-U pairing was also detected, but only in trnL2, trnP and trnS2.
The total length of the two ribosomal RNA genes (16S rRNA = 1390 bp; 12S rRNA = 783 bp) was 2173 bp, accounting for 14.0% of the entire mitogenome (Table 1). The 16S rRNA gene was located between trnL1 and trnV, and the 12S rRNA gene was between trnV and the control region, both without intergenic spacers or overlaps. The predicted secondary structures of the two rRNAs were shown in Supplementary Figs. S2 and S3 respectively. There were five domains (I, II, IV, V, VI) in the 16S rRNA of C. houshuaii, and the domain III was absent as in all Arthropoda species30. There were three domains (I, II, III) in the 12S rRNA. Many mismatches, such as G-U, G-A, or U-U pairs were observed in Both rRNAs.
The control region of C. houshuaii was 406 bp in length, and was located between the 12S rRNA and trnM genes (Table 1). The following transcription or replication related elements were identified (Fig. 4): an ATAGA motif, followed by an 18 bp polyT stretch located at downstream of 12S rRNA; a 9 bp polyA stretch at the upstream of trnM; and a microsatellite-like repeat region (AT)9. However, no tandem repeat sequence or any functional stem-loop structure was found.

The control region in mitochondrial genome of C. houshuaii.
Phylogenetic analyses.
Different combinations of datasets, partitioning models and tree-building methods resulted in nine phylogenetic trees within Zygaenoidea (Fig. 5). Their topologies are largely congruent, especially on family and tribal levels. In all nine trees, the three families (Phaudidae, Limacodidae and Zygaenidae) appeared in our analyses were successfully recovered as monophylies in the superfamily Zygaenoidea with strong nodal supports. The sibling relationship between Phaudidae and Zygaenidae was highly supported, and together being the sister group to Limacodidae. This backbone topology was consistent with other recent phylogenetic analyses concerning Zygaenoidea21,31. Limacodidae was resolved into three major clades, Clade A, B and C, which roughly equaled to the Lineage 3, 5 and 6 respectively in Lin et al.17. Our species C. houshuaii was clustered into Clade B, where all the member Limacodids had nettle type larvae. Noteworthily, C. houshuaii was consistently recovered at the base of Clade B with strong nodal supports in all seven trees produced using site-homogeneous substitution models, regardless of the analytical methods and datasets used. However, in the two BI trees (AA-BI-CAT + GTR and AA-BI-CAT + MtArt) produced from site-heterogeneous models, C. houshuaii was positioned much more terminally, being sibling to Thosea sinensis, only with weak node confidence.

Inferred phylogenetic relationships among Zygaenoidea from the four datasets using different methods and models. Branch in red represents family Phaudidae; branches in green represent family Zygaenidae; branches in blue represent family Limacodidae. Nodes with 90–95 bootstrap support values in ML analyses or 0.90–0.95 posterior probabilities in BI analyses are labelled by light pink circles; 96–99 or 0.96–0.99 by pink circles; 100 or 1.00 by red circles. The species followed by an asterisk is sequenced for this study. “OG” in the black box is short for “outgroup”.
Although all these trees were identical in the high-level backbone topologies, there were still minor discrepancies among them within each family or major clade. Such as mentioned above, C. houshuaii was located either at the base or terminal of Clade B, depending on the type of the substitution models used. The positionings of Rhodopsona rubiginosa and Pidorus atratus were quite unstable as well. In four trees (P123R-ML, P12-BI, AA-ML and AA-BI-CAT + GTR), they were located at more basal positions in Zygaenidae, while in other trees, either of them was located at more terminal positions (Rhodopsona rubiginosa in three trees: P123-ML, P12-ML and AA-BI-CAT + MtArt; Pidorus atratus in two trees: P123R-BI and P123-BI).
Discussion
In this study, the phylogenetic results confirmed the monophylies of Phaudidae, Limacodidae and Zygaenidae. The phylogeny within these three families was Limacodidae + (Phaudidae + Zygaenidae), supporting the results in other mitochondrial genes-based phylogenetic studies21,31. Although, Limacodidae was recovered as monophyletic, two closely related problematic families Chrysopolomidae and Dalceridae had not been included in the present analyses, due to the scarcity of their mitogenomes. Chrysopolomidae and Dalceridae were two small families compared to Limacodidae. Epstein1, based on morphological observations, suggested the degradation of Chrysopolomidae to a subfamily in Limacodidae, and Dalceridae should be included within Limacodidae as well. In addition, two recent multi-gene phylogenetic studies also supported the inclusion of these two families within Limacodidae12,14. Because of the limited samplings of these two families in the above studies, it was immature to jump directly into any changes in their classification, and because of that, Chrysopolomidae and Dalceridae had maintained their familial status13,15. Obviously, many more mitochondrial genomes were demanded to be sequenced in order to better understand the monophyly of Limacodidae as well as its interior phylogeny.
Methods
Sample collection, DNA extraction and sequencing.
The adult specimen of C. houshuaii was collected by light trap from Mountain Jizu, Dali Bai Ethnic Autonomous Prefecture, Yunnan Province, China (25° 56′ 37′′N, 100° 23′′ 14′′ E) on 6 August 2021. The specimen was then deposited in 95% ethanol under − 20 °C in the Insect Collection of Guizhou University of Traditional Chinese Medicine, Guiyang, China, until DNA extraction. The total DNA was extracted from two legs using a MagicMag Genomic DNA Micro Kit (Sangon Biotech Co., Shanghai, China) according to the manufacturer’s protocol. A total amount of 0.2 μg sample DNA was fragmented by sonication to a size of 350 bp, then subjected to high-throughput pair-ended sequencing (PE150) on Illumina 6000 platform in Novogene Bioinformatics Technology Co., Ltd (Tianjin, China).
Mitogenome assembly, annotation, and analyses.
The raw data were processed using Fastp V.0.19.732 to discard low-quality reads and obtain a high-quality clean data of 3.26 g bp. The complete mitogenome was de novo assembled by MitoZ V.3.433 and SPAdes V.3.15.134. The sequence was polished with the aid of Pilon V.1.2435, then annotated using MitoZ software and MITOS WebServer (http://mitos2.bioinf.uni-leipzig.de/index.py)36, and subsequently manually checked. The final mitogenome map was produced using Organellar Genome DRAW (OGDRAW)37.
The statistics of the mitogenome base composition was produced in MEGA V11.0.1138. The asymmetry of the two mitogenome strands was evaluated using the formulae reported by Perna and Kocher28: AT-skew = (A− T)/(A + T), and GC-skew = (G− C)/(G + C). The relative synonymous codon usage (RSCU) of the PCGs was analyzed and visualized in PhyloSuite V.1.2.339,40. The non-synonymous substitutions (Ka), synonymous substitutions (Ks), and their ratios (Ka/Ks) for the 13 PCGs in Limacodidae were calculated through DnaSP V.5.10.0141. The secondary structures of the tRNAs were predicted on MITOS WebServer. The secondary structures of the rRNAs were predicted with R2DT V.1.342. Tandem repeat elements in the control region were detected by using Tandem Repeats Finder (https://tandem.bu.edu/trf/home)43, and the possible helix structures were predicted on Mfold Web Server (http://www.unafold.org/mfold/applications/dna-folding-form.php)44.
Phylogenetic analyses.
We chose 24 taxa, including our newly sequenced one, from three families in the superfamily Zygaenoidea as ingroup, and two other taxa (Phyllonorycter ringoniella and Plutella armoraciae as outgroup) from Gracillarioidea and Yponomeutoidea respectively to analyze the phylogenetic position of C. houshuaii, and the phylogenetic relationships within Zygaenoidea (Table 3). In these analyses, a total of nine Maximum Likelihood (ML) or Bayesian Inference (BI) trees were reconstructed based on four data matrixes, (1) “P123R” matrix: all three positions of 13 PCGs plus two rRNA genes (12,863 sites); (2) “P123” matrix: all three positions of 13 PCGs (11,118 sites); (3) “P12” matrix: 1st and 2nd positions of PCGs (7412 sites); (4) “AA” matrix: amino acid sequences translated from 13 PCGs (3706 sites). The DNA sequences of each gene were first aligned separately using MAFFT V.7.5245 with the L-INS-i algorithm, next trimmed by trimAI V.1.4.146, then concatenated in MEGA 11 software, and last converted into the above-mentioned four datasets. Two site-heterogeneous substitution models (CAT + GTR and CAT + MtArt respectively) were used for the AA matrix in the BI analyses. The site-homogeneous models were used in all our other tree-building analyses, and the optimal partitioning scheme and the corresponding best fit substitution models for each analysis were determined using ModelFinder V.2.2.247 implemented in IQ-TREE2 V.2.2.2.748 (Supplementary Tables S1–S7). The ML trees were constructed in IQ-TREE2 software, and the node confidence values were assessed with 20,000 ultrafast bootstraps49. The BI trees from AA matrix were generated via PhyloBayes-MPI V.1.8c50. For each tree, two independent searches were run for 25 thousand generations until the maxdiff values dropped below 0.1. The BI trees from the other three datesets were generated via MrBayes V.3.2.751 in parallel mode. For each tree, two independent searches were run for five million generations until the average standard deviations of split frequencies dropped below 0.01. In each BI analysis, the initial 25% trees of each run were discarded as burnin, then the consensus trees and the posterior probability (PP) of each node were computed from the remaining trees. The phylogenetic trees were visualized in FigTree V.1.4.452.
Data availability
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at https://www.ncbi.nlm.nih.gov/nuccore/OR619569 under the Accession no. OR619569.
References
Epstein, M. E. Revision and phylogeny of the Limacodid-group families, with evolutionary studies on slug caterpillars (Lepidoptera: Zygaenoidea). Smithson. Contrib. Zool. 582, 1–102 (1996).
Solovyev, A. V. & Giusti, A. Revision of the genus Scopelodes Westwood, 1841 (Lepidoptera, Limacodidae) with description of 16 new species. Insect Syst. Evol. 49, 520–581 (2018).
Solovyev, A. V. & Witt, T. h. J. The Limacodidae of Vietnam. Entomofauna Supplement 16, 33–229 (2009).
Epstein, M. E., Henk, G., Naumann, C. M. & Tarmann, G. M. Chapter 10. the Zygaenidae. In Lepidoptera, Moths and Butterflies.Vol. 1: Evolution, Systematics, and Biogeography (ed. Kristensen, N. P.) 159–180 (Walter de Gruyter, 1999).
Fourie, P. B. & Hull, P. R. Urticaria caused by the slug caterpillar Latoia vivida (Lepidoptera: Limacodidae). S. Afr. J. Anim. Sci. 15, 56 (1980).
Itokawa, H., Kano, R., Nakajima, T. & Yasuhara, T. Examination of the pain producing amines in the venomous hairs or spines of Japanese urticating caterpillars. Jpn. J. Sanit. Zool. 36, 83–86 (1985).
Murphy, S. M., Leahy, S. M., Williams, L. S. & Lill, J. T. Stinging spines protect slug caterpillars (Limacodidae) from multiple generalist predators. Behav. Ecol. 21, 153–160 (2010).
Sourakov, A. Scientific note: Evaluating potential aposematic signals in caterpillars using a fluorescent microscope and spectrometer. Trop. Lepid. Res. 29, 52–55 (2019).
Holloway, J. D. Limacodid pests of tropical Australasia. In Slug and Nettle Caterpillars: The Biology, Taxonomy and Control of the Limacodidae of Economic Importance on Palms in South-East Asia (eds Cock, M. J. W. et al.) 119–121 (CAB International, 1987).
Holloway, J. D., Cock, M., Desmier, J. W. & Chenon, R. Systematic account of South-East Asian pest Limacodidae. In Slug and Nettle Caterpillars: The biology, Taxonomy and Control of the Limacodidae of Economic Importance on Palms in South-East Asia (eds Cock, M. J. W. et al.) 15–117 (Cab International, 1987).
Conant, P., Hara, A. H., Nagamine, W. T., Kishimoto, C. M. & Heu, R. A. Nettle caterpillar Darna pallivitta Moore (Lepidoptera: Limacodidae). New pest Advisory, 01–03. State of Hawaii, Department of Agriculture https://hdoa80b.hawaii.gov/pi/files/2013/01/npa01-03_netcat.pdf (2001).
Zaspel, J. M., Weller, S. J. & Epstein, M. E. Origin of the hungry caterpillar: Evolution of fasting in slug moths (Insecta: Lepidoptera: Limacodidae). Mol. Phylogenet. Evol. 94, 827–832 (2016).
Zolotuhin, V. V., Kurshakov, P. A. & Solovyev, A. V. The use of muscle morphology of Chrysopolomidae (Lepidoptera) genitalic appendages in developing a family system. Entomol. Rev. 94, 181–189 (2014).
Mayer, C. et al. Adding leaves to the Lepidoptera tree: capturing hundreds of nuclear genes from old museum specimens. Syst. Entomol. 46, 649–671 (2021).
Taberer, T. R. Revision of the family Chrysopolomidae Aurivillius, 1895 (Lepidoptera: Zygaenoidea) with the description of three new genera. Bonn Zool. Bull. 72, 1–23 (2023).
Holloway, J. D. The moths of Borneo: Key to families Cossidae, Metarbelidae, Ratardidae, Dudgeoneidae Epipyropidae and Limacodidae. Malay. Nat. J. 40, 1–165 (1986).
Lin, Y. C., Lin, R. J., Braby, M. F. & Hsu, Y. F. Evolution and losses of spines in slug caterpillars (Lepidoptera: Limacodidae). Ecol. Evol. 9, 9827–9840 (2019).
Boore, J. L. Animal mitochondrial genomes. Nucl. Acids Res. 27, 1767–1780 (1999).
Cameron, S. L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 59, 95–117 (2014).
Clayton, D. A. Transcription and replication of animal mitochondrial DNAs. Int. Rev. Cytol. 141, 217–232 (1992).
Zhang, S. P. et al. Complete mitochondrial genome of Parasa sinica: New insights into the phylogeny of Limacodidae. Heliyon 9, e21375 (2023).
Curole, J. P. & Kocher, T. D. Mitogenomics: Digging deeper with complete mitochondrial genomes. Trends Ecol. Evol. 14, 394–398 (1999).
Yang, M. S. et al. First mitochondrial genome from Yponomeutidae (Lepidoptera, Yponomeutoidea) and the phylogenetic analysis for Lepidoptera. ZooKeys 879, 137–156 (2019).
Ji, S. Q., Huang, S. Y., Ma, L. J. & Wang, M. Two new species of the genus Chibiraga Matsumura, 1931 (Lepidoptera: Limacodidae) from China. Zootaxa 4429, 165–172 (2018).
Wolstenholme, D. R. Animal mitochondrial DNA: Structure and evolution. Int. Rev Cytol. 141, 173–216 (1992).
Cao, Y. Q., Ma, C., Chen, J. Y. & Yang, D. R. The complete mitochondrial genomes of two ghost moths, Thitarodes renzhiensis and Thitarodes yunnanensis: The ancestral gene arrangement in Lepidoptera. BMC Genom. 13, 276 (2012).
Park, J. S., Kim, M. J., Jeong, S. Y., Kim, S. S. & Kim, I. Complete mitochondrial genomes of two gelechioids, Mesophleps albilinella and Dichomeris ustalella (Lepidoptera: Gelechiidae), with a description of gene rearrangement in Lepidoptera. Curr. Genet. 62, 809–826 (2016).
Perna, N. T. & Kocher, T. D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 41, 353–358 (1995).
Hurst, L. D. The Ka/Ks ratio: Diagnosing the form of sequence evolution. Trends Genet. 18, 486–487 (2002).
Cannone, J. J. et al. The Comparative RNA Web (CRW) Site: An online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics 3, 2 (2002).
Zhang, R. Y. et al. The first mitochondrial genome for Phaudidae (Lepidoptera) with phylogenetic analyses of Zygaenoidea. Int. J. Biol. Macromol. 149, 951–961 (2020).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Meng, G. L., Li, Y. Y., Yang, C. T. & Liu, S. L. MitoZ: A toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucl. Acids Res. 47, e63 (2019).
Bankevich, A. et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012).
Walker, B. J. et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9, e112963 (2014).
Bernt, M. et al. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319 (2013).
Marc, L., Oliver, D., Sabine, K. & Ralph, B. OrganellarGenomeDRAW—A suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucl. Acids Res. 41, W575–W581 (2013).
Tamura, K., Stecher, G. & Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027 (2021).
Zhang, D. et al. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 20, 348–355 (2020).
Xiang, C. Y. Using PhyloSuite for molecular phylogeny and tree-based analyses. iMeta 2, e87 (2023).
Librado, P. & Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452 (2009).
Sweeney, B. A. et al. R2DT is a framework for predicting and visualizing RNA secondary structure using templates. Nat. Commun. 12, 3494 (2021).
Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucl. Acids Res. 27, 573–580 (1999).
Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucl. Acids Res. 31, 3406–3415 (2003).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. TrimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K., Von Haeseler, A. & Jermiin, L. S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589 (2017).
Minh, B. Q. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Hoang, D. T., Chernomor, O., Von Haeseler, A., Minh, B. Q. & Vinh, L. S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522 (2018).
Lartillot, N., Rodrigue, N., Stubbs, D. & Richer, J. PhyloBayes MPI: Phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Syst. Biol. 62, 611–615 (2013).
Ronquist, F. et al. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542 (2012).
Rambaut, A., Drummond, A. J., Xie, D., Baele, G. & Suchard, M. A. Posterior summarization in Bayesian phylogenetics using tracer 1.7. Syst. Biol. 67, 901–904 (2018).
Acknowledgements
This project is supported by the National Natural Science Foundation of China (No. 31760630).
Author information
Authors and Affiliations
Contributions
All authors took part in material acquisition and preparing samples for DNA analyses. Y.P.C. analyzed the data. A.H.Y. prepared the figures and tables. Y.P.C. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cai, Y., Yin, A. The complete mitochondrial genome of Chibiraga houshuaii (Lepidoptera, Limacodidae) and its phylogenetic implications. Sci Rep 14, 7009 (2024). https://doi.org/10.1038/s41598-024-57709-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-57709-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
