
Abstract
Mutations leading to a reduced or loss of function in genes of the leptin-melanocortin system confer a risk for monogenic forms of obesity. Yet, gain of function variants in the melanocortin-4-receptor (MC4R) gene predispose to a lower BMI. In individuals with reduced body weight, we thus expected mutations leading to an enhanced function in the respective genes, like leptin (LEP) and MC4R. Therefore, we have Sanger sequenced the coding regions of LEP and MC4R in 462 female patients with anorexia nervosa (AN), and 445 healthy-lean controls. In total, we have observed four and eight variants in LEP and MC4R, respectively. Previous studies showed different functional in vitro effects for the detected frameshift and non-synonymous variants: (1) LEP: reduced/loss of function (p.Val94Met), (2) MC4R: gain of function (p.Val103Ile, p.Ile251Leu), reduced or loss of function (p.Thr112Met, p.Ser127Leu, p.Leu211fsX) and without functional in vitro data (p.Val50Leut). In LEP, the variant p.Val94Met was detected in one patient with AN. For MC4R variants, one patient with AN carried the frameshift variant p.Leu211fsX. One patient with AN was heterozygous for two variants at the MC4R (p.Val103Ile and p.Ser127Leu). All other functionally relevant variants were detected in similar frequencies in patients with AN and lean individuals.
Similar content being viewed by others
Introduction
Anorexia nervosa (AN) is marked by a diminished body weight, a pronounced fear of gaining weight and a distorted body image1. Among psychiatric disorders, AN exhibits the highest mortality rates2,3,4,5,6,7. Formal genetic studies revealed a robust genetic component for AN. In fact, twin-based studies have estimated a heritability for AN between 28 and 74%, depending on stringency of its definition and sample size8. In the last decade, genome-wide association studies (GWAS) uncovered eight genetic loci associated with AN9.
Energy homeostasis and consequently, body weight regulation, are modulated by the leptin-melanocortin-system exerting its functions in the hypothalamus. Leptin (LEP) is secreted by the adipose tissue (AT) and relays adaptive signals about the mass of the AT and the nutritional status to the brain. In the hypothalamic arcuate nucleus, leptin binds to the leptin receptor leading to the inhibition of the orexigenic agouti-related peptide (AgRP)/neuropeptide Y (NPY) neurons, while stimulating the anorexigenic pro-opiomelanocortin (POMC)/cocaine and amphetamine regulated transcript (CART) neurons. The released POMC is post-transcriptionally processed into α-melanocyte-stimulating hormone (α-MSH), which subsequently bind to the melanocortin-4-receptor (MC4R). The resultant activation of MC4R leads to the production of satiety signals and consequently to a lower food intake and higher energy expenditure10,11,12,13.
Disruption of this regulation by loss of function (LoF) variants is known to be associated with severe monogenic forms of obesity13,14. The variants include non-synonymous and nonsense LEP variants15,16,17,18,19,20. Yet, gain of function (GoF) variants, e.g. in MC4R21,22,23, predispose to a lower body mass index (BMI). Further, previous studies have reported shared genetic loci for reduced BMI and AN risk24,25,26.
Generally, patients with acute AN have considerably reduced serum leptin levels compared to BMI-matched controls. An increase in weight is accompanied by an increase in leptin27,28,29,30. Besides, a Mendelian randomization study concluded that low leptin levels are correlated with a higher risk of AN31. Treating patients with AN off-label with recombinant leptin improved cognition, behaviour and mood32,33,34.
Despite emerging evidence for the crucial role of the leptin-melanocortin system in the aetiology of AN31,35, no associations of variants in LEP in patients with AN have been described so far9,36,37. In MC4R, the mutant allele of the variant rs2229616 was previously shown to have a BMI-decreasing effect21,38. Previously, we have screened both genes in rather small study groups of patients with AN36,39. In the present study, we expanded our previous findings by Sanger sequencing of the coding regions of LEP and MC4R in larger study groups comprising 462 acutely ill or recovered women with AN and 445 healthy-lean controls. Based on in silico analyses and previously published in vitro and in vivo data, we subsequently assessed the functional consequences of the detected variants.
Results
Variants in the coding regions of LEP
The sequencing of the coding region of LEP revealed a total of four rare (minor allele frequency (MAF) ≤ 0.01%) variants (see Table 1). All identified carriers are heterozygous for the respective variant.
One variant led to a non-conservative amino acid exchange (rs17151919, p.Val94Met), while all others were synonymous variants (rs201523305, p.Cys7=; rs13306517, p.Gln25= and a variant without an rsID, p.Ser91=). The non-synonymous variant (rs17151919) was detected in the heterozygous state in an acutely ill female patient with AN (age: 34.12 years old; BMI: 15.94 kg/m2; BMI-SDSLMS: − 3.62). In silico tools, like MutationTaster202140 and PredictSNP241, hinted at an overall non-pathogenic effect. Yet, the resultant amino acid substitution was predicted to have a destabilizing impact on LEP (see Table 2). Previously, two studies have implied a role of the mutant allele (Met94; in mature protein this equals position 73; see Supplementary Fig. 1) in body weight regulation42,43. Met94 was already reported to be associated with lower leptin levels and a higher childhood BMI in individuals of African ancestry42. At baseline, each mutant allele added approximately 2.6 kg to the carrier’s weight, while the weight increased to 4.8 kg at year 2043. No associations were detected in Europeans, as this variant was extremely rare in this population42. Functional in vitro analyses reported a decreased leptin secretion in human embryonic kidney 293 cells caused by the substitution of valine by methionine42.
Moreover, three synonymous variants, namely rs201523305 (p.Cys7=), rs13306517 (p.Gln25=) and a variant without an assigned rsID (p.Ser91=), were determined (see Table 1). The latter variant (p.Ser91=) had already been detected in our previous mutation screen36. One recovered patient with AN (BMI: 21.23 kg/m2; BMI-SDSLMS: − 1.11) harboured the variant rs201523305 (p.Cys7=) predicted to alter splice sites (see Table 2). The variant leading to p.Ser91 = was found in one acutely ill adolescent with AN (BMI: 15.43 kg/m2; BMI-SDSLMS: − 3.03). Again, in silico tools hinted at a benign impact (see Table 2). The variant rs13306517 (p.Gln25=) was exclusively detected in one lean female control (see Table 1). This variant was implied not to have functional implications (see Table 2). Beyond, no functional studies have previously been published for these variants (see Supplementary Tables 2 and 4).
Variants in the coding region of MC4R
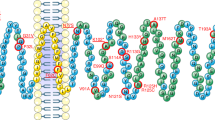
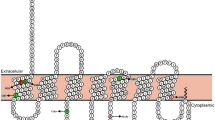
Eight variants located in the coding sequence of MC4R were identified (see Table 1). Five of those are non-synonymous variants (rs121913557, p.Val50Leu; rs2229616, p.Val103Ile; rs13447329, p.Thr112Met; rs13447331, p.Ser127Leu; rs52820871, p.Ile251Leu; for structural positions of non-synonymous variants see Supplementary Fig. 2). Additionally, one frameshift (rs13447338, p.Leu211fsX), one novel synonymous (p.Tyr153=) and one synonymous variant without an assigned rsID (p.Val193=) were determined (see Table 1).
We observed the non-synonymous variants rs2229616 (p.Val103Ile) and rs52820871 (p.Ile251Leu) in similar frequencies in our 462 patients with AN, and 445 healthy-lean controls (see Table 1). Within our control group, which included both sexes, variant rs52820871 (p.Ile251Leu) was detected four times in each sex, while variant rs2229616 (p.Val103Ile) was present in four males and nine females. Our in silico analyses hinted at a putative deleterious effect caused by rs2229616 without affecting splice sites and the protein’s torsion, while rs52828071 was predicted to be overall benign but has the potential to destabilize MC4R (see Table 2). In our literature search, we found numerous studies reporting a reduced risk for obesity and an association with a lower BMI of the infrequent alleles of rs2229616 and rs5282087121,22,44. In 2004, Geller et al.21 detected a reduced transmission rate of the Ile103 allele in an analysis of family-based samples, thus providing initial evidence for an obesity-protective effect of this variant. Functional implications of both frequent variants have been thoroughly studied, as well. For instance, the introduction of the variants in inbred mice, showed no significant difference in food intake and BMI between homozygous or heterozygous mice for each mutant and their wildtype (WT) littermates. Yet, homozygous female mice (MC4RV103I/V103I and MC4RI251L/I251L) had a reduced longitudinal length and their white adipose tissue weighed less in relation to their WT littermates. Albeit, when fed a high-fat diet, these mice’s adiposity resembled those of their littermates not carrying a variant23. Another study showed that variants p.Val103Ile and p.Ile251Leu can be characterised as GoF variants as they have a significant bias towards the ß-arrestin recruitment rather than cyclic adenosine monophosphate (cAMP)-mediated signalling. Beyond, it was determined that the MC4R carrying the mutant Ile103 allele did not internalize like the WT MC4R and consequently exhibited a stable expression on the cell surface upon agonist stimulation22. In accordance, another study supported a deviating signalling behaviour due to p.Val103Ile yielding in GoF characteristics45. Conflicting to these findings, others reported that both variants displayed an elevated presence at the plasma membrane. Upon agonist stimulation, solely p.Ile251Leu led to a reduced internalisation, while the MC4R with the mutant Ile103 allele maintained an unaltered internalisation46. Besides, multiple studies did not ascertain any functional differences between the WT and mutant MC4R for p.Val103Ile and p.Ile251Leu21,47,48,49,50.
In our mutation screen, we have further detected one lean control individual (BMI: 17.00 kg/m2; BMI-SDS: − 2.64) harbouring two frequent variants leading to p.Val103Ile and p.Ile251Leu. Similarly, one patient with AN carried the common rs2229616 (p.Val103Ile) and the rare variant rs13447331 (p.Ser127Leu). This patient had a restrictive AN. At admission the patient had a weight of 45.3 kg being 169.5 cm tall (BMI: 15.67 kg/m2; 4th BMI-percentile). During the 19 weeks in-patient treatment 6 kg were gained resulting in a BMI of 17.75 kg/m2 at discharge. Leptin levels were 1.66 µg/l at admission and increased during treatment up to 5.59 µg/l at discharge. After 2.5 years, the patient had a BMI of 22.66 kg/m2 (weight: 66 kg), while leptin levels were 43.31 µg/l. In vitro studies identified in the subsequent literature search on double mutant receptors with p.Val103Ile and p.Ser127Leu demonstrated that these can cause a reduced efficacy and potency51 and are less abundant on cell surfaces52. An increase in the cAMP accumulation in relation to the single mutant MC4R carrying the Leu127 allele was determined52. Within our mutation analysis, we did not detect the rare variant leading to the non-conservative amino acid substitution from serine to leucine at position 127 in any additional individuals here (see Table 2). Our in silico analyses detected functional implications for this variant, as it was rated as deleterious by MutationTaster2021 and PredictSNP2. Yet, no destabilizing potential on the MC4R protein was indicated (see Table 2).
The frameshift mutation in MC4R (rs13447338) resulting in a truncated protein (p.Leu211fsX) was detected once in a female adolescent with AN, and not in the lean individuals (see Table 1). The female patient with AN displayed the restrictive form of AN. At admission, she weighed 35.10 kg while being 161 cm tall (BMI: 13.54 kg/m2; 0th BMI-percentile). During her 20-week in-patient treatment, she gained 13 kg. At discharge, she had a weight of 48.1 kg and a BMI of 18.56 kg/m2. Leptin levels were 0.85 µg/l at admission and increased to 10.6 µg/l at discharge. After 2.5 years, the patient’s weight was 49 kg yielding in a BMI of 18.29 kg/m2, while leptin levels were 6.61 µg/l. One in vitro study validated the functional impact of the truncated protein reporting that this variant caused a complete loss of function47.
The other two non-synonymous and rare variants rs121913557 (p.Val50Leu) and rs13447329 (p.Thr112Met) were detected in both study groups. While in silico analyses hinted at a putative pathogenic and protein destabilizing impact of rs121913557, no functional implications for rs13447329 were detected (see Table 2). Previous studies analysing rs13447329 effects in vitro resulted in ambiguous findings. Thus, increased agonist potency and affinity for α-MSH were detected53,54. The mutant receptor further showed a decrease in cell surface expression in comparison to the WT53. Yet, multiple studies reported no functional difference of the mutant MC4R caused by this variant and the WT receptor48,51,55. We could not detect any studies in the evaluated literature resources, which have performed functional analyses for the Leu50 allele of rs121913557 so far (see Supplementary Tables 3, 4 and 5).
Both synonymous variants identified (p.Tyr153= and p.Val193=) were detected each in one proband (see Table 1). The variant p.Val193= was detected once in the lean control group and was implied to be benign (see Table 2). The other synonymous and novel variant p.Tyr153= was found in one patient with AN (BMI: 17.29 kg/m2). For this variant, in silico tools revealed a high capacity to affect splicing and to be pathogenic (see Table 2). Within a literature search, no previously published functional in vitro studies were available (see Supplementary Tables 3, 4 and 5).
Discussion
Genetic variants in genes of the leptin-melanocortin system, like LEP and MC4R, are predominantly associated with monogenic forms of obesity13,15,18,20,36,39,56,57,58. Reports emerged indicating that some variants in these genes also predispose to a lower BMI21,22,59. Genetic correlations between BMI, obesity and AN have been uncovered9,24,25,26. In fact, genetic variants for AN are negatively correlated with BMI, leptin levels, body fat percentage, waist circumference, overweight as well as obesity9,26. Previously, we have identified nine SNPs at three independent genetic loci being associated with low BMI and AN24. To expand the findings of our previous mutation screens36,39, we have sequenced the coding regions of LEP and MC4R in 462 female patients with AN, and 445 healthy-lean controls. Collectively, we have detected four variants in the LEP gene and eight variants in MC4R. Strikingly, we detected known variants leading to reduced or even loss of function which are associated with obesity39,60. Yet, the MC4R-located variants rs2229616 (p.Val103Ile) and rs52820871 (p.Ile251Leu) detected in multiple patients with AN and healthy-lean controls were already known to be associated with a lower BMI and a reduced risk for obesity21,22,44,61. One GWAS for BMI performed for both sexes combined and separately revealed that rs2229616 (p.Val103Ile) was genome-wide significantly associated with the BMI in both sexes38. Thus confirming one of our previous studies21. In silico analyses indicated largely benign effects, while certain variants in both genes might impact the stability or torsion of the respective protein.
The relevance of the leptin-melanocortin system in body weight regulation, energy expenditure as well as AN is already known10,29. Patients with AN have reduced serum leptin levels, which typically rise as weight is restored27,28,29. Strikingly, low leptin levels were linked to AN hallmark characteristics, like hyperactivity and amenorrhea62. A recent Mendelian randomization study has reported that low leptin levels are correlated with a higher risk of AN. This correlation was also evident when analysing an exclusively female dataset. Notably, a correlation of AN with leptin levels was not detected31. Beyond, off-label treatment of patients with AN (case studies) with recombinant leptin helped to improve cognition, emotions, and behavioural traits, like hyperactivity and repetitive thoughts of food32,33,34. However, it remains uncertain how variants in the leptin gene have an effect on the etiology of AN. The here detected variant leading to the amino acid exchange of p.Val94Met was already linked to lower leptin levels42, which are known to increase AN risk31. Therefore, one could hypothesise that depending on other genetic variants or environmental exposures, one person may develop AN while another develops obesity. Yet, if this variant had an effect on body weight or reduced body weight, we would expect it to be equally present in underweight but healthy controls and not to be associated with obesity. Given that AN and obesity share some comorbidities, such as major depressive disorder63,64,65 as well as body dissatisfaction1,66,67 and regarding that metreleptin treatment in patients with AN and in one female with congenital leptin deficiency had beneficial effects on the mental traits32,33,34,68, the LEP variant might impact mental factors relevant for both, AN and obesity. Yet, further research is needed.
Evidence for the involvement of leptin’s downstream target MC4R in AN is mostly based on animal models. In fact, the activation of MC4R in rats decreased food intake, while coincidently activating the hypothalamic–pituitary–adrenal axis. This activation eventually led to increased motor activity. It was suggested that genetic variants might trigger this coherence and thus lead to a prolonged stimulation of the leptin-melanocortin-system supporting the development of AN5. Yet, especially as overlaps between genetic loci relevant for low BMI and AN are known24,25, it is feasible that genetic variants in genes of the leptin-melanocortin system, impact the etiology of AN.
However, to date, no significant associations of genetic variants in these genes with AN were reported36,37. Due to our still limited sample size and generally low allele frequencies of the here detected variants, we were also unable to report associations with AN. Nevertheless, this study was able to expand our previously published mutation screens for both genes36,39. Our extended sample size enabled us to discover new variants in LEP and MC4R, which potentially are relevant for AN. This emphasises the ongoing necessity for comprehensive large-scale genetic studies. Here, we did detect variants in LEP and MC4R, which were previously described to cause a partial or even complete loss of function in humans when present in a homozygous state and were thus associated with severe forms of obesity39,51,60. For instance, in 1999, we have described the frameshift mutation (rs13447338) yielding in a truncated MC4R in one adolescent and her mother with severe obesity39. In the present study, we have detected the same frameshift variant in the heterozygous state in one patient with AN. Yet, as no premorbid weight data was available, it is feasible that this patient had premorbid obesity triggering the development of AN. Generally, 2–5% of obesity cases are attributable to MC4R variants13. For the here commonly detected MC4R variants, like p.Val103Ile which was detected in 2% of patients with AN and 1.5% of controls, similar or slightly lower frequencies have been reported in patients with obesity21,61,69,70. Interestingly, this frequency is lower in females with obesity than in males with obesity61. The p.Val103Ile comprises the first polygenic variant for body weight regulation. The infrequent allele is associated with a lower BMI21. Further, we have to note that pathogenic variants typically are present homozygously15,71. For heterozygous MC4R variants, carrier develop obesity later in childhood13, while for heterozygous LEP variants, carriers are mainly unaffected15,71. As we have exclusively detected heterozygous variants in both genes, the relevance remains unknown. Yet, one patient with AN was detected to harbour two variants simultaneously (rs2229616, p.Val103Ile and rs13447331, p.Ser127Leu). Leptin levels at admission were in the expected range72 and increased during in-patient treatment. Yet, after 2.5 years, leptin levels were rather high (43.31 µg/l). The variant leading to p.Val103Ile is known to lead to an elevated function21,22,46, while the amino acid substitution of serine to leucine at the 127th position was described to lead to a LoF in the majority of parameters analysed45,55,73 (see Supplementary Tables 3, 4 and 5). Notably, certain studies also indicated no functional deviations for mutant receptors with either Ile103 or Leu12748,49,54. Yet, functional studies for a double mutant MC4R showed for example a reduced efficacy and potency51. Previously, we have also detected weight reducing variants in MC4R in patients with bulimia nervosa74. Further, the FTO variant rs9939609 associated with obesity75,76 was previously identified in patients with eating disorders77,78. Besides a nominal association with bulimia nervosa and anorexia nervosa77, a synergic effect of this variant on leptin levels with an another variant in ABCA1 was described78.
In addition, a number of studies demonstrated that two types of MC4R variants exist which can be distinguished according to their distinct signalling behaviour22,46. For example, variants such as the rs13447329 (p.Thr112Met) detected here, are characterised by a LoF. Other variants, such as rs2229616 (p.Val103Ile), show a GoF phenotype22,46. The latter GoF variants are associated with a reduced risk for obesity and a lower BMI21,22,44. In fact, heterozygous carriers of GoF variants weighed on overage 0.39 kg/m2 less than non-carriers, while homozygotes even showed an average 0.88 kg/m2 decreased BMI22. Similarly, an animal model with transgenic mice expressing either Val103Ile or Ile251Leu exhibited GoF phenotypes. This was even more pronounced in female than male mice. For instance, female mice carrying the variant Val103Ile homozygous had a 40% lower abdominal white adipose tissue mass than their wildtype littermates. This difference was not detected in the male transgenic mice23.
Based on our data, we are unable to deduce a mechanism explaining how these variants may have a relevance for the phenotypes at the extremes of the weight scale. Yet, a resembling mechanism of action, as FTO’s rs9939609 is feasible78. Functional in vitro and in vivo studies under fasting and starvation conditions are needed to expand the findings of the general in vitro data already published for each variant.
We are aware that in silico tools have their limitations and can vary substantially in their specificity79,80,81. Particularly, predicting functional implications of GoF variants was found to be more challenging by various in silico tools than finding an accurate implication for a LoF variant82. Hence, we have additionally checked the already published in vitro and in vivo data stated in LitVar2 and on the www.mc4r.org.uk website. Nevertheless, no clear-cut results were obtained for most of the variants (see Supplementary Tables 2, 3, 4 and 5). Presumably, this is due to variations in study design as well as the time period in which the studies were conducted. For example, older studies increasingly failed to detect any differences between WT and mutant MC4R. This was particularly evident for studies investigating the common GoF variant rs2229616 (p.Val103Ile) in MC4R. Supposedly, this results from the technical advances and new experimental techniques implemented in recent years. In addition, there are many variants for which no or only very few functional studies have been completed. For example, only 16 studies have been conducted on the rs17151919 variant in the leptin gene, of which just one included functional in vitro analyses (see Supplementary Table 2). Conversely, for the common variant rs2229616 in MC4R, there are over 150 studies, of which numerous also present in vitro or even in vivo data (see Supplementary Tables 3, 4 and 5).
Given the reported genetic overlaps between BMI and AN9,24,25,26 and GoF variants in LEP and MC4R being associated with a lower BMI and a reduced odds for obesity21,22,23,59, we have Sanger sequenced the coding regions of LEP and MC4R in 462 female patients with AN and 445 healthy-lean controls. We have detected four variants in LEP and eight variants in the coding sequence of MC4R. Here we detected variants with a partial or complete LoF in vitro in heterozygous patients with AN. Homozygous carriers of these variants typically develop obesity.
Methods
Study groups
To detect variants in the coding regions of LEP and MC4R, 462 female patients with AN (acutely ill or recovered; age: 20.83 ± 8.63 years old; BMI: 16.48 ± 2.77 kg/m2; BMI-SDSLMS: − 2.55 ± 1.58; see Table 3) and 445 healthy-lean individuals without a diagnosed eating disorder (age: 26.04 ± 5.77 years old; BMI: 18.08 ± 1.13 kg/m2; BMI-SDSLMS: − 2.77 ± 0.58; see Table 3) were included. In previous studies we have already screened 49 patients with AN for LEP and 51 patients with AN and 25 lean individuals for MC4R36,39. Written informed consent was given by all participants and in case of minors by their parents. This study was approved by the Ethics committee of the respective Universities and was performed in accordance with the Declaration of Helsinki.
Mutation screen
The coding regions of LEP (chr7: 128,241,278–128,257,629; GRCh38; ENSG00000174697) and MC4R (chr18: 60,371,062–60,372,775; GRCh38; ENSG00000166603) were Sanger sequenced in 462 patients with AN, and 445 healthy-lean individuals (see Table 3) to detect genetic variants. These were identified by performing polymerase chain reactions (PCR; Veriti 96-well Thermal Cycler, Applied Biosystems, Foster City, CA, USA) with coding sequence specific primers (see Supplementary Table 1). PCR products were sent for sequencing to MicroSynth Seqlab GmbH (Göttingen, Germany). At least two independent scientists performed the sequence analysis and genotype assignment using the SeqMan Pro software (v.11.0.0, DNAstar Inc., Madison, WI, USA). Discrepancies were solved by either reaching consensus or by re-sequencing.
Hardy–Weinberg-Equilibrium
All detected variants were checked for compliance with the Hardy–Weinberg-Equilibrium (HWE). All variants fulfilled the HWE.
In silico analyses and assessment of functional implications by published data
To uncover putative functional implications of the detected variants, in silico analyses applying various tools ensued. The detected variants were analysed pertaining their potential to alter splice sites (ESEfinder83) and their general pathogenicity (MutationTaster202140; PredictSNP241). Effects of non-synonymous variants on the protein structure of LEP or MC4R were examined using PANTHER-PSEP84, the Cologne University Protein Stability Analysis Tool85 (CUPSAT) and the tool Sorting from Intolerant to Tolerant86 (SIFT). The required protein structures were extracted from the Research Collaboratory for Structural Bioinformatics protein data bank (RCSB PDB): LEP (1AX887) and MC4R (7AUE88).
To extend the functional implications derived from the in silico analyses with previously published in vitro and in vivo data, the LitVar289 (accessed on Sep. 26th 2022) database was screened for each variant in LEP and MC4R detected in this study. For MC4R variants, the website www.mc4r.org.uk (accessed on Sep. 26th 2022) summarizing publications for each variant, was additionally analysed. This website is provided by the University of Cambridge Metabolic Research Laboratories. A complete list of all studies provided by LitVar2 and www.mc4r.org.uk can be found in the Supplementary Table 5.
Data availability
The generated and analysed Sanger-sequencing data are available from the corresponding author on reasonable request. Data which was extracted from public websites or databases are included in the Supplementary Information files.
References
Association, A. P. Diagnostic and Statistical Manual of Mental Disorders (DSM-5®) (American Psychiatric Publishing, 2013).
Arcelus, J., Mitchell, A. J., Wales, J. & Nielsen, S. Mortality rates in patients with anorexia nervosa and other eating disorders. A meta-analysis of 36 studies. Arch. Gen. Psychiatry 68, 724–731. https://doi.org/10.1001/archgenpsychiatry.2011.74 (2011).
Chesney, E., Goodwin, G. M. & Fazel, S. Risks of all-cause and suicide mortality in mental disorders: A meta-review. World Psychiatry 13, 153–160. https://doi.org/10.1002/wps.20128 (2014).
Moskowitz, L. & Weiselberg, E. Anorexia nervosa/atypical anorexia nervosa. Curr. Probl. Pediatr. Adolesc. Health Care 47, 70–84. https://doi.org/10.1016/j.cppeds.2017.02.003 (2017).
Paolacci, S. et al. Genetic contributions to the etiology of anorexia nervosa: New perspectives in molecular diagnosis and treatment. Mol. Genet. Genom. Med. 8, e1244. https://doi.org/10.1002/mgg3.1244 (2020).
Yao, S. et al. Familial liability for eating disorders and suicide attempts: Evidence from a population registry in Sweden. JAMA Psychiatry 73, 284–291. https://doi.org/10.1001/jamapsychiatry.2015.2737 (2016).
Zipfel, S., Lowe, B., Reas, D. L., Deter, H. C. & Herzog, W. Long-term prognosis in anorexia nervosa: Lessons from a 21-year follow-up study. Lancet 355, 721–722. https://doi.org/10.1016/S0140-6736(99)05363-5 (2000).
Bulik, C. M. et al. Genetics and neurobiology of eating disorders. Nat. Neurosci. 25, 543–554. https://doi.org/10.1038/s41593-022-01071-z (2022).
Watson, H. J. et al. Genome-wide association study identifies eight risk loci and implicates metabo-psychiatric origins for anorexia nervosa. Nat. Genet. 51, 1207–1214. https://doi.org/10.1038/s41588-019-0439-2 (2019).
Friedman, J. M. Leptin and the endocrine control of energy balance. Nat. Metab. 1, 754–764. https://doi.org/10.1038/s42255-019-0095-y (2019).
Hinney, A., Volckmar, A. L. & Antel, J. Genes and the hypothalamic control of metabolism in humans. Best Pract. Res. Clin. Endocrinol. Metab. 28, 635–647. https://doi.org/10.1016/j.beem.2014.04.007 (2014).
Jais, A. & Bruning, J. C. Hypothalamic inflammation in obesity and metabolic disease. J. Clin. Invest. 127, 24–32. https://doi.org/10.1172/JCI88878 (2017).
Hinney, A., Korner, A. & Fischer-Posovszky, P. The promise of new anti-obesity therapies arising from knowledge of genetic obesity traits. Nat. Rev. Endocrinol. https://doi.org/10.1038/s41574-022-00716-0 (2022).
Loos, R. J. F. & Yeo, G. S. H. The genetics of obesity: From discovery to biology. Nat. Rev. Genet. https://doi.org/10.1038/s41576-021-00414-z (2021).
Montague, C. T. et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature 387, 903–908. https://doi.org/10.1038/43185 (1997).
Gibson, W. T. et al. Congenital leptin deficiency due to homozygosity for the Delta133G mutation: Report of another case and evaluation of response to four years of leptin therapy. J. Clin. Endocrinol. Metab. 89, 4821–4826. https://doi.org/10.1210/jc.2004-0376 (2004).
Fatima, W. et al. Leptin deficiency and leptin gene mutations in obese children from Pakistan. Int. J. Pediatr. Obes. 6, 419–427. https://doi.org/10.3109/17477166.2011.608431 (2011).
Saeed, S., Butt, T. A., Anwer, M., Arslan, M. & Froguel, P. High prevalence of leptin and melanocortin-4 receptor gene mutations in children with severe obesity from Pakistani consanguineous families. Mol. Genet. Metab. 106, 121–126. https://doi.org/10.1016/j.ymgme.2012.03.001 (2012).
Mazen, I., El-Gammal, M., Abdel-Hamid, M. & Amr, K. A novel homozygous missense mutation of the leptin gene (N103K) in an obese Egyptian patient. Mol. Genet. Metab. 97, 305–308. https://doi.org/10.1016/j.ymgme.2009.04.002 (2009).
Wabitsch, M. et al. Severe early-onset obesity due to bioinactive leptin caused by a p.N103K mutation in the leptin gene. J. Clin. Endocrinol. Metab. 100, 3227–3230. https://doi.org/10.1210/jc.2015-2263 (2015).
Geller, F. et al. Melanocortin-4 receptor gene variant I103 is negatively associated with obesity. Am. J. Hum. Genet. 74, 572–581. https://doi.org/10.1086/382490 (2004).
Lotta, L. A. et al. Human gain-of-function MC4R variants show signaling bias and protect against obesity. Cell 177, 597–607. https://doi.org/10.1016/j.cell.2019.03.044 (2019).
Rojo, D., McCarthy, C., Raingo, J. & Rubinstein, M. Mouse models for V103I and I251L gain of function variants of the human MC4R display decreased adiposity but are not protected against a hypercaloric diet. Mol. Metab. 42, 101077. https://doi.org/10.1016/j.molmet.2020.101077 (2020).
Hinney, A. et al. Evidence for three genetic loci involved in both anorexia nervosa risk and variation of body mass index. Mol. Psychiatry 22, 321–322. https://doi.org/10.1038/mp.2016.126 (2017).
Zheng, Y. et al. PTBP2—a gene with relevance for both Anorexia nervosa and body weight regulation. Transl. Psychiatry 12, 241. https://doi.org/10.1038/s41398-022-02018-5 (2022).
Bulik-Sullivan, B. et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 47, 1236–1241. https://doi.org/10.1038/ng.3406 (2015).
Seitz, J. et al. Leptin levels in patients with anorexia nervosa following day/inpatient treatment do not predict weight 1 year post-referral. Eur. Child. Adolesc. Psychiatry 25, 1019–1025. https://doi.org/10.1007/s00787-016-0819-4 (2016).
Haas, V. et al. Leptin and body weight regulation in patients with anorexia nervosa before and during weight recovery. Am. J. Clin. Nutr. 81, 889–896. https://doi.org/10.1093/ajcn/81.4.889 (2005).
Hebebrand, J. et al. Leptin levels in patients with anorexia nervosa are reduced in the acute stage and elevated upon short-term weight restoration. Mol. Psychiatry 2, 330–334. https://doi.org/10.1038/sj.mp.4000282 (1997).
Hebebrand, J. et al. The role of hypoleptinemia in the psychological and behavioral adaptation to starvation: Implications for anorexia nervosa. Neurosci. Biobehav. Rev. 141, 104807. https://doi.org/10.1016/j.neubiorev.2022.104807 (2022).
Peters, T. et al. Suggestive evidence for causal effect of leptin levels on risk for anorexia nervosa: Results of a mendelian randomization study. Front. Genet. 12, 733606. https://doi.org/10.3389/fgene.2021.733606 (2021).
Milos, G. et al. Short-term metreleptin treatment of patients with anorexia nervosa: Rapid on-set of beneficial cognitive, emotional, and behavioral effects. Transl. Psychiatry 10, 303. https://doi.org/10.1038/s41398-020-00977-1 (2020).
Antel, J. et al. Rapid amelioration of anorexia nervosa in a male adolescent during metreleptin treatment including recovery from hypogonadotropic hypogonadism. Eur. Child. Adolesc. Psychiatry 31, 1573–1579. https://doi.org/10.1007/s00787-021-01778-7 (2022).
Gradl-Dietsch, G. et al. Rapid emergence of appetite and hunger resulting in weight gain and improvement of eating disorder symptomatology during and after short-term off-label metreleptin treatment of a patient with anorexia nervosa. Obes. Facts 16, 99–107. https://doi.org/10.1159/000527386 (2023).
Ehrlich, S. et al. Promoter specific DNA methylation and gene expression of POMC in acutely underweight and recovered patients with anorexia nervosa. J. Psychiatr. Res. 44, 827–833. https://doi.org/10.1016/j.jpsychires.2010.01.011 (2010).
Hinney, A. et al. No evidence for involvement of the leptin gene in anorexia nervosa, bulimia nervosa, underweight or early onset extreme obesity: Identification of two novel mutations in the coding sequence and a novel polymorphism in the leptin gene linked upstream region. Mol. Psychiatry 3, 539–543. https://doi.org/10.1038/sj.mp.4000394 (1998).
Yilmaz, Z. et al. The role of leptin, melanocortin, and neurotrophin system genes on body weight in anorexia nervosa and bulimia nervosa. J. Psychiatr. Res. 55, 77–86. https://doi.org/10.1016/j.jpsychires.2014.04.005 (2014).
Pulit, S. L. et al. Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry. Hum. Mol. Genet. 28, 166–174. https://doi.org/10.1093/hmg/ddy327 (2019).
Hinney, A. et al. Several mutations in the melanocortin-4 receptor gene including a nonsense and a frameshift mutation associated with dominantly inherited obesity in humans. J. Clin. Endocrinol. Metab. 84, 1483–1486. https://doi.org/10.1210/jcem.84.4.5728 (1999).
Steinhaus, R. et al. MutationTaster2021. Nucleic Acids Res. 49, W446–W451. https://doi.org/10.1093/nar/gkab266 (2021).
Bendl, J. et al. PredictSNP2: A unified platform for accurately evaluating SNP effects by exploiting the different characteristics of variants in distinct genomic regions. PLoS Comput. Biol. 12, e1004962. https://doi.org/10.1371/journal.pcbi.1004962 (2016).
Yaghootkar, H. et al. Genetic studies of leptin concentrations implicate leptin in the regulation of early adiposity. Diabetes 69, 2806–2818. https://doi.org/10.2337/db20-0070 (2020).
Friedlander, Y. et al. Candidate molecular pathway genes related to appetite regulatory neural network, adipocyte homeostasis and obesity: Results from the CARDIA Study. Ann. Hum. Genet. 74, 387–398. https://doi.org/10.1111/j.1469-1809.2010.00596.x (2010).
Stutzmann, F. et al. Non-synonymous polymorphisms in melanocortin-4 receptor protect against obesity: The two facets of a Janus obesity gene. Hum. Mol. Genet. 16, 1837–1844. https://doi.org/10.1093/hmg/ddm132 (2007).
Paisdzior, S. et al. Differential signaling profiles of MC4R mutations with three different ligands. Int. J. Mol. Sci. 21, 1224. https://doi.org/10.3390/ijms21041224 (2020).
Brouwers, B. et al. Human MC4R variants affect endocytosis, trafficking and dimerization revealing multiple cellular mechanisms involved in weight regulation. Cell Rep. 34, 108862. https://doi.org/10.1016/j.celrep.2021.108862 (2021).
Hinney, A. et al. Melanocortin-4 receptor gene: Ccase-control study and transmission disequilibrium test confirm that functionally relevant mutations are compatible with a major gene effect for extreme obesity. J. Clin. Endocrinol. Metab. 88, 4258–4267. https://doi.org/10.1210/jc.2003-030233 (2003).
Gu, W. et al. Identification and functional analysis of novel human melanocortin-4 receptor variants. Diabetes 48, 635–639. https://doi.org/10.2337/diabetes.48.3.635 (1999).
Ho, G. & MacKenzie, R. G. Functional characterization of mutations in melanocortin-4 receptor associated with human obesity. J. Biol. Chem. 274, 35816–35822. https://doi.org/10.1074/jbc.274.50.35816 (1999).
Thearle, M. S. et al. Greater impact of melanocortin-4 receptor deficiency on rates of growth and risk of type 2 diabetes during childhood compared with adulthood in Pima Indians. Diabetes 61, 250–257. https://doi.org/10.2337/db11-0708 (2012).
Melchior, C. et al. Clinical and functional relevance of melanocortin-4 receptor variants in obese German children. Horm. Res. Paediatr. 78, 237–246. https://doi.org/10.1159/000343816 (2012).
Rovite, V. et al. The role of common and rare MC4R variants and FTO polymorphisms in extreme form of obesity. Mol. Biol. Rep. 41, 1491–1500. https://doi.org/10.1007/s11033-013-2994-4 (2014).
Nijenhuis, W. A., Garner, K. M., van Rozen, R. J. & Adan, R. A. Poor cell surface expression of human melanocortin-4 receptor mutations associated with obesity. J. Biol. Chem. 278, 22939–22945. https://doi.org/10.1074/jbc.M211326200 (2003).
Xiang, Z. et al. Pharmacological characterization of 40 human melanocortin-4 receptor polymorphisms with the endogenous proopiomelanocortin-derived agonists and the agouti-related protein (AGRP) antagonist. Biochemistry 45, 7277–7288. https://doi.org/10.1021/bi0600300 (2006).
Valli-Jaakola, K. et al. Identification and characterization of melanocortin-4 receptor gene mutations in morbidly obese finnish children and adults. J. Clin. Endocrinol. Metab. 89, 940–945. https://doi.org/10.1210/jc.2003-031182 (2004).
Fischer-Posovszky, P. et al. A new missense mutation in the leptin gene causes mild obesity and hypogonadism without affecting T cell responsiveness. J. Clin. Endocrinol. Metab. 95, 2836–2840. https://doi.org/10.1210/jc.2009-2466 (2010).
Farooqi, I. S. et al. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N. Engl. J. Med. 348, 1085–1095. https://doi.org/10.1056/NEJMoa022050 (2003).
Saeed, S. et al. Genetic variants in LEP, LEPR, and MC4R explain 30% of severe obesity in children from a consanguineous population. Obes. (Silver Spring) 23, 1687–1695. https://doi.org/10.1002/oby.21142 (2015).
Murray, P. G. et al. Reduced appetite and body mass index with delayed puberty in a mother and son: Association with a rare novel sequence variant in the leptin gene. Eur. J. Endocrinol. 164, 521–527. https://doi.org/10.1530/EJE-10-0656 (2011).
Dubern, B. et al. Mutational analysis of melanocortin-4 receptor, agouti-related protein, and alpha-melanocyte-stimulating hormone genes in severely obese children. J. Pediatr. 139, 204–209. https://doi.org/10.1067/mpd.2001.116284 (2001).
Heid, I. M. et al. Association of the 103I MC4R allele with decreased body mass in 7937 participants of two population based surveys. J. Med. Genet. 42, e21. https://doi.org/10.1136/jmg.2004.027011 (2005).
Hebebrand, J., Muller, T. D., Holtkamp, K. & Herpertz-Dahlmann, B. The role of leptin in anorexia nervosa: Clinical implications. Mol. Psychiatry 12, 23–35. https://doi.org/10.1038/sj.mp.4001909 (2007).
Blinder, B. J., Cumella, E. J. & Sanathara, V. A. Psychiatric comorbidities of female inpatients with eating disorders. Psychosom. Med. 68, 454–462. https://doi.org/10.1097/01.psy.0000221254.77675.f5 (2006).
Marucci, S. et al. Anorexia nervosa and comorbid psychopathology. Endocr. Metab. Immune Disord. Drug Targets 18, 316–324. https://doi.org/10.2174/1871530318666180213111637 (2018).
Rajan, T. M. & Menon, V. Psychiatric disorders and obesity: A review of association studies. J. Postgrad. Med. 63, 182–190. https://doi.org/10.4103/jpgm.JPGM_712_16 (2017).
Weinberger, N. A., Kersting, A., Riedel-Heller, S. G. & Luck-Sikorski, C. Body dissatisfaction in individuals with obesity compared to normal-weight individuals: A systematic review and meta-analysis. Obes. Facts 9, 424–441. https://doi.org/10.1159/000454837 (2016).
Moradi, M., Mozaffari, H., Askari, M. & Azadbakht, L. Association between overweight/obesity with depression, anxiety, low self-esteem, and body dissatisfaction in children and adolescents: A systematic review and meta-analysis of observational studies. Crit. Rev. Food Sci. Nutr. 62, 555–570. https://doi.org/10.1080/10408398.2020.1823813 (2022).
Hebebrand, J. et al. First account of psychological changes perceived by a female with congenital leptin deficiency upon treatment with metreleptin. Obes. Facts 15, 730–735. https://doi.org/10.1159/000526169 (2022).
Young, E. H. et al. The V103I polymorphism of the MC4R gene and obesity: Population based studies and meta-analysis of 29 563 individuals. Int. J. Obes. (Lond.) 31, 1437–1441. https://doi.org/10.1038/sj.ijo.0803609 (2007).
Loos, R. J. The genetic epidemiology of melanocortin 4 receptor variants. Eur. J. Pharmacol. 660, 156–164. https://doi.org/10.1016/j.ejphar.2011.01.033 (2011).
Wabitsch, M. et al. Biologically inactive leptin and early-onset extreme obesity. N. Engl. J. Med. 372, 48–54. https://doi.org/10.1056/NEJMoa1406653 (2015).
Hebebrand, J. et al. Plasma concentrations of obese protein in anorexia nervosa. Lancet 346, 1624–1625. https://doi.org/10.1016/s0140-6736(95)91955-4 (1995).
Fan, Z. C. & Tao, Y. X. Functional characterization and pharmacological rescue of melanocortin-4 receptor mutations identified from obese patients. J. Cell Mol. Med. 13, 3268–3282. https://doi.org/10.1111/j.1582-4934.2009.00726.x (2009).
Hebebrand, J. et al. Genetic predisposition to obesity in bulimia nervosa: A mutation screen of the melanocortin-4 receptor gene. Mol. Psychiatry 7, 647–651. https://doi.org/10.1038/sj.mp.4001053 (2002).
Hunt, S. C. et al. Association of the FTO gene with BMI. Obes. (Silver Spring) 16, 902–904. https://doi.org/10.1038/oby.2007.126 (2008).
Hotta, K. et al. Variations in the FTO gene are associated with severe obesity in the Japanese. J. Hum. Genet. 53, 546–553. https://doi.org/10.1007/s10038-008-0283-1 (2008).
Muller, T. D. et al. Fat mass and obesity-associated gene (FTO) in eating disorders: Evidence for association of the rs9939609 obesity risk allele with bulimia nervosa and anorexia nervosa. Obes. Facts 5, 408–419. https://doi.org/10.1159/000340057 (2012).
Genis-Mendoza, A. D. et al. Interaction of FTO rs9939609 and the native American-origin ABCA1 p.Arg230Cys with circulating leptin levels in Mexican adolescents diagnosed with eating disorders: Preliminary results. Psychiatry Res. 291, 113270. https://doi.org/10.1016/j.psychres.2020.113270 (2020).
Niroula, A. & Vihinen, M. How good are pathogenicity predictors in detecting benign variants?. PLoS Comput. Biol. 15, e1006481. https://doi.org/10.1371/journal.pcbi.1006481 (2019).
Pshennikova, V. G. et al. Comparison of predictive in silico tools on missense variants in GJB2, GJB6, and GJB3 genes associated with autosomal recessive deafness 1A (DFNB1A). Sci. World J. 2019, 5198931. https://doi.org/10.1155/2019/5198931 (2019).
Cubuk, C. et al. Clinical likelihood ratios and balanced accuracy for 44 in silico tools against multiple large-scale functional assays of cancer susceptibility genes. Genet. Med. 23, 2096–2104. https://doi.org/10.1038/s41436-021-01265-z (2021).
Flanagan, S. E., Patch, A. M. & Ellard, S. Using SIFT and PolyPhen to predict loss-of-function and gain-of-function mutations. Genet. Test Mol. Biomark. 14, 533–537. https://doi.org/10.1089/gtmb.2010.0036 (2010).
Cartegni, L., Wang, J., Zhu, Z., Zhang, M. Q. & Krainer, A. R. ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res. 31, 3568–3571. https://doi.org/10.1093/nar/gkg616 (2003).
Tang, H. & Thomas, P. D. PANTHER-PSEP: Predicting disease-causing genetic variants using position-specific evolutionary preservation. Bioinformatics 32, 2230–2232. https://doi.org/10.1093/bioinformatics/btw222 (2016).
Parthiban, V., Gromiha, M. M. & Schomburg, D. CUPSAT: Prediction of protein stability upon point mutations. Nucleic Acids Res. 34, W239-242. https://doi.org/10.1093/nar/gkl190 (2006).
Sim, N. L. et al. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 40, W452-457. https://doi.org/10.1093/nar/gks539 (2012).
Zhang, F. et al. Crystal structure of the obese protein leptin-E100. Nature 387, 206–209. https://doi.org/10.1038/387206a0 (1997).
Israeli, H. et al. Structure reveals the activation mechanism of the MC4 receptor to initiate satiation signaling. Science 372, 808–814. https://doi.org/10.1126/science.abf7958 (2021).
Allot, A. et al. LitVar: A semantic search engine for linking genomic variant data in PubMed and PMC. Nucleic Acids Res. 46, W530–W536. https://doi.org/10.1093/nar/gky355 (2018).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. https://doi.org/10.1038/s41586-020-2308-7 (2020).
Acknowledgements
We thank all participants for their participation in our study and we are further indebted to Sieglinde Düerkop for her excellent technical support. This study was funded by the Deutsche Forschungsgemeinschaft (DFG, HI 865/2-1, DFG Research Unit FOR2488), the BMBF (01GS0820; PALGER 2017-33: 01DH19010) and the Stiftung Universitätsmedizin Essen. We further acknowledge support by the Open Access Publication Fund of the University of Duisburg-Essen.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
BHD, JS, MdZ, WH, SE, SZ, KG, KE, RB, MF, JH and AH recruited participants and provided the samples. LSR, YZ, PFP, JH and AH conceptualized the study. LSR and YZ performed the mutation screen and in silico analyses. LSR, YZ, JA and AH interpreted the data. LSR did the database research and wrote the manuscript. All authors read and approved the submitted version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rajcsanyi, L.S., Zheng, Y., Herpertz-Dahlmann, B. et al. Unexpected identification of obesity-associated mutations in LEP and MC4R genes in patients with anorexia nervosa. Sci Rep 14, 7067 (2024). https://doi.org/10.1038/s41598-024-57517-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-57517-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
