
Abstract
Small ruminant lentiviruses (SRLVs), are grouped in Retroviridae family, remain a significant loss in the small ruminant husbandry. As a result of unavailability of vaccine and effective treatment, the diagnosis plays a crucial role for the control of SRLV infection. However, the major challenge of diagnosis of SRLV infection is the genetic and antigenic variability of the viruses that can lead to a failure in serological detection. This study investigated the circulating strains of the viruses in goats in Thailand and an in-house ELISA was developed. The coding sequences for gag protein were optimized, synthesized, and expressed in Escherichia coli for increasing the sensitivity of ELISA test. A total of 365 serum samples were examined against the recombinant protein in an in-house ELISA. The results showed that the recombinant gag achieves 96.67% sensitivity and 93.18% specificity as compared with the commercially available ELISA test kit.
Similar content being viewed by others
Introduction
Small ruminant lentiviruses (SRLVs) are classified among the genus Lentivirus in Retroviridae family. These viruses can induce persistent infection in sheep and goats. The SRLV genome comprise of two linear positive-sense single-stranded RNA subunits, which compose of gag (group-specific antigens), pol (polymerase), env (envelope), and long terminal repeats (LTRs) genes in addition to a group of regulatory genes. Both single-stranded RNA subunits contain 3 main genes: gag and env codify structural protein while pol codified enzymatic protein1,2,3. SRLV can be categorized into 5 groups: A, B, C, D and E. While genotype A and B, occasionally introduced to as Maedi-visna virus-like and Caprine arthritis encephalitis virus-like, respectively, are widely distributed, whereas genotypes C and E are found in the confined areas. Genotype D was described only pol sequences while phylogenetic analysis on gag sequences of genotype D was classified these sequences into group A3,4. Previous studies have provided evidence supporting the inter-species transmission of viruses between goats and sheep5,6,7,8.
The SRLV infection has been reported worldwide with high variation of prevalence, including in Egypt (8.52%)9, Mexico (0.4%)10, Southern Spain (23.22%)11, India (19.58%)12, Japan (10.0%)13, South Korea (2.73%)14, and Thailand (2.60%)15.
The major route of SRLV transmission is through infected colostrum or milk ingestion or inhalation of the respiratory secretion16. Once SRLVs are transmitted, they preferably infect macrophages and monocytes, where they incorporate their genome into the host genome. By means of the circulation, precursor cells in bone marrow are infected by the circulating infected macrophages and monocytes leading to a life-long infection2. Most infected animals show no clinical manifestation and SRLV infection is mostly in the form of subclinical infection7. However, some infected animals can exhibit pathological manifestations like arthritis, encephalitis, pneumonia, and mastitis17,18.
Currently, there is no effective vaccines or medication available in the prevention or treatment of SRLV infection. Accordingly, the precise diagnosis has an important role to avoid viral dissemination. The most common methods of routine diagnosis are the enzyme-linked immunosorbent assay (ELISA) and the agar gel immunodiffusion (AGID) test. Nonetheless, the delayed seroconversion in some infected animals can result in the false negative from the ELISA19,20.
In addition to serological tests, molecular methods such as PCR were developed for the detection of the viral nucleic acid. However, SRLV infection diagnosis by using molecular methods may be unreliable because of the low quantity of RNA or provirus in the infected animals and the high genetic variability of the viruses1,21. Thus, knowledge about circulating strains of the virus is crucial for disease diagnosis. Nonetheless, there are several reports showed that the combination of PCR and serologic testing is crucial to the development of an effective control program22,23. For the serological methods, the antibody titers can vary throughout the course of infection, so the antibody detection depends upon the sensitivity of the test. Due to the intermittent appearance of antibody, the detection of the viral nucleic acid, such as PCR, prior to seroconversion is essential for a more accurate diagnosis24.
In general, the conserved gene is a favorable candidate for diagnosis. Both gag and pol genes are relatively preserved in SRLV genome, making them optimal sites for PCR primers design. Gag proteins tended to have higher antigenicity. Although SRLV infections have been reported in small ruminants in Thailand for a long time, the studies about the molecular characterization of this virus in goats are limited. In this study, the phylogenetic tree of the gag gene of SRLVs was developed from the sequences obtained from circulating viruses in Thailand and the in-house ELISA based on the results of local strain of SRLVs was developed.
Materials and methods
This study was approved by the Kasetsart University’s Institutional Animal Care and Use Committee. The approved number is ACKU64-VET-007. The authors confirmed that all experiments were performed in accordance with relevant guidelines and regulations of the animal care and use under the Animals for Scientific Purposes Act, B.E. 2558 (2015).
Sample collection and proviral DNA extraction
The samples were taken from whole blood, spleen, and brain of CAE suspected goat. DNA was isolated using a commercially available DNA extraction kit (FavorPrep™ Tissue Genomic DNA Extraction Mini Kit, FAVORGEN Biotech Corporation, Taiwan) according to the manufacturer’s manual. Afterward, nucleic acids were subsequently quantified and verified by the NANODROP 2000 spectrophotometer (ThermoFisher Scientific, USA) and stored at − 20 °C until they were used.
Nested PCR primers
Nested PCR was performed for amplification of proviral DNA from the extracted DNA using the specific primers of the gag gene of SRLVs25. ACTB gene was amplified by using specific primers26 to confirm the quality of DNA samples (Table 1).
General protocol for nested PCR and DNA sequencing
The DNA samples were amplified a fragment of the gag gene by using nested PCR. The target fragment amplifications were carried out with a final volume of 20 μL containing 0.4 U of DNA polymerase (Phusion™ High–Fidelity DNA Polymerase), 200 μM of each dNTP, and 0.5 μM of each primer. The following PCR conditions were applied: initial denaturation at 98 °C for 1 min, 40 cycles of denaturation at 98 °C for 15 s, annealing at 58 °C for 30 s, and extension at 72 °C for 1 min, and a final extension at 72 °C for 10 min. For the second round, 2 μL of product from the first round was added as a DNA template, and the nested round of PCRs were performed in the same condition. Then, products were visualized on 1.5% agarose gel containing RedSafe™ Nucleic Acid Staining Solution (iNtRON Biotechnology, Gyeonggi-do, Korea). The nested PCR positive samples in agarose gel were purified by using FavorPrep™ GEL/PCR Purification Kit (FAVORGEN Biotech Corporation, Taiwan) and sequenced using the Sanger sequencing method (Sequencer ABI3730XL, Bionics Co., Ltd., South Korea).
Multiple sequence alignment and phylogenetic tree construction
Multiple alignments of the partial gag gene were generated. Phylogenetic construction was accomplished by aligning the newly obtained sequences with the GenBank reference sequences using the Neighbour-Joining (NJ method) with the Tamura-Nei gamma distance achieved in MEGA version X27.
Expression of recombinant protein
The partial part of the gag gene of 2 genotypes of SRLVs (KU-003 and KU-009) was selected, optimized, synthesized, and cloned into pET-28b ( +) vectors at a specific restriction site (HindIII and NotI) and transformed into BL21 (DE3) competent E. coli. The transformed E. coli was spread on the 25 μg/ml kanamycin supplemented LB agar and incubated at 37 °C. Then, a colony was picked up and grown with 180 rpm shake in kanamycin supplemented LB broth at 37 °C. The culture was streaked on LB agar and incubated overnight at 37 °C. The single colony on LB media were confirmed the existence of gag gene by using PCR. The colony which contains the target gene was selected and subsequently cultivated in kanamycin-supplemented LB broth at 37 °C with agitation at 180 rpm overnight. Afterwards, the 100 μl of overnight culture were added into 5 ml of the same antibiotic supplemented LB broth and shaken for 3 h (or until an O.D. of 0.5 was reached at 600 nm). The induction of protein production was done using IPTG to a final concentration of 0.1 μM and incubated with agitation at 180 rpm for 4 h at 37 °C. The cells were taken using 10000 × g centrifugation for 5 min at 4 °C and underwent protein purification.
Recombinant protein purification
The recombinant protein tagged with histidine was purified by an anti-His-tag immune-affinity chromatography (ÄKTA™ start protein purification system, GE Healthcare Bio-Sciences AB) in accordance with the manufacturer’s manual.
Western blot analysis of recombinant protein
The western blotting was accomplished to figure out the immunoreactivities of the recombinant protein. After SDS-PAGE, the recombinant protein was transferred to a nitrocellulose membrane. The protein-transferred-membrane was blocked using blocking buffer (10% horse serum and 5% skimmed milk in PBS) for 60 min at 37 °C. Then, the membrane was incubated with positive SRLV goat serum diluted 1: 500 in primary antibody buffer (10% horse serum, 5% skimmed milk, and 10% E. coli lysate) for 1 h at 37 °C. After 3 times of washing process with 0.05% Tween 20 in PBS for 3 min, the membrane was incubated with the rabbit anti-goat IgG (H + L) (KPL Peroxidase Labeled Affinity Purified Antibody to Goat IgG (H + L) [Rabbit]) diluted 1: 1000 in secondary antibody buffer (10% horse serum and 1% skimmed milk in PBS) at 37 °C for 60 min. Then, the membrane was washed 3 times for 5 min. The recombinant protein was detected using TMB substrate as chromogenic substrate.
Samples for enzyme-linked immunosorbent assay (ELISA) development
In this study, the serum samples were obtained from 365 goats. All samples were divided by using a commercial ELISA test kit (IDEXX CAEV/MVV total Ab test®, IDEXX Laboratories, Inc., Maine, USA) into 2 groups: the positive samples and negative samples.
ELISA general protocol
100 μl of 12.5 μg/ml SRLV-gag protein in coating buffer (1.5% Na2CO3 and 2.93% NaHCO3 in distilled water) was coated into 96-well ELISA plates (Nunc-Immuno™ MicroWell™ 96 well solid plates) at 37 °C for 1 h. The coated plates were then washed with 300 μl of wash buffer (0.05% Tween-20 in PBS) for 5 times by means of an automatic plate washer (Tecan HydroSpeed™ plate washer). Then they were blocked by adding 100 μl/well of blocking buffer (10% horse serum and 5% skimmed milk in PBS) for 30 min at 37 °C, then, the blocking buffer was discarded. The samples were diluted (1: 40, based on the result from checkerboard analysis) in dilution buffer (10% horse serum, 5% skimmed milk, and 10% E. coli lysate in PBS). The 100 μl/well of diluted samples were added to the blocked plates and incubated for 30 min at 37 °C. Then, five-time washing was done as described above. Afterward, 100 μl/well of 1: 25 (based on the result of checkerboard analysis) diluted rabbit anti-goat IgG (H + L) in secondary antibody buffer was added to the well and incubated for 30 min at 37 °C. Thereafter, 5 times of washing procedure as described were done. 100 μl of KPL TMB microwell peroxidase substrate (SureBlue™ TMB 1-Component Microwell Peroxidase Substrate) was added to each well and then incubated for 15 min at room temperature. The reaction was stopped by means of 100 μl of the stop solution (0.25 M HCl). The optical density (O.D.) was measured at 450 nm.
ELISA optimization
To define the optimal concentration of recombinant protein for coating on the wells, the titration checkerboard was performed. The concentration of the purified antigen was 0.5 mg/ml. The plates were coated with 100 μl of two-fold serial dilution of recombinant gag protein and incubated for 1 h at 37 °C. Washing of the coated plates was performed 5 times using wash buffer. Then, the blocking buffer (100 μl) was added to each well for 30 min at 37 °C. The two-fold serial diluted serum samples in dilution buffer were added to the blocked plates and incubated for 30 min at 37 °C. Then, the general protocol of ELISA was done as described above. The 334 negative samples and 31 positive samples were tested, and the results were used for sensitivity and specificity calculation.
Statistical analyses
Statistical calculations were done in Stata statistical software (Release 16, StataCorp, College Station, TX). The optimal cut off value, sensitivity, and specificity for in-house ELISA were established. The expected test performance was evaluated using the receiver operating characteristic (ROC) curve. The area under the ROC curve was obtained for accuracy evaluation. The degree of agreement between the results of commercial ELISA test kit (IDEXX CAEV/MVV total Ab test®, IDEXX Laboratories, Inc., Maine, USA) and developed in-house ELISA was measured using Cohen’s Kappa statistic, which the degree of agreement was interpreted according to Landis and Koch28.
Ethics approval
The experimental protocol of this study was conducted under the ethical approval of Kasetsart University’s Institutional Animal Care and Use Committee. The approved number is ACKU64-VET-007. The authors confirm that the ethical policies of the journal, as noted on the journal’s author guidelines page, have been adhered to and the appropriate ethical review committee approval has been received. The study was also reported according to the ARRIVE guidelines.
Results
Phylogenetic tree construction of SRLVs based on gag fragment
The nested PCR of the partial gag gene was carried out. The results of nested PCR were shown in Fig. 1. There were 11 samples showed positive results and all positive samples were sequenced. The DNA obtained from KU-002 was isolated from brain tissue whereas the DNA from KU-001, KU-003 to KU-011 were extracted from spleen. The sequences of SRLV isolates were analyzed in comparison with reference sequences retrieved from databases. The strain of SRLVs circulating in Thailand is B2. A phylogenetic tree was shown in Fig. 2. The phylogenetic tree showed the sequences reported in this study are different from the Thai sequences reported previously. The similarities of nucleotide sequences range between 84.81% and 88.76%, whereas the similarities of amino acid sequences are 82.01–88.65% in comparison to MH827520.1 which was reported in 2018.

Result of nested PCR of shows the band of viral nucleic acid which the target size is ~ 1300 bps. NTC stand for no template control.

Neighbor-Joining (NJ) phylogenetic tree based on partial gag gene sequences showing genetic relationship between the Thai SRLV isolates obtained in this study and other strains. The analysis involved a total of 550 base pairs from 40 nucleotide sequences. The sequences reported in this study were different from Thai sequences reported in 2018 (MH827520.1, MH827521.1). The numbers displayed on the node represent the percentage of bootstrap values obtained from 1000 replicates. The lengths of the branches indicate the number of substitutions per site. The nucleotide sequences exhibit similarities ranging from 84.81% to 88.76% compared to the reference sequence (MH827520.1) which was previously reported in Thailand. The sequences found in this study are similar to B1 group which differs from the previous report that closer to B2 group.
Western blot analysis
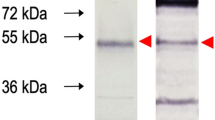
The western blotting was achieved for the detection of an immunological reaction between natural antibodies and recombinant protein which the size around 28 kDa. The results were shown in Fig. 3. A positive serum sample was able to bind the recombinant gag protein while a negative serum sample was unable to bind the protein.

Results of western blotting assay of recombinant gag protein of SRLVs; (a) displayed a specific band upon utilizing the anti-histidine antibody as the primary antibody, indicating the protein expression is not inhibited. (b) a specific band was observed when confirmed positive serum was used as the primary antibody. This indicated the capacity of the natural antibody to effectively bind to the recombinant gag protein. (c) the negative result when negative serum was used as the primary antibody.
ELISA optimization and evaluation
Checkerboard analysis result
Eight concentrations of the antigen were tested. Two-fold dilutions of 0.5 mg/ml of antigen were tested. The optimal coating concentration was 12.5 μg/ml. While the two-fold dilutions of the detecting antibody were tested. The suitable dilution of the detecting antibody was 1:25 in the primary antibody buffer.
Statistical analysis result
The receiver operating characteristic (ROC) curve with area under the curve (0.9856) is shown in Fig. 4. The developed in-house ELISA can be considered as “excellent” according to the AUC29. The optimal cut off value is 0.321 with 96.67% sensitivity and 93.18% specificity. The Cohen’s Kappa statistic, is 0.66 (95% confidence interval = 0.55–0.78), indicated that developed in-house ELISA showed “substantial” agreement with commercial ELISA test kit (IDEXX CAEV/MVV total Ab test®, IDEXX Laboratories, Inc., Maine, USA).

(a) The receiver operating characteristic (ROC) curve with area under the curve, (b) the relationship between sensitivity, specificity and O.D.
Discussion
Small ruminant lentiviruses (SRLVs) are known to induce chronic progressive inflammation in small ruminants. Despite posing a significant challenge due to the lack of an effective vaccine and specific treatment, disease control heavily relies on diagnosis. Nowadays, the gold standard for SRLV infection diagnosis remains undetermined30. However, the serological investigation could decrease the prevalence of SRLV infection in goats15,31,32. The ELISAs are appropriate for the large numbers of samples screening. The antigen detection methods such as conventional PCR, nested PCR, or qPCR have been designed to detect SRLVs in seronegative animals. However, the low quantity of viral load among infected animals and the antigenic variation of the virus can lead to failure of either antigen or antibody detection. As shown in the phylogenetic tree (Fig. 2), the sequences of partial gag gene of the virus reported in this study were different from Thai sequences reported previously in 2018. Since the high mutation rate of retroviruses, the phylogenetic data should be employed to select the proper epitopes from the circulating genotypes of the virus for use in ELISAs. The gag protein is the choice protein for the development of immunoassays for antibody detection due to its conservative and antigenic properties of this protein33,34. The recombinant gag polypeptide produced in this study was expressed in the soluble fraction. The hydrophilic property causes the polypeptide to be easily recoverable from the culture system without denaturing agents. Absence of denaturing agents while purification processes could reduce the misfolding of the polypeptide which may cause the false negative results.
Use of the combination of different genotypes of SRLV in the same ELISA well can improve the detection throughout the course of infection, as compared to using of a commercial ELISA test kit35. The use of combination recombinant protein derived from various strains within the same well can improve the sensitivity of ELISA. This was attributed to their capability to detect diverse strain of virus. In this study, two genotypes of the partial gag gene of SRLVs were selected and coated into ELISA wells for reducing the false negative result. This notion was based on the identification of the most hydrophobic segment within the gag gene from a total of 11 samples, which were subsequently categorized into two distinct groups. The result from the ELISA test developed using recombinant gag protein achieved a specificity and sensitivity of 93.18% and 96.67%, respectively, compared to a commercial test kit. This indicates that the test could be effectively applied for SRLV infection diagnosis in goats.
Conclusions
The phylogenetic analysis of SRLVs shows that the circulating viruses found in this study have significantly changed their nucleic acid sequences from the previous report. The indirect ELISA developed with the partial part of gag protein based on circulating strain of SRLVs as an antigen for detection of anti-SRLV IgG has high sensitivity (96.67%) and specificity (93.18%).
Data availability
All data collected and analyzed in this research are included in the article.
References
de Azevedo, D. A. A. et al. Molecular characterization of circulating strains of small ruminant lentiviruses in Brazil based on complete gag and pol genes. Small Rumin. Res. 177, 160–166. https://doi.org/10.1016/j.smallrumres.2019.06.011 (2019).
Blacklaws, B. A. Small ruminant lentiviruses: Immunopathogenesis of visna-maedi and caprine arthritis and encephalitis virus. Comp. Immunol. Microbiol. Infect. Dis. 35, 259–269. https://doi.org/10.1016/j.cimid.2011.12.003 (2012).
Ramirez, H., Reina, R., Amorena, B., de Andres, D. & Martinez, H. A. Small ruminant lentiviruses: Genetic variability, tropism and diagnosis. Viruses 5, 1175–1207. https://doi.org/10.3390/v5041175 (2013).
Grego, E. et al. Viral load, tissue distribution and histopathological lesions in goats naturally and experimentally infected with the small ruminant lentivirus genotype E (subtype E1 Roccaverano strain). Res. Vet. Sci. 118, 107–114. https://doi.org/10.1016/j.rvsc.2018.01.008 (2018).
Leroux, C., Chastang, J., Greenland, T. & Mornex, J. F. Genomic heterogeneity of small ruminant lentiviruses: Existenceof heterogeneous populations in sheep and of the samelentiviral genotypes in sheep and goats. Arch. Virol. 142, 1125–1137. https://doi.org/10.1007/s007050050147 (1997).
Leroux, C., Vuillermoz, S., Mornex, J.-F. & Greenland, T. Genomic heterogeneity in the pol region of ovine lentiviruses obtained from bronchoalveolar cells of infected sheep from France. J. Gen. Virol. 76, 1533–1537. https://doi.org/10.1099/0022-1317-76-6-1533 (1995).
Leroux, C., Cruz, J. C. M. & Mornex, J. F. SRLV: A genetic continuum of lentiviral species in sheep and goats with cumulative evidence of cross species transmission. Curr. HIV Res. 8, 94–100 (2010).
Shah, C. et al. Phylogenetic analysis and reclassification of caprine and ovine lentiviruses based on 104 new isolates: Evidence for regular sheep-to-goat transmission and worldwide propagation through livestock trade. Virology 319, 12–26. https://doi.org/10.1016/j.virol.2003.09.047 (2004).
Baraka, E. et al. Sero-epidemiological study on Caprine arthritis-encephalitis virus infection in goats in two localities in Egypt. Alex. J. Vet. Sci. 59, 68. https://doi.org/10.5455/ajvs.8048 (2018).
Torres-Acosta, J. F. J. et al. Serological survey of caprine arthritis-encephalitis virus in 83 goat herds of Yucatan, Mexico. Small Rumin. Res. 49, 207–211. https://doi.org/10.1016/s0921-4488(03)00093-2 (2003).
Barrero Dominguez, B. et al. Seroprevalence and risk factors of exposure to caprine arthritis-encephalitis virus in southern Spain. Vet. Rec. 180, 226. https://doi.org/10.1136/vr.104014 (2017).
Kumar, K. et al. Detection and immune cell response of natural maedi visna virus (MVV) infection in Indian sheep and goats. Microb. Pathog. 165, 105467. https://doi.org/10.1016/j.micpath.2022.105467 (2022).
Konishi, M. et al. Serological survey of caprine arthritis-encephalitis virus infection in Japan. J. Vet. Med. Sci. 78, 447–450. https://doi.org/10.1292/jvms.15-0357 (2016).
Oem, J. K. et al. Large-scale serological survey of caprine arthritis-encephalitis virus (CAEV) in Korean black goats (Capra hircus aegagrus). J. Vet. Med. Sci. 74, 1657–1659. https://doi.org/10.1292/jvms.12-0103 (2012).
Mongkonwattanaporn, T., Lertwatcharasarakul, P., Intaravichai, P. & Rukkwamsuk, T. Seroprevalence of small ruminant lentivirus infection in goats in Thailand. Pol. J. Vet. Sci. 24, 313–317. https://doi.org/10.24425/pjvs.2021.137668 (2021).
Blacklaws, B. A. et al. Transmission of small ruminant lentiviruses. Vet. Microbiol. 101, 199–208. https://doi.org/10.1016/j.vetmic.2004.04.006 (2004).
Minguijon, E. et al. Small ruminant lentivirus infections and diseases. Vet. Microbiol. 181, 75–89. https://doi.org/10.1016/j.vetmic.2015.08.007 (2015).
Smith, M. C. & Sherman, D. M. Goat Medicine 2nd edn. (John Wiley and Sons Ltd, 2009).
Michiels, R. et al. Comparative analysis of different serological and molecular tests for the detection of small ruminant lentiviruses (SRLVs) in Belgian sheep and goats. Viruses 10, 696. https://doi.org/10.3390/v10120696 (2018).
Rimstad, E. et al. Delayed seroconversion following naturally acquired caprine arthritis-encephalitis virus infection in goats. Am. J. Vet. Res. 54, 1858–1862 (1993).
Zhang, Z., Watt, N. J., Hopkins, J., Harkiss, G. & Woodall, C. J. Quantitative analysis of maedi-visna virus DNA load in peripheral blood monocytes and alveolar macrophages. J. Virol. Methods 86, 13–20. https://doi.org/10.1016/S0166-0934(99)00169-X (2000).
Brinkhof, J. M., Moll, L., van Maanen, C. & Houwers, D. J. Use of serology and polymerase chain reaction for the rapid eradication of small ruminant lentivirus infections from a sheep flock: A case report. Res. Vet. Sci. 88, 41–43. https://doi.org/10.1016/j.rvsc.2009.05.014 (2010).
Konishi, M. et al. Combined eradication strategy for CAE in a dairy goat farm in Japan. Small Rumin. Res. 99, 65–71. https://doi.org/10.1016/j.smallrumres.2011.03.051 (2011).
Dolfini, T., Conrad, L. F., Flores, I. V. C. & Ravazzolo, A. P. Comparison of primer pairs: Greater degeneracy improves small ruminant lentivirus (SRLVs) detection by seminested PCR. Small Rumin. Res. 123, 189–192. https://doi.org/10.1016/j.smallrumres.2014.10.007 (2015).
L’Homme, Y. et al. Molecular characterization and phylogenetic analysis of small ruminant lentiviruses isolated from Canadian sheep and goats. Virol. J. https://doi.org/10.1186/1743-422X-8-271 (2011).
Zhang, Y. et al. Reference gene screening for analyzing gene expression across goat tissue. Asian-Australas. J. Anim. Sci. 26, 1665–1671. https://doi.org/10.5713/ajas.2013.13199 (2013).
Tamura, K. & Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 10, 512–526. https://doi.org/10.1093/oxfordjournals.molbev.a040023 (1993).
Landis, J. R. & Koch, G. G. The measurement of observer agreement for categorical data. Biometrics 33, 159–174. https://doi.org/10.2307/2529310 (1977).
Li, F. & He, H. Assessing the accuracy of diagnostic tests. Shanghai Arch. Psychiatry 30, 207–212. https://doi.org/10.11919/j.issn.1002-0829.218052 (2018).
Panneum, S. & Rukkwamsuk, T. Diagnosis of caprine arthritis encephalitis virus infection in dairy goats by ELISA, PCR and viral culture. Pol. J. Vet. Sci. 20, 347–353. https://doi.org/10.1515/pjvs-2017-0042 (2017).
Peterhans, E. et al. Routes of transmission and consequences of small ruminant lentiviruses (SRLVs) infection and eradication schemes. Vet. Res. 35, 257–274. https://doi.org/10.1051/vetres:2004014 (2004).
Tavella, A. et al. Achievements of an eradication programme against caprine arthritis encephalitis virus in South Tyrol, Italy. Vet. Rec. 182, 51. https://doi.org/10.1136/vr.104503 (2018).
Lacerenza, D., Genovese, F., Profiti, M., Nucera, D. & Rosati, S. Characterization of an immunodominant epitope of small ruminant lentivirus (SRLV) nucleoprotein. Vet. Immunol. Immunopathol. 125, 361–367. https://doi.org/10.1016/j.vetimm.2008.05.013 (2008).
Nenci, C. et al. Vaccination with a T-cell-priming Gag peptide of caprine arthritis encephalitis virus enhances virus replication transiently in vivo. J. Gen. Virol. 88, 1589–1593. https://doi.org/10.1099/vir.0.82800-0 (2007).
Sanjose, L. et al. Diagnosing infection with small ruminant lentiviruses of genotypes A and B by combining synthetic peptides in ELISA. Vet. J. 204, 88–93. https://doi.org/10.1016/j.tvjl.2015.01.012 (2015).
Acknowledgements
This study was financially supported by the joint funding between the National Research Council of Thailand for Royal Golden Jubilee Ph.D. Programme, (RGJ.PHD/0140/2561) and Kasetsart University.
Author information
Authors and Affiliations
Contributions
T.R. supervised the project. T.M. and T.R. formulated the conceptual framework of the study and the hypothesis being tested. T.M. conducted the experiment. T.M. and P.L. performed the sample analysis. T.M. and T.R. analyzed the data. T.M. prepared the draft of manuscript. P.L. and T.R. edited the manuscript and all authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mongkonwattanaporn, T., Lertwatcharasarakul, P. & Rukkwamsuk, T. Development of in-house ELISA based on recombinant gag proteins of small ruminant lentiviruses isolated from goats in Thailand. Sci Rep 14, 3636 (2024). https://doi.org/10.1038/s41598-024-54360-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-54360-x
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
