
Abstract
The anthropogenic impact on wildlife is ever increasing. With shrinking habitats, wild populations are being pushed to co-exist in proximity to humans leading to an increased threat of infectious diseases. Therefore, understanding the immune system of a species is key to assess its resilience in a changing environment. The innate immune system (IIS) is the body’s first line of defense against pathogens. High variability in IIS genes, like toll-like receptor (TLR) genes, appears to be associated with resistance to infectious diseases. However, few studies have investigated diversity in TLR genes in vulnerable species for conservation. Large predators are threatened globally including leopards and cheetahs, both listed as 'vulnerable' by IUCN. To examine IIS diversity in these sympatric species, we used next-generation-sequencing to compare selected TLR genes in African leopards and cheetahs. Despite differences, both species show some TLR haplotype similarity. Historic cheetahs from all subspecies exhibit greater genetic diversity than modern Southern African cheetahs. The diversity in investigated TLR genes is lower in modern Southern African cheetahs than in African leopards. Compared to historic cheetah data and other subspecies, a more recent population decline might explain the observed genetic impoverishment of TLR genes in modern Southern African cheetahs. However, this may not yet impact the health of this cheetah subspecies.
Similar content being viewed by others

Introduction
The innate immune system (IIS) is the genetically predetermined response to foreign substances in most multicellular organisms1. As the body’s first line of defense, its ability to recognize pathogens is crucial to initiate countermeasures and induce defensive reactions2. Nevertheless, foreign substances are also detected by the adaptive immune system (AIS), e.g., by T cell antigen receptors, but this defense mechanism is more specific and only found in jawed vertebrates3. Among IIS-specific receptors that recognize conserved patterns on microorganisms are the toll-like receptors (TLRs)4. TLRs are type I transmembrane proteins either located on the cell surface or within the cell compartment and are grouped by their protein sequence similarity. Structurally, they consist of a leucine-repeat rich ectodomain (LRR), which detects pathogen-associated molecular patterns (PAMPs), a transmembrane domain, and a cytoplasmic domain Toll-IL1 receptor (TIR) that initiates the intracellular signaling cascade5. In mammals, there are at least 13 members of the TLR protein family, each with a designated role in recognizing specific pathogens6. Most TLRs detect bacterial components with a varying specificity to different ligands, e.g., TLR1 and TLR6 respond to different lipopeptides of the bacterial lipoprotein7, while TLR4 in particular recognizes lipopolysaccharides in Gram-negative bacteria4. Most TLRs located within the cell compartment respond to foreign nucleotides, and thus can detect viruses and parthenogenic protozoans8,9. Therefore, TLRs are essential in signal amplification, induce protein trafficking pathways to trigger inflammatory responses and induct the adaptive immune system (AIS)10.
Generally, a species’ resilience against infectious diseases is linked to high genetic diversity11,12. Correspondingly, genes associated with the immune system are considered to be among the most polymorphic due to adaptive evolution13,14,15. In contrast to major parts of the AIS, standing genetic variations is the only source of diversity for the IIS2. It is assumed that the number of polymorphism in innate immunity receptors affects a species’ ability to adapt to future environmental changes16,17. Especially the variability in TLR genes seems to be connected to resistance against infectious diseases because increased variation enhances the potential for binding a larger variety of PAMPs17,18. Though important, only few studies have investigated diversity in TLR genes in non-model organisms and drawn conclusions for conservation19,20,21 while often lacking comparisons to a related species. Studies on felids are especially rare and either focus exclusively on the domestic cat (Felis catus)22,23,24 or are limited by their extent if other species are included25. However, a species’ immune fitness can only be drawn from the context, as a detached evaluation of genetic diversity does not easily allow meaningful conclusions26.
Throughout Africa, many essential niches, especially in the case of large carnivores, are occupied by big cats such as lions (Panthera leo), leopards (Panthera pardus), and cheetahs (Acinonyx jubatus), which serve important ecosystem functions27,28. Savannas, in particular, profit from those apex predators because the trophic impact exacted by carnivores strongly shapes and maintains an ecosystem’s balance29,30,31. In Sub-Saharan Africa, cheetahs and leopards inhabit similar open habitats and often share a sympatric distribution32,33. Their coexistence is a result of niche partitioning. While leopards are opportunistic hunters of larger prey, cheetahs are specialized in small fast-running antelopes and lagomorphs34,35. Due to increasing anthropogenic pressure they frequently occur near settlements, with leopards more prone to direct human contact36,37 while cheetahs rather interact with livestock38,39. Nevertheless, both species occasionally hunt stock animals40,41,42 and therefore, face more active persecution than, e.g., lions which rarely occur outside protected areas43. As a direct consequence of this ever-growing human-wildlife conflict and other factors such as habitat destruction, leopard and cheetah are listed as ‘vulnerable’ on the International Union for Conservation of Nature (IUCN) Red List of Threatened Species44. Still, the conservation status of different subspecies varies greatly and negatively correlates with increasing human impact45,46. Three of the nine leopard subspecies and two of the five classical cheetah subspecies are considered ‘critically endangered’44,47,48. With fewer than 50 individuals in the wild, the Asiatic cheetah (A. j. venaticus) is already facing extinction49,50.
Beyond these threats, however, proximity to humans poses a less apparent danger: infectious diseases. These pathogens, either transferred by vectors or directly by domestic animals like feral cats, further pressure wild populations51,52. Because of a similar solitary lifestyle, it can be assumed that infectious diseases might affect both species similarly. However, leopards and cheetahs are less similar on the genetic diversity level. Especially the African leopard is known to be among the most genetically diverse big cat species, with high heterozygosity and no apparent structure despite its decreasing populations53. On the contrary, cheetahs are classically portrayed as very homogeneous and overall genetically impoverished54,55 due to a proposed bottleneck event 10,000 years ago56,57. Nonetheless, recent studies observe more diversity than previously thought, e.g., for immune gene loci such as the major histocompatibility complex (MHC)58,59. Yet, cheetahs show strong geographical differentiation47,60,61 as well as low genome-wide diversity61. Here, we compare TLR2, TLR4, TLR6 and TLR8 of modern Southern African cheetahs (A. j. jubatus; n = 49) to sympatric occurring African leopards (P. p. pardus; n = 41) and incorporate historic cheetah samples and samples of different cheetah subspecies (A. j. ssp.: n = 15) into our study as a temporal-spatial reference. All four TLRs genes play a major role in the initial detection and defense against infectious diseases by primarily detecting bacterial (TLR2, TLR4, TLR6) and viral (TLR8) PAMPs. Therefore, their underlying genetic diversity can be an indication of a species resilience against emerging infectious threats.
Results
Despite successful preliminary testing of all five primer sets of our target TLR exons using three modern cheetah samples, we excluded exon 1 of TLR4, which failed in subsequent amplifications in most samples. From our 61 modern Southern African cheetah samples (Supplementary Table 1) we discarded 12 samples that failed initial amplification. All results are based on exon data of TLR2, TLR4.2, TLR6, and TLR8, deposited in the Phaidra public repository (https://doi.org/https://doi.org/10.34876/9KFD-2A38).
Genetic diversity in TLR exons of African leopards and cheetahs
All investigated TLR exons showed different haplotype diversity between African leopards and cheetahs varying in total nucleotide allele count, resulting amino acid sequences, phylogenetic structure and diversity (Table 1, Figs. 1, 2, 3, 4).

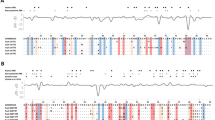
Maximum-likelihood phylogeny of the TLR2 exon sequences (a) and the resulting amino acid sequences (b) for leopard (blue framed) and cheetah (orange framed), faded tip labels indicate nucleotide alleles/resulting amino acid sequences only occurring in historic samples. The scale indicates the number of substitutions per side. Spotted hyena (Crocuta Crocuta, aaCrCr) and striped hyena (Hyaena hyaena, aaHyHy) were used as an outgroup.

Maximum-likelihood phylogeny of the TLR4.2 exon sequences (a) and the resulting amino acid sequences (b) for leopard (blue framed) and cheetah (orange framed), faded tip labels indicate nucleotide alleles/resulting amino acid sequences only occurring in historic samples. The scale indicates the number of substitutions per side. Spotted hyena (Crocuta Crocuta, aaCrCr) and striped hyena (Hyaena hyaena, aaHyHy) were used as an outgroup.

Maximum-likelihood phylogeny of the TLR6 exon sequences (a) and the resulting amino acid sequences (b) for leopard (blue framed) and cheetah (orange framed), faded tip labels indicate nucleotide alleles/resulting amino acid sequences only occurring in historic samples. Crossed-out tip labels indicate likely functionless amino acid sequences due to deletions resulting in preliminary stop codons. The scale indicates the number of substitutions per side. Spotted hyena (Crocuta Crocuta, aaCrCr) and striped hyena (Hyaena hyaena, aaHyHy) were used as an outgroup.

Maximum-likelihood phylogeny of the TLR8 exon sequences (a) and the resulting amino acid sequences (b) for leopard (blue framed) and cheetah (orange framed), faded tip labels indicate nucleotide alleles/resulting amino acid sequences only occurring in historic samples. The scale indicates the number of substitutions per side. Spotted hyena (Crocuta Crocuta, aaCrCr) and striped hyena (Hyaena hyaena, aaHyHy) were used as an outgroup.
Although fewer samples were available for the comparison, African leopards exhibited higher diversity (0.8617–0.9996) in the investigated TLR exons on the nucleotide level than cheetahs (0–0.9606). However, the diversity on the nucleotide level did not necessarily result in a higher amino acid diversity, e.g., the 23 nucleotide alleles for exon TLR4.2 (Table 1, Fig. 2) in the African leopards resulted in just four different amino acid sequences compared to the six nucleotide alleles in cheetahs that also resulted in four amino acid sequences.
The proportion of synonymous nucleotide alleles varied between TLR exons and species. The highest number of synonymous alleles were found in exon TLR4.2 for African leopards (Fig. 2) and exon TLR8 for cheetahs (Fig. 4). Furthermore, TLR8 was the least diverse exon in both species. Interestingly, TLR6 showed nucleotide alleles with a frameshift mutation in both leopards (two base pair deletion) and cheetahs (four base pair deletion) causing early stop codons (Fig. 3). We created three-dimensional models for the shortened polypeptides for both species using AlphaFold262 to visualize the structural alteration. As per-residue confidence estimates, pLDDT (predicted Local Distance Difference Test) scores were extracted and included in the highest ranked structure (Supplementary Fig. 1). Homozygote individuals regarding the deletion in question occurred in both species and were likely unable to synthesize the full-length protein.
Genetic diversity in TLR exons of historic cheetah samples
Historic cheetah samples showed additional nucleotide alleles (Figs. 1, 2, 3, 4) and historic haplotype diversity was always higher compared to modern Southern African cheetahs (Table 1). While no structure within the cheetah was apparent, unique alleles were found in all historic samples irrespective of their subspecies (Supplementary Table 2). In some cases, however, insufficient read coverage after mapping did not allow for accurate variant calling, and samples with less than 6 × coverage were removed from subsequent analyses. Especially, statements about historic diversity in TLR8 must be treated with caution, since less than half of all historic samples passed variant calling due to insufficient coverage.
Phylogenetic structure of TLR2, TLR4, TLR6 and TLR8 in African leopards and cheetahs
Both species’ TLRs were phylogenetically separated at the nucleotide level and, to a lesser degree, at the amino acid level. In the case of TLR6, cheetah amino acid sequences were nested within sequences from African leopards (Fig. 3). While more basal nodes separating species were mostly well supported (bootstrap support > 80), internal nodes (within species) were predominantly uncertain, resulting in poorly resolved topologies (bootstrap support < 50).
Episodic positive selection
In general, few sites of selection were detected (Supplementary Table 3). The codon-based analysis of selection signals in MEME only revealed four sites showing positive diversifying selection in African leopards and none in cheetahs. On the other hand, FEL indicated slightly more sites under positive diversifying selection in both species, notably in TLR6 (13 sites in leopards, 6 sites in cheetahs). Every TLR showed sites under purifying selection ranging from 5 sites in TLR4.2 to 22 sites in TLR6 in leopards, and from 1 site in TLR4.2 to 23 sites in TLR6 in cheetahs.
Discussion
Resources for conservation are limited and studies on endangered species must meaningfully evaluate the standing genetic diversity. High levels of polymorphism in immune genes, especially in IIS-genes, directly affect a species’ ability to react to and counter infectious diseases. Consequently, a species’ resilience against diverse pathogens and its ability to resist future environmental changes are directly linked to genetic diversity. Leopards and cheetahs are threatened by human activity and several subspecies already face the consequences of genetic impoverishment and small population sizes50,63. Surviving in the Anthropocene means coping with strongly altered environments and living close to humans is inevitable, which increases the exposure risk to pathogens from life stock and domestic pets. To identify populations of concern, evaluating a species’ innate immunity is key to understanding its resilience to environmental change and maximizing conservation efforts. Within this study, we shed some light on the still poorly understood innate immunity of two large carnivores of the African Savana, African leopards and Southern African cheetahs, and provide a first assessment of genetic diversity in four selected TLR exons.
Leopards and cheetahs possess different levels of genetic diversity in the investigated TLR exons. Yet, they share some structural and haplotypic similarities. In direct comparison, modern African leopard’s TLR genes are much more diverse than those of modern Southern African cheetahs. This is especially true for TLR2 and TLR8; e. g., modern African leopards exhibit 32 different nucleotide alleles in TLR8, while modern Southern African cheetahs only exhibit one allele and historic cheetah only two (Table 1). Both species show comparatively higher variability in TLR2 and TLR6, which are known to form heterodimers essential for recognizing a large variety of bacterial PAMPs7,64. Leopards consistently showed higher variability than cheetahs. Interestingly, several TLR6 alleles in modern leopards and cheetahs showed reading frame shifts. Correspondingly, shortened polypeptides will be synthesized (Supplementary Fig. 1), similar to an already known reading frame shift in TLR265, indicating a potential loss of function. In addition to TLR2-TLR6-heterodimers, TLR2 can also dimerize with TLR1, resulting in almost identical signal pathways, but also in slightly altered PAMP recognition66. Therefore, a potential loss of function in TLR6 might be compensated by increased diversity in TLR1 and should be considered in future studies. Both of our modern sample sets of Southern African cheetahs and African leopards included homozygote individuals that possessed those frame shift mutations in TLR6, which might indicate that deleterious alleles are already abundant in both species. Notably, similar deleterious alleles are absent in our historic cheetah sample set, which could indicate a recent accumulation of harmful mutations in TLR6 in modern Southern African cheetahs. Unfortunately, we lack historic data for comparison in African leopards. In addition, TLR6 was the only TLR that showed some sites under positive diversifying selection in both species and a more extensive historic sample set might help to understand its evolutionary trend in the future.
In general, our historic cheetah samples are more heterozygous compared to modern Southern African cheetahs. This holds true even excluding other subspecies, the three historic Southern African cheetahs were more heterozygous (TLR2, TLR6) and exhibited more unique haplotypes (e.g., 3 vs. 2 in TLR2; 6 vs. 3 in TLR6) than our 49 contemporary samples (Table 1). We are aware that Illumina short-read data is not directly comparable to PacBio long-read data and variant calling from short reads is more difficult due to the additional phasing. Therefore, we discarded samples with less than 6× coverage and manually verified each allele. Still, we have identified new unique loci present in different subspecies of historic cheetahs that are absent in modern South African individuals.
In comparison, TLR4.2 and TLR8 seem to be less diverse than the other exons of TLR2 and TLR6 in historic and modern cheetahs. This supports previous studies that identified TLR4 and TLR8 as more conserved among non-closely related species67,68. However, as both TLR exons were more difficult to amplify in modern samples and also less covered in the historic samples, we would need to investigate more individuals to understand the functional evolution of TLR4 and TLR8 in the species.
Historic cheetah samples exhibited consistently lower haplotype diversity in the investigated TLR genes compared to modern African leopards, but higher than modern Southern African cheetah. Therefore, loss in genetic diversity and low heterozygosity in contemporary Southern African cheetahs could be more recent and the result of a population decline within the last 150 years. Even if only this cheetah subspecies is considered, the historic genetic diversity is visible higher. It is known that increasing human activity in Africa in the previous century reduced the numbers of large carnivores drastically69,70, and both leopards and cheetahs became extinct in large parts of their former ranges44,48. However, the African leopard appeared to be highly diverse regarding the investigated TLRs (this study) and on the genomic level53 and was seemingly far less affected by human activity. While we chose both species due to their similar lifestyle and sympatric distribution in Savanna habitats, there are still noticeable differences that might explain a deviating innate immune response gene diversity. Cheetahs are highly specialized carnivores that only occur in open habitats, African leopards, on the other hand, are less tied to a specific biome. Except for desserts, they occur in mountain ridges and open grasslands as well as in tropical forests and marshlands71. Therefore, current leopard populations are larger and less fragmented than modern cheetah populations44,72. Habitat diversity and population size might explain the higher genetic diversity in the leopards’ TLR genes because they are likely more exposed to different and more diverse pathogens. Additionally, persecution might threaten cheetahs more because of their diurnal lifestyle and the recently increasing illegal pet trade73.
Due to easy accessibility, most immunological studies on cheetahs and African leopards only included their Southern and Southeastern African distribution (this study included). Hence, only two classical cheetah subspecies (A. j. jubatus and A. j. raineyi) and less than half of the African leopard’s current distribution are covered. Yet, conclusions about the immune response in both species should consider subspecies and local populations because the imposed environmental pressure might differ significantly; a problem only to be solved by extended modern and historic sample sets throughout both species’ distribution. By extending our sampling to the other cheetah subspecies and also including historic material, we aimed to account for the exposure to different environmental pressures. Although, modern Southern African cheetahs exhibit lower heterozygosity and lower diversity in TLR exons than African leopards, this might not be representative of TLR genes as a whole in the species. We mostly investigated anti-bacterial TLRs that are known to differ, e.g., in selection patterns, from virus detecting TLRs74,75. Furthermore, anti-viral TLRs are assumed to be under stronger evolutionary constrain than other TLR-families76,77. Nevertheless, it was shown that the constitutive innate immune response in Southern African cheetahs is stronger than in sympatric leopards78. Thus, relevant levels of genetic diversity might occur in other IIS-genes. However, genetic deprivation might already similarly affect African leopards despite apparently high genetic diversity. Moreover, MHC diversity in cheetahs is not as low as previously expected58,59,61 and the species’ health seems not to be affected at all in the wild78,79. This hints at a sufficiently high adaptive AIS activity in cheetahs, which might enable the species to cope with its low diversity in specific IIS genes80. Further studies should investigate co-evolution between more IIS and AIS genes in cheetahs and leopards and include additional historic data.
In conclusion, like all large carnivores, leopards and cheetahs in Africa are threatened less by their genetic heritage but more by recent habitat loss and persecution. So far, the genetic diversity of African leopards does not appear to have been greatly affected by human activity, while the genetic impoverishment of cheetahs is the direct result of the recent population decline in this highly specialized species. Our study observed a drastic reduction in genetic diversity and heterozygosity in four TLR exons of modern Southern African cheetahs, which may worsen this subspecies’ resilience to future infectious diseases. However, the health of the subspecies does not yet appear to be affected by its reduced genetic diversity. Still, the growing human-livestock-wildlife interface will further increase the risk of infectious diseases for wild populations.
Material and methods
Sample collection
To study the diversity in TLR genes in big cats, we used 61 modern Southern African cheetah samples (Acinonyx jubatus jubatus) from the South African National Biodiversity Institute (SANBI) Biobanks (Pretoria, South Africa; approved project number P2021/12) that had been collected from wild populations between 1998 and 2014 during different translocation and monitoring projects, including 14 individuals from Botswana, 12 from Namibia and 35 from South Africa. To retrieve immune gene diversity information from all cheetah subspecies we added 15 samples from museums’ collections, including five Asiatic cheetahs (A. j. venaticus), one East African cheetah (A. j. raineyi), four Northeast African cheetahs (A. j. soemmeringii), three Southern African cheetahs and three West African cheetahs (A. j hecki). In addition, we included short-read data of 41 previously sequenced African leopards (Panthera pardus pardus)49, compromising four individuals from Ghana, eight from Namibia, 14 from Tanzania, and 15 from Zambia. For detailed sample information see Supplementary Table 1. Samples collected after 1975 were imported under the following CITES permits: AT 16-E-0753, 16SG006329CR, 15JP001990/TE, 11US761881/9, AT 15-E-1769, D79/DFF or transferred between CITES-registered institutions (Supplementary Table 4).
Genomic DNA was extracted from modern samples using the Quick-DNA Miniprep Plus Kit (Zymo Research, Irvine, California, USA) and a DNA salting out method81 for historic samples. Historic samples were rehydrated in nuclease-free water for 24 h in an attempt to remove potential secondary preservatives before DNA extraction.
Library preparation and next-generation sequencing
In preparation for long-read amplicon sequencing (LRS) we created primer sets to investigate the five exons of the selected toll-like receptor genes TLR2, TLR4, TLR6 and TLR8 (Supplementary Table 5) using the available cheetah genome from NCBI (GCF_003709585.1)82 with primer383. We amplified our modern A. j. jubatus samples using long-range Polymerase Chain Reaction and targeted specific primers tailed with M13 adapter sequences. Per individual sample and exon, we used a total reaction volume of 10 µl consisting of 5 µl GoTaq® Long PCR Master Mix (Promega, Madison, Wisconsin, USA), 2.5 µl ddH2O, 0.5 µl of both forward and reverse primers (10 µM) and 1.5 µl of template DNA (50–250 ng). The following PCR protocol was used: Initial denaturation at 95 °C for 2 min following 35 cycles (denaturation at 95 °C for 30 s, annealing at 57 °C for 30 s and elongation at 72 °C for 5 min), and a final elongation at 72 °C for 10 min. Amplicons containing PacBio M13 adaptor sequences (Pacific Biosciences of California, Menlo Park, California, USA) were sent to Inqaba Biotechnical Industries (Pretoria, South Africa) for indexing. A PacBio circular consensus sequencing (CCS) library was prepared using the SMRTbell® express template prep kit 2.0 (PacBio), indexed and the resulting amplicons were sequenced on the PacBio Sequel IIe platform (PacBio).
Whole genomes of historic cheetahs (A. jubatus ssp.) were sequenced as part of a larger project, and in-depth analyses are still ongoing due to necessary re-sequencing of some particularly difficult samples. In brief, we prepared 150 bp paired-end libraries for short-read sequencing using the NEBNext® Ultra™ II DNA Library Prep Kit for Illumina® (New England Biolabs, Ipswich, Massachusetts, USA) with varying insert sizes depending on DNA quality. All libraries were sequenced on the NovaSeq 6000 platform (Illumina, San Diego, California, USA) at Novogene (Cambridge, England, UK).
Data processing
Long-read sequences were received as high fidelity (HIFI) consensus reads and no additional trimming/filtering was necessary. Short reads were trimmed with fastp version 0.20.184 and filtered with base correction enabled and a low complexity filter. All sequencing adapters and polyG tails at the end of reads were removed and a sliding window of 4 bp was applied to detect poor-quality regions (Phred score < 15). Short reads were discarded if they were shorter than 36 bp, had > 40% low-quality bases, or had more than five undetermined bases. We mapped our long reads to the TLR reference sequences derived from the cheetah assembly (GCF_003709585.1) using bwa-mem85 and removed duplicates with Picard version 2.22.386. The variant calling of both alleles was performed with BCFtools mpileup version 1.987 (flags: -s LowQual -e '%QUAL < 20 || DP > 10') in two consecutive steps using the first called consensus as reference for the second variant. The short reads were first mapped to the TLR consensus sequences using bwa-mem and sorted by name using SAMtools sort88. Unmapped reads were removed, and the reads in the resulting mapping file were converted to a new fastq file using SAMtools view. The reduced reads were mapped a second time using bowtie2 version 2.4.589 without any clipping (flags: -end-to-end -x -S) to avoid miss called Single Nuclear Polymorphisms (SNPs) due to over-representation caused by falsely clipped reads. Variant calling followed the same steps described for the long reads above. The script used for mapping and variant calling is provided in the Supplementary material. Insertions and deletions (indels) were curated manually, and each called SNP was re-checked for validity using Tablet version 1.21.02.0890.
We used the PHASE function implemented in DnaSP v.6.12.0391 to derive the alleles of the short-read consensus sequences with a threshold of 0.6, allowing for recombination. No additional phasing for long-read sequences was needed. All further analyses were performed using phased allele sequences.
Comparative analysis
DNA sequences were aligned using the aligner integrated in AliView92. Identical sequences were removed keeping only a single sequence per allele and translated into amino acid sequences. Amino acid and underlying nucleotide alleles were named according to the naming scheme described in Supplementary Fig. 2. Maximum likelihood phylogenies were generated from aligned DNA sequences and the resulting amino acid sequences of each TLR exon using IQ-TREE version 1.6.1293. TLR sequences of the spotted hyena (Crocuta crocuta) (GCA_008692635.1 BGI_CrCroc_1.0) and striped hyena (Hyena hyena) (GCA_003009895.1 ASM300989v1) were used as an outgroup. We used the term “allele” to refer to full-length allele sequences defined by LRS and not to individual SNPs within the exon sequence.
SNP-sites were investigated for recombination with the Genetic Algorithm for Recombination Detection tool (GARD)94. We used the resulting NEXUS files to detect episodic positive selection based on the Mixed Effects Model of Evolution (MEME)95, both implemented in the online tool datamonkey96.
Ethics approval and consent to participate
The study was approved by the institutional review board of the South African National Biodiversity Institute (SANBI) with the project number P2021/12. No animal experiments were conducted in this study. No human participants, tissue, or data were used in this study.
Data availability
All data generated or analyzed during this study are included within this article’s Supplementary material. Additionally, the newly generated TLR sequencing reads are uploaded as FASTQ files to NCBI (PRJNA1005947) and the corresponding TLR 2, 4, 6, and 8 alignments for both species are available on the Phaidra public repository: https://doi.org/https://doi.org/10.34876/9KFD-2A38.
References
Ausubel, F. M. Are innate immune signaling pathways in plants and animals conserved?. Nat. Immunol. 6, 973–979 (2005).
Medzhitov, R. & Janeway, C. A. Innate immunity: The virtues of a nonclonal system of recognition. Cell 91, 295–298 (1997).
Eason, D. D. et al. Mechanisms of antigen receptor evolution. Semin. Immunol. 16, 215–226 (2004).
Lien, E. et al. Toll-like receptor 4 imparts ligand-specific recognition of bacterial lipopolysaccharide. J. Clin. Investig. 105, 497–504 (2000).
Muzio, M. & Mantovani, A. Toll-like receptors. Microbes Infect. 2, 251–255 (2000).
Takeda, K. & Akira, S. Toll-like receptors. Curr. Protoc. Immunol. 109, 14121–141210 (2015).
Buwitt-Beckmann, U. et al. TLR1-and TLR6-independent recognition of bacterial lipopeptides. J. Biol. Chem. 281, 9049–9057 (2006).
Gazzinelli, R. T. & Denkers, E. Y. Protozoan encounters with Toll-like receptor signalling pathways: Implications for host parasitism. Nat. Rev. Immunol. 6, 895–906 (2006).
Moresco, E. M. Y., LaVine, D. & Beutler, B. Toll-like receptors. Curr. Biol. 21, R488–R493 (2011).
Fitzgerald, K. A. & Kagan, J. C. Toll-like receptors and the control of immunity. Cell 180, 1044–1066 (2020).
Hawley, D. M., Sydenstricker, K. V., Kollias, G. V. & Dhondt, A. A. Genetic diversity predicts pathogen resistance and cell-mediated immunocompetence in house finches. Biol. Lett. 1, 326–329 (2005).
Morris, K. M., Wright, B., Grueber, C. E., Hogg, C. & Belov, K. Lack of genetic diversity across diverse immune genes in an endangered mammal, the Tasmanian devil (S arcophilus harrisii). Mol. Ecol. 24, 3860–3872 (2015).
Hughes, A. L. & Hughes, M. K. Natural selection on the peptide-binding regions of major histocompatibility complex molecules. Immunogenetics 42, 233–243 (1995).
Spielman, D., Brook, B. W., Briscoe, D. A. & Frankham, R. Does inbreeding and loss of genetic diversity decrease disease resistance?. Conserv. Genet. 5, 439–448 (2004).
Tonteri, A., Vasemägi, A., Lumme, J. & Primmer, C. Beyond MHC: Signals of elevated selection pressure on Atlantic salmon (Salmo salar) immune-relevant loci. Mol. Ecol. 19, 1273–1282 (2010).
Downing, T., Lloyd, A. T., O’Farrelly, C. & Bradley, D. G. The differential evolutionary dynamics of avian cytokine and TLR gene classes. J. Immunol. 184, 6993–7000 (2010).
Villasenor-Cardoso, M. & Ortega, E. Polymorphisms of innate immunity receptors in infection by parasites. Parasite Immunol. 33, 643–653 (2011).
Netea, M. G., Wijmenga, C. & O’neill, L. A. Genetic variation in Toll-like receptors and disease susceptibility. Nat. Immunol. 13, 535–542 (2012).
Abrantes, J., Areal, H. & Esteves, P. J. Insights into the European rabbit (Oryctolagus cuniculus) innate immune system: Genetic diversity of the toll-like receptor 3 (TLR3) in wild populations and domestic breeds. BMC Genet. 14, 73 (2013).
Dalton, D. L., Vermaak, E., Smit-Robinson, H. A. & Kotze, A. Lack of diversity at innate immunity Toll-like receptor genes in the Critically Endangered White-winged Flufftail (Sarothrura ayresi). Sci. Rep. 6, 36757 (2016).
Grueber, C. E., Wallis, G. P., King, T. M. & Jamieson, I. G. Variation at innate immunity toll-like receptor genes in a bottlenecked population of a New Zealand robin. PLOS ONE 7, e45011 (2012).
Liman, N., Alan, E. & Apaydın, N. The expression and localization of Toll-like receptors 2, 4, 5 and 9 in the epididymis and vas deferens of a adult tom cats. Theriogenology 128, 62–73 (2019).
Whitney, J., Haase, B., Beatty, J. & Barrs, V. R. Breed-specific variations in the coding region of toll-like receptor 4 in the domestic cat. Vet. Immunol. Immunopathol. 209, 61–69 (2019).
Robert-Tissot, C. et al. The innate antiviral immune system of the cat: Molecular tools for the measurement of its state of activation. Vet. Immunol. Immunopathol. 143, 269–281 (2011).
Loots, A. K. et al. The role of toll-like receptor polymorphisms in susceptibility to canine distemper virus. Mammalian Biol. 88, 94–99 (2018).
Odewahn, R., Wright, B. R., Czirják, G. Á. & Higgins, D. P. Differences in constitutive innate immunity between divergent Australian marsupials. Dev. Comp. Immunol. 132, 104399 (2022).
Ripple, W. J. et al. Status and ecological effects of the world’s largest carnivores. Science 343, 1241484 (2014).
Tshabalala, T. et al. Leopards and mesopredators as indicators of mammalian species richness across diverse landscapes of South Africa. Ecol. Indicators 121, 107201 (2021).
Atkins, J. L. et al. Cascading impacts of large-carnivore extirpation in an African ecosystem. Science 364, 173–177 (2019).
Ford, A. T. et al. Large carnivores make savanna tree communities less thorny. Science 346, 346–349 (2014).
Suraci, J. P., Clinchy, M., Dill, L. M., Roberts, D. & Zanette, L. Y. Fear of large carnivores causes a trophic cascade. Nat.Commun. 7, 10698 (2016).
Myers, N. Leopard and cheetah in Ethiopia. Oryx 12, 197–205 (1973).
Verschueren, S. et al. Spatiotemporal sharing and partitioning of scent-marking sites by cheetahs and leopards in north-central Namibia. Afr. J. Ecol. 59, 605–613 (2021).
Hemami, M.-R. et al. Using ecological models to explore niche partitioning within a guild of desert felids. Hystrix 29, 216 (2018).
Vogel, J. T., Somers, M. J. & Venter, J. A. Niche overlap and dietary resource partitioning in an African large carnivore guild. J. Zool. 309, 212–223 (2019).
Seoraj-Pillai, N. & Pillay, N. A meta-analysis of human–wildlife conflict: South African and global perspectives. Sustainability 9, 34 (2016).
Viollaz, J. S., Thompson, S. T. & Petrossian, G. A. When human–wildlife conflict turns deadly: Comparing the situational factors that drive retaliatory leopard killings in South Africa. Animals 11, 3281 (2021).
Patterson, B. D., Kasiki, S. M., Selempo, E. & Kays, R. W. Livestock predation by lions (Panthera leo) and other carnivores on ranches neighboring Tsavo National Parks, Kenya. Biol. Conserv. 119, 507–516 (2004).
Wykstra, M. & Fund, C. C. Cheetah Conservation and Human Impact in Kenya. Cheetah Conservation (2005).
Constant, N., Bell, S. & Hill, R. The impacts, characterisation and management of human–leopard conflict in a multi-use land system in South Africa. Biodivers. Conserv. 24, 2967–2989 (2015).
Marker, L. L., Muntifering, J., Dickman, A., Mills, M. & Macdonald, D. Quantifying prey preferences of free-ranging Namibian cheetahs. South African J. Wildlife Res. 33, 43–53 (2003).
Thuo, D. et al. An insight into the prey spectra and livestock predation by cheetahs in Kenya using faecal DNA metabarcoding. Zoology 143, 125853 (2020).
Cushman, S. A., Elliot, N. B., Macdonald, D. W. & Loveridge, A. J. A multi-scale assessment of population connectivity in African lions (Panthera leo) in response to landscape change. Landscape Ecol. 31, 1337–1353 (2016).
Durant, S. M. et al. The global decline of cheetah Acinonyx jubatus and what it means for conservation. Proc. Natl. Acad. Sci. 114, 528–533 (2017).
Belbachir, F., Pettorelli, N., Wacher, T., Belbachir-Bazi, A. & Durant, S. M. Monitoring rarity: The critically endangered Saharan cheetah as a flagship species for a threatened ecosystem. PLoS One 10, e0115136 (2015).
Marker, L. Cheetahs race for survival: Ecology and conservation. in Wildlife population monitoring (IntechOpen, 2019).
Charruau, P. et al. Phylogeography, genetic structure and population divergence time of cheetahs in Africa and Asia: Evidence for long-term geographic isolates. Mol. Ecol. 20, 706–724 (2011).
Jacobson, A. P. et al. Leopard (Panthera pardus) status, distribution, and the research efforts across its range. PeerJ 4, e1974 (2016).
Farhadinia, M. S. et al. The critically endangered Asiatic cheetah Acinonyx jubatus venaticus in Iran: A review of recent distribution, and conservation status. Biodivers. Conserv. 26, 1027–1046 (2017).
Khalatbari, L., Jowkar, H., Yusefi, G. H., Brito, J. C. & Ostrowski, S. The current status of Asiatic cheetah in Iran. (2018).
Gerhold, R. W. & Jessup, D. A. Zoonotic Diseases associated with free-roaming cats. Zoonoses and Public Health 60, 189–195 (2013).
Wiethoelter, A. K., Beltrán-Alcrudo, D., Kock, R. & Mor, S. M. Global trends in infectious diseases at the wildlife–livestock interface. Proc. Natl. Acad. Sci. 112, 9662–9667 (2015).
Pečnerová, P. et al. High genetic diversity and low differentiation reflect the ecological versatility of the African leopard. Curr. Biol. 31, 1862-1871.e5 (2021).
O’Brien, S. J. et al. Genetic basis for species vulnerability in the Cheetah. Science 227, 1428–1434 (1985).
O’Brien, S. J., Wildt, D. E. & Bush, M. The Cheetah in genetic Peril. Sci. Am. 254, 84–95 (1986).
Hedrick, P. W. Bottleneck (s) or metapopulation in cheetahs. Conserv. Biol. 10, 897–899 (1996).
Menotti-Raymond, M. & O’Brien, S. J. Dating the genetic bottleneck of the African cheetah. Proc. Natl. Acad. Sci. 90, 3172–3176 (1993).
Castro-Prieto, A., Wachter, B. & Sommer, S. Cheetah paradigm revisited: MHC diversity in the world’s largest free-ranging population. Mol. Biol. Evol. 28, 1455–1468 (2011).
Drake, G. et al. The use of reference strand-mediated conformational analysis for the study of cheetah (Acinonyx jubatus) feline leucocyte antigen class II DRB polymorphisms. Mol. Ecol. 13, 221–229 (2004).
Meißner, R. et al. The potential and shortcomings of mitochondrial DNA analysis for cheetah conservation management. Conservation Genetics 1–12 (2022).
Prost, S. et al. Genomic analyses show extremely perilous conservation status of African and Asiatic cheetahs (Acinonyx jubatus). Mol. Ecol. 31, 4208–4223 (2022).
Mirdita, M. et al. ColabFold: Making protein folding accessible to all. Nat. Methods 19, 679–682 (2022).
Sugimoto, T. et al. Noninvasive genetic analyses for estimating population size and genetic diversity of the remaining Far Eastern leopard (Panthera pardus orientalis) population. Conserv. Genet. 15, 521–532 (2014).
Wetzler, L. M. The role of Toll-like receptor 2 in microbial disease and immunity. Vaccine 21, S55–S60 (2003).
Heine, H. et al. Cutting edge: Cells that carry a null allele for toll-like receptor 2 are capable of responding to endotoxin1. J. Immunol. 162, 6971–6975 (1999).
Farhat, K. et al. Heterodimerization of TLR2 with TLR1 or TLR6 expands the ligand spectrum but does not lead to differential signaling. J. Leukocyte Biol. 83, 692–701 (2008).
Ma, Y., Haynes, R. L., Sidman, R. L. & Vartanian, T. TLR8: An innate immune receptor in brain, neurons and axons. Cell Cycle 6, 2859–2868 (2007).
Smirnova, I., Poltorak, A., Chan, E. K., McBride, C. & Beutler, B. Phylogenetic variation and polymorphism at the Toll-like receptor 4 locus (TLR4). Genome Biol. 1, research002.1 (2000).
Creel, S. et al. Changes in African large carnivore diets over the past half-century reveal the loss of large prey. J. Appl. Ecol. 55, 2908–2916 (2018).
Woodroffe, R. Predators and people: Using human densities to interpret declines of large carnivores. Anim. Conserv. 3, 165–173 (2000).
Searle, C. E. et al. Leopard population density varies across habitats and management strategies in a mixed-use Tanzanian landscape. Biol. Conserv. 257, 109120 (2021).
Morris, D. R. et al. Gene flow connects key leopard (Panthera pardus) populations despite habitat fragmentation and persecution. Biodiversity and Conservation 1–19 (2022).
Tricorache, P., Yashphe, S. & Marker, L. Global dataset for seized and non-intercepted illegal cheetah trade (Acinonyx jubatus) 2010–2019. Data in Brief 35, 106848 (2021).
Tong, M. et al. Transcript profiling of toll-like receptor mRNAs in selected tissues of mink (Neovison vison). J. Microbiol. Biotechnol. 26, 2214–2223 (2016).
Wlasiuk, G. & Nachman, M. W. Adaptation and constraint at toll-like receptors in primates. Mol. Biol. Evol. 27, 2172–2186 (2010).
Bagheri, M. & Zahmatkesh, A. Evolution and species-specific conservation of toll-like receptors in terrestrial vertebrates. Int. Rev. Immunol. 37, 217–228 (2018).
Vasseur, E. et al. The selective footprints of viral pressures at the human RIG-I-like receptor family. Hum. Mol. Genet. 20, 4462–4474 (2011).
Heinrich, S. K. et al. Cheetahs have a stronger constitutive innate immunity than leopards. Sci. Rep. 7, 44837 (2017).
Schwensow, N., Castro-Prieto, A., Wachter, B. & Sommer, S. Immunological MHC supertypes and allelic expression: How low is the functional MHC diversity in free-ranging Namibian cheetahs?. Conserv. Genet. 20, 65–80 (2019).
McDade, T. W., Georgiev, A. V. & Kuzawa, C. W. Trade-offs between acquired and innate immune defenses in humans. Evol. Med. Public Health 2016, 1–16 (2016).
De Volo, S. B., Reynolds, R. T., Douglas, M. R. & Antolin, M. F. An improved extraction method to increase DNA yield from molted feathers. The Condor 110, 762–766 (2008).
Dobrynin, P. et al. Genomic legacy of the African cheetah, Acinonyx jubatus. Genome Biol. 16, 1–20 (2015).
Kõressaar, T. et al. Primer3_masker: Integrating masking of template sequence with primer design software. Bioinformatics 34, 1937–1938 (2018).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Institute, B. Picard toolkit. Broad Institute, GitHub repository (2019).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Milne, I. et al. Using Tablet for visual exploration of second-generation sequencing data. Brief. Bioinform. 14, 193–202 (2013).
Librado, P. & Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452 (2009).
Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 30, 3276–3278 (2014).
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Kosakovsky Pond, S. L., Posada, D., Gravenor, M. B., Woelk, C. H. & Frost, S. D. W. Automated phylogenetic detection of recombination using a genetic algorithm. Mol. Biol. Evol. 23, 1891–1901 (2006).
Murrell, B. et al. Detecting individual sites subject to episodic diversifying selection. PLOS Genet. 8, e1002764 (2012).
Weaver, S. et al. Datamonkey 2.0: A modern web application for characterizing selective and other evolutionary processes. Mol. Biol. Evol. 35, 773–777 (2018).
Funding
RM acknowledges funding from the Central European Science Partnership (CEUS) project Austrian Science Fund (FWF) I5081-B/ GACR Czech Republic 21-28637 L (to SP. PH, and PAB), as well as to the Scientific & Technology Cooperation Austria, South Africa grant ZA02/2019 from the Austrian Agency for Education and Internationalization (OeAD) and the National Research Foundation, South Africa (to DD and PAB).
Author information
Authors and Affiliations
Contributions
All the authors contributed to the study’s conception and design. Material preparation was carried out by P.M., C.P., R.M., K.L. and A.K. Data collection was performed by R.M., S.W., S.P., D.D., P.A.B. and P.H. R.M. analyzed the data and wrote the first draft of the manuscript. D.D., S.P., P.H. and P.A.B. supervised the project and provided funding. All authors reviewed and commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Meißner, R., Mokgokong, P., Pretorius, C. et al. Diversity of selected toll-like receptor genes in cheetahs (Acinonyx jubatus) and African leopards (Panthera pardus pardus). Sci Rep 14, 3756 (2024). https://doi.org/10.1038/s41598-024-54076-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-54076-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
