
Abstract
Computational materials research is vital in improving our understanding of various class of materials and their properties, contributing valuable information that helps predict innovative structures and complement empirical investigations. In this context, DHQ-graphene recently emerged as a stable two-dimensional carbon allotrope composed of decagonal, hexagonal, and quadrilateral carbon rings. Here, we employ density functional theory calculations to investigate the mechanical, electronic, and optical features of its boron nitride counterpart (DHQ-BN). Our findings reveal an insulating band gap of 5.11 eV at the HSE06 level and good structural stability supported by phonon calculations and ab initio molecular dynamics simulations. Moreover, DHQ-BN exhibits strong ultraviolet (UV) activity, suggesting its potential as a highly efficient UV light absorber. Its mechanical properties, including Young’s modulus (230 GPa) and Poisson’s ratio (0.7), provide insight into its mechanical resilience and structural stability.
Similar content being viewed by others

Introduction
Ever since the groundbreaking synthesis of graphene in 2004 by Novoselov et al.1, extensive research efforts have been devoted to exploring the vast potential of two-dimensional (2D) materials, seeking to exploit their unique optoelectronic, structural, and thermal properties for applications in flat electronics2,3,4. Graphene’s exceptional attributes have opened doors to new possibilities in nanotechnology5,6,7,8. However, its intrinsic zero-bandgap nature presents limitations for specific optoelectronic applications, prompting the exploration of new 2D materials capable of addressing this challenge9,10,11,12,13,14.
To fill this gap, both theoretically and experimentally, researchers have been actively engaged in identifying 2D materials with tailored bandgaps that are conducive to optoelectronic applications, such as solar cells and light-emitting diodes15,16. Promising materials emerging from these efforts include transition metal dichalcogenides17, black phosphorus (phosphorene)18, boron nitride monolayers19, and various hybrid organic-inorganic perovskite structures20, each possessing distinct electronic properties, tunable bandgaps, and exceptional optoelectronic characteristics.
In computational materials science, quantum, and classical approaches have become indispensable for materials modeling and design, often preceding the synthesis of novel nanostructured materials by decades21,22,23,24. Recent advances in material modeling have paved the way for the theoretical exploration of 2D materials of boron nitride, which exhibit topological similarities to their carbon-based counterparts25,26,27,28,29,30. Some examples of these boron nitride structures are hexagonal boron nitride (h-BN)31, binary monolayer amorphous boron nitride32 (MABN), pentagraphene-like BN (penta-BN)32, T-graphene-like BN (T-BN)33, phagraphene-like BN (pha-BN)34, and the BN biphenylene network (BN-BPN)35,36,37,38.
In this context, DHQ-BN, the counterpart to boron nitride of DHQ-graphene39, stands out as a solution to the challenge of zero-band-gap limitations in optoelectronic applications. Its potential lies in enabling to more effective light absorption and emission within specific energy ranges. DHQ-graphene, a recently discovered two-dimensional carbon allotrope characterized by a unique arrangement of decagonal, hexagonal, and quadrilateral carbon rings, has garnered attention. So far, DHQ-BN’s structural, mechanical, electronic, and optical properties remain untested.
DHQ-graphene is a metastable material exhibiting anisotropic mechanical properties. Its in-plane Young’s modulus is comparable to that of penta-graphene32. DHQ-graphene demonstrates metallic behavior with a significantly higher electronic density of states, approximately 0.193 eV per state per atom. The bridge site at the edge shared by six- and ten-membered rings in DHQ-graphene is the most favorable adsorption site, displaying adsorption energy for oxygen (− 0.27 eV). A charge transfer of 0.834e from a DHQ-graphene sheet to an O atom, 3.35% higher than the one for perfect graphene, suggests its potential for controlled oxygen adsorption.
The nomenclature of 2D carbon and boron nitride allotropes has become significant due to their growing diversity and intricacy40. Ensuring clarity and scientific precision in the characterization of these materials is essential40. Hence, we have retained the original name of the carbon-based structure under study (the DHQ-graphene) while referring to its boron nitride variant with analogous topology.
This study used density functional theory (DFT) calculations to thoroughly examine DHQ-BN, delving into its mechanical, electronic, and optical properties. We establish the material’s structural stability by observing an absence of negative frequencies in the phonon spectrum. Moreover, the material exhibits exceptional thermal stability, as evidenced by its lack of topology reconfiguration at 1000 K in ab initio molecular dynamics (MD) simulations. The results also unveil the direct bandgap of DHQ-BN at 5.11 eV and its significant optical absorption capacity within the ultraviolet (UV) range. We explored its mechanical behavior, which varies depending on the applied strain direction, yielding Young’s Modulus values ranging from 30 to 230 GPa.
Results and discussion
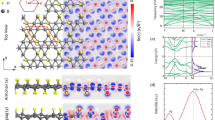
We begin by analyzing the optimized lattice arrangement of DHQ-BN, as illustrated in Fig. 1. Geometry optimization calculations, performed with the PBE and HSE06 methods, produce comparable lattice parameters. However, for the sake of this discussion, we focus on the results obtained using the HSE06 method, which is highlighted in Fig. 1 and summarized in Table 1. The lattice vectors a and b are determined to be 9.13 Å and 6.78 Å, as indicated in Fig. 1. The crystal structure of DHQ-BN aligns with the P2/C (C2H-4) space group, pointing to its orthorhombic nature.

Illustration depicting the DHQ-BN topology, presented from both a top view (a) and a side view (b). Emphasized within the diagram is the unit cell, with boron and nitrogen atoms represented by the colors pink and blue, respectively. The red numbers pinpoint the atoms responsible for forming distinct B-N bonds.
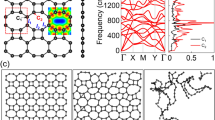
Figure 2a illustrates the dispersion of phonons in the DHQ-BN network. No imaginary frequencies are present, suggesting the material’s dynamic stability. The absence of a band gap between the acoustic and optical modes indicates a significant scattering rate and brief phonon lifetime, contributing to the material’s moderate lattice thermal conductivity. Furthermore, the formation energy of DHQ-BN is − 7.71 eV.

(a) Phonon dispersion and (b) computed formation energy as a function of the nitrogen concentration for DHQ-BN, calculated at PBE level.
When the formation energy of a structure aligns with the convex hull, it signifies thermodynamic stability and feasibility for experimental realization. We have pinpointed a unique global minimum structure within this two-dimensional parameter space by analyzing the relationship between formation energy and nitrogen concentrations, as illustrated in Fig. 2b. This specific minimum is situated precisely at the midpoint, characterized by an N:B ratio of 1:1. This composition resembles a structure reminiscent of the all-carbon DHQ-BN.
The convex hull plot was generated by systematically varying the concentration of N in the structure that was initially composed solely of B atoms while keeping the overall composition constant. This allowed us to assess the energetic stability of different configurations with distinct proportions between B and N atoms. As a result, one can note that the most favorable structure for the proposed lattice is indeed the one with B10N10 proportion.
We performed ab initio molecular dynamics (AIMD) simulations to assess the material’s dynamic and thermal behavior. Spanning a timeframe of 5000 fs, these simulations subjected the material to an elevated temperature of 1000 K. The selected time frame was deemed sufficient for our purposes, given the high-temperature regime adopted in our simulations. Our primary focus during these simulations was the continuous monitoring of the temporal evolution of the total energy per atom, a critical indicator of thermal stability. Employing a supercell measuring \(2 \times 2 \times 1\), containing 80 atoms, we scrutinized the fluctuations in energy.
The results, showcased in Fig. 3, unveil that the DHQ-BN lattice presents good thermal stability. The total energy exhibits only minimal fluctuations during the simulation, underscoring the material’s ability to maintain a balanced energy distribution. The insets in the figure provide a closer look at the DHQ-BN topology at the end of the simulation. One can note that no planarity deviations are noted. Importantly, the DHQ-BN does not entail bond breakage or reconstruction at 1000 K. The lattice pattern at 1000 K mirrors that of the optimized structure (refer to Fig. 1), reinforcing the assertion of DHQ-BN’s good thermal stability.

The evolution of the total energy per atom within the DHQ-BN lattice was tracked during AIMD simulations conducted at 1000 K, employing the PBE computational method. To visually represent the lattice arrangement of DHQ-BN at 5000 fs, we present insets displaying its top and side views.
Turning our attention to the electronic properties, Fig. 4a depicts the band structure profiles generated through the PBE (in black) and HSE06 (in red) methods. These calculations yield band gap values of 3.84 eV and 5.11 eV for the PBE and HSE06 levels, respectively. It is important to note that PBE calculations tend to underestimate band gap values, necessitating the adoption of the HSE06 hybrid functional to determine DHQ-BN’s electronic and optical characteristics precisely. The HSE06 band structure establishes DHQ-BN as an insulator, with energy bands near the Fermi level displaying dispersion. This dispersion signifies the delocalized nature of electronic states within the material.

In (a), we present the electronic band structure, while (b) provides insight into the partial density of states (PDOS) within the DHQ-BN monolayer. The band structures were computed using the PBE (in black) and HSE06 (in red) methods. The PDOS analysis in (b) was conducted at the HSE06 level. (c) Illustrates the highest occupied crystal orbital (HOCO, in red), and the lowest unoccupied crystal orbital (LUCO, in green). (d) Schematically depicts the electron localization function. These properties were also calculated using the HSE06 approach.
It is important to note that variations in the predicted band gap values for DHQ-BN arise from the inherent limitations and approximations of computational methods. The HSE06 approach, classified as a hybrid functional, introduces a fraction of exact exchange in combination with the exchange-correlation functional. The inclusion of non-local exchange interactions serves to improve the precision of electronic structure predictions, especially in cases involving substantial electron localization or delocalization, such as semiconductors. Therefore, the band gap determined through HSE06 is frequently regarded as offering a more accurate portrayal of electronic properties.
Similarly, h-BN is characterized by an insulating band gap of approximately 5.95 eV41. The comparatively smaller band gap observed in DHQ-BN, compared to h-BN, can be attributed to its unique ring topology. These factors introduce distinct pathways for electronic transitions, contributing to the altered electronic properties of DHQ-BN.
It is important to acknowledge that variations in the predicted band gap values for DHQ-BN are rooted in computational methodologies’ inherent limitations and approximations. The HSE06 approach, characterized by its hybrid functional nature, incorporates a fraction of exact exchange and the exchange-correlation functional. Incorporating non-local exchange interactions significantly improves the precision of electronic structure predictions, especially for systems where electron localization or delocalization plays a crucial role, such as semiconductors. As a result, the band gap computed using the HSE06 method is frequently regarded as a more dependable representation of electronic properties.
In Fig. 4b, we delve into the partial density of states (PDOS) within DHQ-BN, employing the HSE06 hybrid functional for precise analysis. One can note a prevalence of p-states of nitrogen in the electronic structure. This prominence of p-orbitals indicates their pivotal role in driving electronic transitions and interactions within the material. They are often associated with directional bonding phenomena.
Further insights into chemical interactions within DHQ-BN are provided through the examination of the highest occupied crystal orbital (HOCO), lowest unoccupied crystal orbital (LUCO), and electron localization function (ELF) analyses, as illustrated in Fig. 4c,d. These analyses reveal distinctive orbital localizations within DHQ-BN. In Fig. 4c, one can note that HOCO predominantly localizes on nitrogen atoms of the tetragonal rings. At the same time, the LUCO primarily resides in boron atoms in the tetragonal rings. This trend is consistent with the ELF pattern, as discussed below, once there is a pronounced electron localization/delocalization on boron/nitrogen atoms. The electronegativity disparity between nitrogen and boron atoms results in an induced dipole towards the former, favoring the localization of LUCO on them. The electron localization function (ELF) assigns a numerical value between 0 and 1 to every point within space, allowing us to gain insights into the nature of electron interactions. Typically, regions where ELF values approach 1 indicate the presence of strong covalent interactions or lone pair electrons. Conversely, lower ELF values, around 0.5, suggest delocalization, potentially pointing to ionic bonds or weak Van der Waals interactions. The ELF map in Fig. 4d highlights the prevalence of localized electrons on boron atoms and delocalized electrons on nitrogen atoms, aligning with the earlier discussed charge distribution concept.
Figure 5 provides insights into the optical properties of the DHQ-BN lattice, explicitly focusing on the polarization of light along the x (E//X), y (E//Y), and z (E//Z) directions. These calculations were carried out utilizing the HSE06 method. The solid and dashed lines in this figure correspond to the dielectric function’s real (Re) and imaginary (Im) parts. The optical properties of DHQ-BN are marked by strong optical activity, predominantly in the UV region. This characteristic aligns with the behavior observed in h-BN, which exhibits optical activity across a broad range of wavelengths, including UV and visible light41.

(a) absorption coefficient (\(\alpha\)), (b) reflectivity, and (c) Refractive index for polarized light along the x (E//X), y (E//Y), and z (E//Z) directions at the HSE06 level are depicted in this figure. The solid and dashed lines represent the dielectric function’s real (Re) and imaginary (Im) components.
It is important to note that the band gap of h-BN typically falls within the UV range, rendering it an insulator with limited electronic conductivity. The transparent nature of both DHQ-BN and h-BN can be attributed to their insulating band gaps, preventing the absorption of photons with energies below their respective band gap values.
The DHQ-BN optical absorption is substantial and limited to the UV region, as shown in Fig. 5a. These optical characteristics suggest that this material could serve as a UV collector. DQH-BN’s low reflectivity is primarily confined to the UV region, as Fig. 5b depicts. Similarly to h-BN, DHQ-BN exhibits relatively high refractive index values, as seen in Fig. 5c. This trend indicates its ability to polarize in response to external electric fields, implying potential applications in electronics and photonics.
The elastic properties of DHQ-BN play a pivotal role in understanding microcrack behavior and its overall durability. To evaluate the anisotropy inherent in its mechanical characteristics, Poisson’s ratio (\(\nu (\theta )\)) and Young’s modulus (\(Y(\theta )\)) were determined under pressure within the xy plane, as defined by the following equations42,43:
and
In this context, \(\alpha =\cos (\theta )\) and \(\beta =\sin (\theta )\).
Table 2 presents the DHQ-BN elastic properties, and Fig. 6 shows the 2D representation of (a) Poisson’s ratio and (b) Young’s modulus (GPa) in the xy plane. Poisson’s ratio of DHQ-BN reaches a maximum value (\(\nu _{MAX}\)) of approximately 0.7, while the associated Young’s modulus remains below 30 GPa. This trend indicates the incompressibility of the material under biaxial strains.

2D representation of (a) Poisson’s ratio and (b) Young’s modulus (GPa) in the xy plane for the DHQ-BN monolayer.
The anisotropic behavior of DHQ-BN is evident in its Young’s modulus values, measured at approximately 330 GPa and 230 GPa for strains along the x and y directions, respectively. These values reflect the material’s resistance to deformation under tension, with lower Young’s modulus values indicating greater deformability. The Poisson’s ratio values for uniaxial strains along the x and y directions are approximately 0.18, smaller than the h-BN one (about 0.29)44. The diminished Poisson ratio in DHQ-BN can be attributed to its unique structural arrangement, particularly the inclusion of decagonal rings, which enhance flexibility and extensive deformation under tension, resulting in lower Poisson ratios.
The observed anisotropy in mechanical properties in DHQ-BN stems from its unique topology. The distinctive ring distribution along different plane directions under uniaxial strain plays a crucial role. When subjected to uniaxial tensile strain in the y-direction, the aligned sequence of 10-atom porous rings facilitates easier deformation. In contrast, the tensile strain in the x-direction presents a sequence of fused hexagonal rings, making DHQ-BN stiffer in this direction. This variation in ring distribution directly influences the material’s response to mechanical stress, resulting in anisotropic mechanical behavior.
Finally, we conducted calculations on the bilayer configuration of DHQ-BN, where we observed that the minimum energy state occurred with an AA stacking arrangement, as shown in Fig. 7. Figure 7a illustrates the optimized structure of the bilayer material in the AA stacking configuration. This arrangement involves aligning the atoms in one layer directly on top of the atoms in the adjacent layer.

Schematic representation of (a) DHQ-BN AA stacking arrangement at the minimum distance, (b) potential energy curve as a function of interlayer distance, and (c) phonon spectrum for the AA-stacked configuration of DHQ-BN.
For the sake of comparison, we investigate here only the AA staking of DHQ-BN since previous studies consistently demonstrate the stability of the AA arrangement in bilayer h-BN systems compared to other stacking patterns45,46,47,48. Given this established precedence, we believe that investigating the AA stacking of DHQ-BN is sufficient to draw meaningful comparisons with its analogous h-BN.
To gain insights into the interlayer interactions and their impact on the material’s stability, we examined the potential energy curve as a function of interlayer distance for the AA-stacked configuration, as shown in Fig. 7b. We identified a specific interlayer energy, denoted as \(D_0=0.95\), where the potential energy reaches its minimum value around 3.4 Å. This equilibrium distance represents the most stable separation between the two layers. It signifies that the atoms in the bilayer material stay at this specific distance due to the balance between attractive and repulsive forces.
We observed that the bilayer’s energy is higher than the monolayer’s. This energy difference highlights the significant influence of stacking arrangements on the material’s overall energy landscape. Adding the second layer introduces interactions contributing to a slightly higher energy state, emphasizing the importance of considering layer-to-layer interactions in understanding the material’s behavior.
To further assess the stability of the AA-stacked bilayer, we examined its phonon spectrum, as displayed in Fig. 7c. The absence of imaginary frequencies within the phonon spectrum indicates the stacked system’s stability. As mentioned, imaginary frequencies suggest vibrational modes that could lead to structural instabilities or phase transitions. The absence of such modes reinforces our confidence in the structural integrity of the AA-stacked bilayer.
Methods
We conducted a series of DFT simulations to comprehensively investigate DHQ-BN’s mechanical, electronic, and optical characteristics. The lattice configuration of this material is illustrated in Fig. 1. The simulations were carried out with the CASTEP49 code.
Exchange and correlation effects were treated using the generalized gradient approximation (GGA), with two specific functionals applied: the Perdew–Burke–Ernzerhof (PBE)50 and the Heyd–Scuseria–Ernzerhof (HSE06)51 hybrid functional. To accurately account for the interactions among the nuclear electrons of boron and nitrogen, norm-conserving pseudopotentials tailored for these elements were employed, as integrated into the CASTEP framework.
Electronic self-consistency was achieved through the Broyden–Fletcher–Goldfarb–Shannon (BF GS) unrestricted nonlinear iterative algorithm52,53. We considered a plane-wave basis set with an energy cutoff of 700 eV and convergence criteria for energy as 1.0 \(\times 10^{-5}\) eV. Periodic boundary conditions were applied to ensure a fully relaxed DHQ-BN lattice. The relaxation process aimed to maintain residual forces on each atom below 1.0 \(\times 10^{-3}\) eV/Å, while keeping the pressure below 0.01 GPa. During lattice optimization, the basis vector in the z-direction remained fixed, and a k-grid of \(10 \times 10 \times 1\) was employed. For electronic and optical calculations, k-grids of \(15 \times 15 \times 1\) and \(5 \times 5 \times 1\) were used for the PBE and HSE06 approaches, respectively. Additionally, Partial Density of States (PDOS) analyses were conducted at the HSE06 level, employing a k-grid of \(20 \times 20 \times 1\).
Elastic properties of DHQ-BN were determined using the LDA/CA-PZ method54,55, and a vacuum region of 20 Å was incorporated to mitigate artificial interactions among periodic images.
It is worth noting that the choice of a reduced k-grid resolution in HSE06 calculations, compared to the PBE approach, was primarily driven by practical computational considerations, given the inherent computational expense associated with the HSE06 method. Rigorous convergence tests were conducted to validate the chosen k-grid resolution for HSE06 calculations, ensuring the required precision and reliability within an acceptable error range.
To analyze phonon properties, the linear response methodology was employed with a grid spacing of 0.05 1/Å and a convergence tolerance of 10\(^{-5}\) eV/Å\(^2\). For the assessment of the mechanical properties of DHQ-BN, the stress–strain method, based on the Voigt–Reuss–Hill approximation56,57, was utilized. Additionally, it’s noteworthy that the stability of the material was confirmed through ab initio molecular dynamics (MD) simulations, demonstrating its resistance to reconfiguration even at temperatures as high as 1000 K.
Conclusions
In summary, our comprehensive exploration of the DHQ-BN lattice, which serves as the boron nitride counterpart to DHQ-graphene, has affirmed its structural stability. We have demonstrated the DHQ-BN dynamical and thermal stability through DFT and AIMD simulations. Our study revealed insulting band gap properties for DHQ-BN with computed values of 3.84 eV (PBE) and 5.11 eV (HSE06).
On the optical characteristics, our investigation has uncovered the lattice’s potent UV activity, hinting at its potential as an effective UV collector. The lattice’s high refractive index values have implications for applications in electronics and photonics. Examining the material’s mechanical behavior has shed light on its anisotropic traits, encompassing Poisson’s ratio and Young’s modulus. The anisotropic behavior of DHQ-BN is evident in its Young’s modulus values, measured at approximately 330 GPa and 230 GPa for strains along the x and y directions, respectively.
Possessing a broad semiconducting bandgap and electronic states with notable delocalization, DHQ-BN once manufactured can present a potential for implementation in nanoelectronics and photonics applications. Its favorable thermal stability and reduced lattice thermal conductivity position it as a candidate for addressing thermal management challenges in microelectronics. The distinctive anisotropic mechanical properties of DHQ-BN open avenues for enhancing the strength of lightweight materials. Additionally, its significant UV absorption coupled with low reflectivity positions DHQ-BN as a promising candidate for efficient UV collection, particularly relevant in sensing and detection applications.
Data availability
Data supporting this study’s findings are available upon reasonable request from the first author K.A.L.L.
References
Novoselov, K. S. et al. Electric field effect in atomically thin carbon films. Science 306, 666–669 (2004).
Wang, Z. et al. Electronic and optical properties of novel carbon allotropes. Carbon 101, 77–85 (2016).
Allen, M. J., Tung, V. C. & Kaner, R. B. Honeycomb carbon: A review of graphene. Chem. Rev. 110, 132–145 (2010).
Cassiano, T. S. A., Monteiro, F. F., de Sousa, L. E., e Silva, G. M. & de Oliveira Neto, P. H. Smooth gap tuning strategy for cove-type graphene nanoribbons. RSC Adv. 10, 26937–26943 (2020).
Renteria, J. D., Nika, D. L. & Balandin, A. A. Graphene thermal properties: Applications in thermal management and energy storage. Appl. Sci. 4, 525–547 (2014).
Asha, A. B. & Narain, R. Nanomaterials properties. In Polymer Science and Nanotechnology (eds Asha, A. B. & Narain, R.) 343–359 (Elsevier, 2020).
Lue, J. T. Physical properties of nanomaterials. Encycl. Nanosci. Nanotechnol. 10, 1–46 (2007).
Saleh, T. A. Nanomaterials: Classification, properties, and environmental toxicities. Environ. Technol. Innov. 20, 101067 (2020).
Kumar, R., Singh, R. K. & Singh, D. P. Natural and waste hydrocarbon precursors for the synthesis of carbon based nanomaterials: Graphene and cnts. Renew. Sustain. Energy Rev. 58, 976–1006 (2016).
Lima, K. A. L. et al. Adsorption of carbon dioxide and ammonia in transition metal-doped boron nitride nanotubes. J. Mol. Model. 25, 1–7 (2019).
Nasir, S., Hussein, M. Z., Zainal, Z. & Yusof, N. A. Carbon-based nanomaterials/allotropes: A glimpse of their synthesis, properties and some applications. Materials 11, 295 (2018).
Zhu, C.-Y., Ye, Y.-W., Guo, X. & Cheng, F. Design and synthesis of carbon-based nanomaterials for electrochemical energy storage. New Carbon Mater. 37, 59–92 (2022).
Monteiro, F. F., Azevedo, D. L., da Silva, E. C., Ribeiro, L. A. & de Almeida Fonseca, A. L. Theoretical insight into the stabilization dynamics. Encapsulated β-carotene in ZnO nanotubes. Chem. Phys. Lett. 636, 62–66 (2015).
Ng, Y. H., Ikeda, S., Matsumura, M. & Amal, R. A perspective on fabricating carbon-based nanomaterials by photocatalysis and their applications. Energy Environ. Sci. 5, 9307–9318 (2012).
Murad, A. R., Iraqi, A., Aziz, S. B., Abdullah, S. N. & Brza, M. A. Conducting polymers for optoelectronic devices and organic solar cells: A review. Polymers 12, 2627 (2020).
Park, N.-M., Kim, T.-S. & Park, S.-J. Band gap engineering of amorphous silicon quantum dots for light-emitting diodes. Appl. Phys. Lett. 78, 2575–2577 (2001).
Manzeli, S., Ovchinnikov, D., Pasquier, D., Yazyev, O. V. & Kis, A. 2d transition metal dichalcogenides. Nat. Rev. Mater. 2, 1–15 (2017).
Morita, A. Semiconducting black phosphorus. Appl. Phys. A 39, 227–242 (1986).
Tran, T. T., Bray, K., Ford, M. J., Toth, M. & Aharonovich, I. Quantum emission from hexagonal boron nitride monolayers. Nat. Nanotechnol. 11, 37–41 (2016).
Li, W. et al. Chemically diverse and multifunctional hybrid organic–inorganic perovskites. Nat. Rev. Mater. 2, 1–18 (2017).
Louie, S. G., Chan, Y.-H., da Jornada, F. H., Li, Z. & Qiu, D. Y. Discovering and understanding materials through computation. Nat. Mater. 20, 728–735 (2021).
Bauer, B., Wecker, D., Millis, A. J., Hastings, M. B. & Troyer, M. Hybrid quantum-classical approach to correlated materials. Phys. Rev. X 6, 031045 (2016).
Panchal, J. H., Kalidindi, S. R. & McDowell, D. L. Key computational modeling issues in integrated computational materials engineering. Comput. Aided Des. 45, 4–25 (2013).
Butler, K. T., Frost, J. M., Skelton, J. M., Svane, K. L. & Walsh, A. Computational materials design of crystalline solids. Chem. Soc. Rev. 45, 6138–6146 (2016).
Pacile, D., Meyer, J., Girit, Ç. & Zettl, A. The two-dimensional phase of boron nitride: Few-atomic-layer sheets and suspended membranes. Appl. Phys. Lett. 92, 133107 (2008).
Jin, C., Lin, F., Suenaga, K. & Iijima, S. Fabrication of a freestanding boron nitride single layer and its defect assignments. Phys. Rev. Lett. 102, 195505 (2009).
Felix, L. C., Tromer, R. M., Autreto, P. A., Ribeiro Junior, L. A. & Galvao, D. S. On the mechanical properties and thermal stability of a recently synthesized monolayer amorphous carbon. J. Phys. Chem. C 124, 14855–14860 (2020).
Liu, L., Feng, Y. & Shen, Z. Structural and electronic properties of h-bn. Phys. Rev. B 68, 104102 (2003).
Wang, Z. et al. Phagraphene: A low-energy graphene allotrope composed of 5-6-7 carbon rings with distorted dirac cones. Nano Lett. 15, 6182–6186 (2015).
Fan, Q. et al. Biphenylene network: A nonbenzenoid carbon allotrope. Science 372, 852–856 (2021).
Song, L. et al. Large scale growth and characterization of atomic hexagonal boron nitride layers. Nano Lett. 10, 3209–3215 (2010).
Zhang, Y.-T. et al. Structure of amorphous two-dimensional materials: Elemental monolayer amorphous carbon versus binary monolayer amorphous boron nitride. Nano Lett. 22, 8018–8024 (2022).
Siqi, J., Shasha, Y., Xiao, W. & Gu, W. T-graphene and its boron nitride analogue as versatile drug delivery systems. Mol. Phys. 118, e1757775 (2020).
Pontes, J., Frazão, N., Azevedo, D. L. & Lima, J. R. Electronic, optical, vibrational and thermodynamic properties of phabn structure: A first principles study. Comput. Mater. Sci. 188, 110210 (2021).
Ma, X.-D., Tian, Z.-W., Jia, R. & Bai, F.-Q. Bn counterpart of biphenylene network: A theoretical investigation. Appl. Surf. Sci. 598, 153674 (2022).
Shahrokhi, M., Mortazavi, B. & Berdiyorov, G. R. New two-dimensional boron nitride allotropes with attractive electronic and optical properties. Solid State Commun. 253, 51–56 (2017).
Rublev, P., Tkachenko, N. V. & Boldyrev, A. I. Overlapping electron density and the global delocalization of π-aromatic fragments as the reason of conductivity of the biphenylene network. J. Comput. Chem. 44, 168 (2022).
Karaush, N. N. et al. Computational study of the structure, UV–Vis absorption spectra and conductivity of biphenylene-based polymers and their boron nitride analogues. RSC Adv. 6, 49505–49516 (2016).
Wang, X. et al. Dhq-graphene: A novel two-dimensional defective graphene for corrosion-resistant coating. J. Mater. Chem. A 7, 8967–8974 (2019).
Bianco, A. et al. All in the graphene family—A recommended nomenclature for two-dimensional carbon materials. Carbon 65, 1–6 (2013).
Cassabois, G., Valvin, P. & Gil, B. Hexagonal boron nitride is an indirect bandgap semiconductor. Nat. Photonics 10, 262–266 (2016).
Wang, B., Wu, Q., Zhang, Y., Ma, L. & Wang, J. Auxetic b4n monolayer: A promising 2d material with in-plane negative Poisson’s ratio and large anisotropic mechanics. ACS Appl. Mater. Interfaces 11, 33231–33237. https://doi.org/10.1021/acsami.9b10472 (2019).
Zhao, Y., Li, X., Liu, J., Zhang, C. & Wang, Q. A new anisotropic dirac cone material: A b2s honeycomb monolayer. J. Phys. Chem. Lett. 9, 1815–1820. https://doi.org/10.1021/acs.jpclett.8b00616 (2018).
Yi, J., Wang, L. & Zhang, Y. Vibration of two-dimensional hexagonal boron nitride. Theor. Appl. Mech. Lett. 8, 408–414 (2018).
Wang, J., Ma, F. & Sun, M. Graphene, hexagonal boron nitride, and their heterostructures: Properties and applications. RSC Adv. 7, 16801–16822 (2017).
Gilbert, S. M. et al. Alternative stacking sequences in hexagonal boron nitride. 2D Mater. 6, 021006 (2019).
Lebedev, A. V., Lebedeva, I. V., Knizhnik, A. A. & Popov, A. M. Interlayer interaction and related properties of bilayer hexagonal boron nitride: Ab initio study. RSC Adv. 6, 6423–6435 (2016).
Kang, J.-X., Harrath, K. & Chen, X. Theoretical study on hydrogen storage of pristine bilayer hexagonal boron nitride. Theoret. Chem. Acc. 140, 72 (2021).
Clark, S. J. et al. First principles methods using castep. Z. Kristallogr. Crystall. Mater. 220, 567–570 (2005).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Heyd, J., Scuseria, G. E. & Ernzerhof, M. Hybrid functionals based on a screened coulomb potential. J. Chem. Phys. 118, 8207–8215 (2003).
Head, J. D. & Zerner, M. C. A Broyden–Fletcher–Goldfarb–Shanno optimization procedure for molecular geometries. Chem. Phys. Lett. 122, 264–270 (1985).
Pfrommer, B. G., Côté, M., Louie, S. G. & Cohen, M. L. Relaxation of crystals with the quasi-newton method. J. Comput. Phys. 131, 233–240. https://doi.org/10.1006/jcph.1996.5612 (1997).
Ceperley, D. M. & Alder, B. J. Ground state of the electron gas by a stochastic method. Phys. Rev. Lett. 45, 566–569. https://doi.org/10.1103/PhysRevLett.45.566 (1980).
Perdew, J. P. & Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 23, 5048–5079. https://doi.org/10.1103/PhysRevB.23.5048 (1981).
Zuo, L., Humbert, M. & Esling, C. Elastic properties of polycrystals in the Voigt–Reuss–Hill approximation. J. Appl. Crystallogr. 25, 751–755. https://doi.org/10.1107/S0021889892004874 (1992).
Chung, D. H. & Buessem, W. R. The Voigt–Reuss–Hill approximation and elastic moduli of polycrystalline MgO, CaF2, \(\beta\)-ZnS, ZnSe, and CdTe. J. Appl. Phys. 38, 2535–2540. https://doi.org/10.1063/1.1709944 (2004).
Acknowledgements
L.A.R.J acknowledges the financial support from Brazilian Research Council FAP-DF Grants, 00193.00001808/2022-71, 00193-00001857/2023-95, 00193-00000857/2021-14, CNPq Grant and 350176/2022-1, and FAP-DF-PRONEM Grant 00193.00001247/2021-20. W.F.G acknowledges the financial support from FAP-DF Grant 00193-00000811/2021-97. This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES)—Finance Code 88887.691997/2022-00. RTDS is supported by CNPq-Brazilian National Research Council (Grant 310941/2022-9 PQ-1D), FAPDF-Brazilian Federal District Research Support Foundation (Grant 625/2022 SIST\(\varepsilon\)R City), and the University of Brasilia (Grant 7129 UnB COPEI).
Author information
Authors and Affiliations
Contributions
K.A.L.L. and F.L.L.M.: Data curation, Formal analysis, Methodology, Prepared Figures, and Writing—Original draft preparation. W.F.G., R.T.S.J., and L.A.R.J.: Conceptualization, Funding acquisition and Writing—Reviewing and Editing. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lopes Lima, K.A., Lopes Mendonça, F.L., Giozza, W.F. et al. Insights into the DHQ-BN: mechanical, electronic, and optical properties. Sci Rep 14, 2510 (2024). https://doi.org/10.1038/s41598-024-52347-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-52347-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
