
Abstract
In this study, a two pyrazole derivatives; 2-(5-methyl-1H-pyrazole-3-carbonyl)-N-phenylhydrazine-1-carboxamide (Pyz-1) and 4-amino-5-(5-methyl-1H-pyrazol-3-yl)-4H-1,2,4-triazole-3-thiol (Pyz-2) were synthesized and characterized by 13C-NMR, 1H-NMR, FT-IR, and mass spectrometry. A complete molecular structures optimization, electronic and thermodynamic properties of Pyz-1 and Pyz-2 in gas phase and aqueous solution were predicted by using hybrid B3LYP method with the 6-311++G** basis sets. Pyz-1 and Pyz-2 were evaluated in vitro for their anti-diabetic, antioxidant and xanthine oxidase inhibition activities. For anti-diabetic activity, Pyz-1 and Pyz-2 showed a potent α-glucosidase and α-amylase inhibition with IC50 values of 75.62 ± 0.56, 95.85 ± 0.92 and 119.3 ± 0.75, 120.2 ± 0.68 µM, respectively, compared to Acarbose (IC50(α-glucosidase) = 72.58 ± 0.68 µM, IC50(α-amylase) = 115.6 ± 0.574 µM). In xanthine oxidase assay, Pyz-1 and Pyz-2 exhibited remarkable inhibitory ability with IC50 values 24.32 ± 0.78 and 10.75 ± 0.54 µM, respectively. The result of antioxidant activities showed that the title compounds have considerable antioxidant and radical scavenger abilities. In addition, molecular docking simulation was used to determine the binding modes and energies between the title compounds and α-glucosidase and α-amylase enzymes.
Similar content being viewed by others
Introduction
Type 2 diabetes mellitus (T2DM) is characterized by high blood glucose levels, which can lead to major complications such as cardiovascular disease, neuropathy, retinopathy and kidney disease1. The disease affects 4.9 million people worldwide, with 90% of diabetes cases attributable to T2DM. It is one of the most common public health problems2,3,4,5. Controlling postprandial hyperglycemia in T2DM by inhibiting dietary carbohydrate digestion (α-amylase and α-glucosidase) is considered one of the most effective therapeutic approaches for lowering blood glucose levels. Thus, α-amylase and α-glucosidase are two enzymes essential for breaking down and degrading dietary carbohydrates such as starch in the digestive tract into simple monosaccharides, in particular, glucose, which passes into the bloodstream after absorption. Consequently, enzymatic inhibition of α-amylase and α-glucosidase can suppress carbohydrate digestion, delay glucose absorption and, consequently, lead to a reduction in blood glucose levels6,7,8. On the other hand, several chronic diseases such as T2DM have been related to oxidative stress which entails the production of reactive oxygen species (ROS) such as the hydroxyl radical (OH–), hydrogen peroxide (H2O2) and superoxide anion radical (O2–)9. On other hand, the increase of malondialdehyde (MDA) concentration by lipid peroxidation in pancreatic tissue of diabetic animal models confirmed the role of ROS in the pathogenesis and progression of diabetes. Thus, in vivo animal studies have shown that potent inhibition of oxidative stress by specific anti-oxidants under experimental diabetic conditions has shown preventive effects on the progression of diabetic complications10,11. Therefore, it would be very beneficial to develop new compounds with both anti-diabetic and antioxidant activities to minimize adverse effects.
Indeed, pyrazole constitute a large class of heterocycles which can be explored for the development of new drug substances. An extensive study of this class has demonstrated that pyrazole can be present in various known drugs of different classes with different therapeutic activities12,13. In the last few years, pyrazole chemistry and its derivatives have attracted considerable interest due to their broad spectrum of biological activities, such as antidiabetic14, antibacterial15,16, antioxidant and analgesic17, anticancer18, antiviral19 and antituberculosis agents20. Considering the biological and pharmacological properties of these derivatives, it is essential to study their electronic and structural properties to know the influence of the different groups on their biological properties21,22. For this purpose, computational approaches are known for their efficiency and accuracy when it comes to predict and understand the properties of novel synthesized organic molecules23. In continuation of our efforts to develop new potential anti-diabetic and antioxidant agents13,19,20, we report herein the synthesis and characterization of two pyrazole derivatives Pyz-1 and Pyz-2 as new potent antiadiabetic and antioxidant agents. The molecular geometry and electronic characters of these molecules were explored by using DFT calculations. Thus, the title compounds were evaluated in vitro for their α-glucosidase and α-amylase enzymes inhibitory. Antioxidant activities of Pyz-1 and Pyz-2 were determined by DPPH, ABTS, FRAP, H2O2 radical scavenging activity assays and Xanthine Oxidase (XO) inhibition assay. In addition, molecular docking and ADMET studies were also performed.
Experimental
Reagents and instruments
Chemical reagents (Methanol, Ethanol, Phenyl isocyante, Potassium hydroxide, hydrazine hydrate, Carbon disulfide and Hydrochloric acid) were purchased from Fluka, Sigma and Aldrich chemicals. Melting points were measured using a Buchi B-545 digital capillary melting point apparatus. TLC with silica gel 60 F254 were used to check the reactions. The IR spectra were recorded by using Perkin-Elmer VERTEX 70 FT-IR spectrometer covering field 400–4.000 cm−1. The 1H NMR and 13C NMR spectra were recorded using JNM-ECZ500R/S1 FT NMR SYSTEM (JEOL) (1H 500 MHz/13C 125 MHz) spectrometer by using dry deuterated DMSO as solvent. The Mass spectra were obtained using an API 3200 LC/MS/MS spectrometer.
Chemistry
Procedure for the synthesis of 2-(5-methyl-1H-pyrazole-3-carbonyl)-N-phenylhydrazine-1-carboxamide (Pyz-1)
5-methyl-1H-pyrazole-3-carbohydrazide (1) was synthesized according the previously reported method14,17. Phenyl isocyanate (0.5 g, 4.2 mmol) was added dropwise to a solution of 5-methyl-1H-pyrazole-3-carbohydrazide (1) (0.5 g, 3.75 mmol) in methanol (20 ml), and the mixture was refluxed for 8 h. The solid was filtered and recrystallized from ethanol to afford pure product (Pyz-1). White solid; Yield = 81%; m.p = 252–254 °C; FT-IR (ATR, cm−1): 3129–3295 (NH), 2934–3000 (CH), 1703, 1650 (C = O), 1602 (C = N); 1H NMR: (500 MHz, DMSO-d6, δ(ppm)): 2.23 (3H, s, CH3), 6.42 (1H, s, CH-pyrazole), 6.89–7.43 (m, 5H, H-Ar), 8.04 (s, 1H, NH), 8.73 (s, 1H, NH), 9.75 (s, 1H, NH), 12.99 (1H, s, NH-pz); 13C NMR: (125 MHz, DMSO-d6, δ (ppm)): 10.82, 105.06, 118.87, 122.31, 129.18, 140.17, 140.27, 145.89, 156.04, 162.61. ESI-HRMS: m/z calcd. for C12H13N5O2 [M-H]–: 258.1102, found 258.0986.
Procedure for the synthesis of 4-amino-5-(5-methyl-1H-pyrazol-3-yl)-4H-1,2,4-triazole-3-thiol (Pyz-2)
To an ice cooled solution of 5-methyl-1H-pyrazole-3-carbohydrazide (1) (0.5 g, 3.75 mmol) and KOH (0.2 g, 3.75 mmol) in absolute ethanol (15 mL), was added CS2 (0.65 ml, 10.71 mmol) dropwise, and the mixture was stirred at room temperature for 12 h. The separated solid was filtered and washed with ethanol. The obtained pyrazole-potassium salt was used in the next reaction without further purification. To a solution of the pyrazole carbodthionate (0.5 g, 1.97 mmol) in ethanol (10 ml) was added hydrazine hydrate (1 ml) and the mixture was refluxed for 8 h. The completion of the reaction was monitored by TLC. The reaction mixture was diluted with cold water and neutralized with concentrated HCl. The solid was filtered and recrystallized from ethanol to afford pure product (Pyz-2). White solid; Yield = 67%; m.p = 216–218 °C; FT-IR (ATR, cm−1): 3144–3367 (NH, NH2), 2732 (SH); 1H NMR: (500 MHz, DMSO-d6, δ(ppm)): 2.25 (3H, s, CH3), 5.89 (2H, s, NH2), 6.60 (1H, s, CHpyrazole), 13.09 (1H, s, NH-pz), 13.66 (1H, s, SH); 13C NMR: (125 MHz, DMSO-d6, δ (ppm)): 10.71, 105.08, 138.78, 139.97, 145.46, 165.17; ESI-HRMS: m/z calcd. for C6H8N6S [M + H]+: 197.0545, found 197.0586.
Computational details
Two series of calculations were conducted in an attempt to deduce and interpret the experimental data. First, The DFT computations were carried out in order to gather information regarding the reactivity of the synthesized compounds. Then, a molecular docking simulation was used to determine the binding modes and energies between the compounds (Pyz-1 and Pyz-2) and α-glucosidase and α-amylase enzymes. A complete molecular structures optimization was performed using the B3LYP hybrid functional24,25 in conjunction with the 6-311G + + (d,p)26,27,28,29,30 basis set in order to determine the respective total energy and the most stable gas-phase geometry. Throughout the geometry optimization, no symmetry constraints were imposed. The absence of imaginary frequencies in the calculation of the vibrational modes indicates that these structures correspond to real local minima on the potential energy surface. The effects of water as a solvent (ε = 46.7) were examined using a self-consistent reaction field (SCRF)31 based on the polarizable continuum model (PCM) of the Thomasi group32,33 via a single-point calculation of the optimized geometries performed in the phase gas. Molecular electrostatic potential (MEP) surfaces were obtained using the Multiwfn program34 at the same level of theory. The obtained results were visualized using the VMD 1.9 software35. Gaussian NBO Version 3.136,37,38 was used to perform the natural bond orbital (NBO) calculations. To analyze and evaluate the tendency of the drug for charge transfer processes and drug-receptor interactions39, several theoretical reactivity parameters40 based on the conceptual density functional theory (CDFT)41. All calculations were performed using the Gaussian09 suite of program42 and GaussView graphical interface43.
Molecular docking
To investigate possible binding modes of selected chemical entities, a docking simulation employing the AutodockVina v1.5.644 was carried out targeting the α-glucosidase and α-amylase enzyme active sites45. Pyz-1, Pyz-2 and Acarbose structures have been designed by using ChemDraw softaware package and were optimized using the molecular building module implemented in ChemDraw. Thus, crystal structure of α-glucosidase (PDB Id: 3A4A) and α-amylase (PDB Id: 2GJP) were downloaded from the PDB database (https://www.rcsb.org/pdb). Potential binding sites of α-amylase and α-glucosidase were identified using Discovery Studio software45, based on the receptor structure without prior introduction of the ligand. The preparation of the binding site geometry was performed using AutoGrid, employing a grid with multiple points defined by coordinates (x, y, z) = (20, 20, 20), with a grid spacing of 0.375 Å. The different interactions within the protein–ligand complexes were analyzed using the Discovery 2016 Studio Visualizer45 and PyMol molecular graphing system46. Pharmacokinetic and pharmacodynamic parameters of Pyz-1 and Pyz-2 were computed by means of the ADMETLab2.0 server47.
Biology
α-Glucosidase and α-amylase inhibitory assays
The α-glucosidase and α-amylase inhibitory activities of compounds Pyz-1 and Pyz-2 were determined using the method described in our previous works48,49.
Antioxidant activities
The antioxidant activities of the title compound were determined in vitro by DPPH, ABTS, FRAP and hydrogen peroxide activity (H2O2) methods according to the procedures described in our previous works49.
Xanthine oxidase inhibition assay (XO)
The xanthine oxidase inhibitory activity of Pyz-1 and Pyz-2 was performed following the method described by Kostić et al.50, with minor modifications. The samples were prepared at series of concentrations. 1.95 ml of 5 mM phosphate buffer pH 7.5 and 50 µl of enzyme solution was added to the mixtures. After pre-incubation at 25 °C for 15 min. 1 ml of substrate was added. After 30 min add 0.5 ml of HCl to stop the reaction the absorbance was determined spectrophotometrically at 295 nm associated with uric acid formation.
Statistical analysis
Statistical analyze was performed using GraphPad Prism8 program. One-way ANOVA was used to determine the significant difference. Quantitative data were presented as mean ± standard error of the mean (SEM) and p < 0.0001 was considered statistically highly significant.
Results and discussion
Chemistry
First, the synthesis protocol of Pyz-1 and Pyz-2 compounds was performed according to Scheme 1. The starting hydrazide acid (1) was synthesized according the previously reported method49. The compound (1) was treated with CS2 in ethanol in the presence of KOH as base at room temperature to afford the intermediate potassium salt in quantitative yield. Thus, treatment of this intermediate with hydrazine hydrate under ethanol reflux afforded a pure product (Pyz-1) in good yield. Subsequently, 2-(5-methyl-1H-pyrazole-3-carbonyl)-N-phenylhydrazine-1-carbothioamide (Pyz-2) was prepared in excellent yield by condensation of pyrazole hydrazide (1) and phenyl isocyanate under reflux of methanol.

Synthetic route for preparation of compounds Pyz-1 and Pyz-2.
The molecular structures of Pyz-1 and Pyz-2 were confirmed by using 1H-NMR, 13C-NMR and ESI–MS spectrometry. The 1H NMR spectrum of Pyz-1 shows a singlet at 2.18 ppm characteristic of methyl group, a singlet at 6.42 ppm due to the proton in position 3 of the pyrazole ring. Aromatic protons of phenyl group resonate as multiplet at 6.92–7.44 ppm. The NH protons of carboxamide group appear as three singlets at 8.02, 8.72 and 9.74 ppm. The singlet at 12.99 ppm due to the NH group of pyrazole. The 13C NMR spectrum of Pyz-1, shows a signal at 10.75 ppm characteristic to carbon of the CH3 group, a signal at 105.97 ppm characteristic to the tertiary carbon of pyrazole, followed by the carbons of the aromatic part which appear between 118.78 and 140.9 ppm. The two quaternary carbons of pyrazole resonate at 140.19 and 145.81 ppm. The two signals observed at 155.94 and 162.51 ppm are attributed to the two C = O groups.
The 1H NMR spectrum of Pyz-2 shows five singlet at 2.25, 5.89, 6.60, 13.09 and 13.66 ppm characteristic of methyl, NH2, H4 of pyrazole, NH and SH groups, respectively. The 13C NMR spectrum of Pyz-2, shows a signal at 10.69 ppm associated with the carbon of the CH3 group, a signal at 05.08 ppm characteristic of the tertiary carbon of the pyrazole, followed by two signals of the quaternary carbons of pyrazole ring, which appear at 138.64 and 139.90 ppm, and the two characteristic signals of carbons 1,2,4-triazole resonate at 155.40 and 165.06 ppm.
The mass spectra (ESI) show a peak related to the molecular ions at m/z = 260.2 and 197.0 thus confirming the proposed structures of Pyz-1 and Pyz-2, respectively.
Computational results
Electronic properties
Electronic and thermodynamic parameters are an effective way to explain the stability and reactivity of molecules. The physical quantities of Pyz-1 and Pyz-2 calculated at B3LYP/6–311G + + (d,p) level of theory are given in Table 1, the optimized structures of the most stable conformer of Pyz-1 and Pyz-2 with the numbering of atoms are shown in Fig. 1.

Optimized molecular structures and FMO’s density distributions of (a) Pyz-1 and (b) Pyz-2.
The total energies of title molecules have been calculated for Pyz-1 as − 889.560233 au in gas phase and − 889.580733 au in solvated phase and for Pyz-2 as − 960.268510 au in gas phase and − 960.287960 au in solvated phase. The thermodynamic and kinetic preferability is reduced with the introduction of solvation resulting in higher stability in aqueous phase relative to the gas phase. The quantity of heat needed to increase a substance's temperature by one degree is referred to as the heat capacity, the Cv quantities of the compounds were computed as 65.152 cal/molK in gas phase and 65.480 cal/molK in water phase for Pyz-1 and 45.239 cal/molK in gas phase and 43.155 cal/molK in water phase. The randomness of the system is quantitatively measured by the entropy. The computed values for Pyz-1 and Pyz-2 in gas and water phases are respectively 142.552 cal/molK, 143.766 cal/molK, and 115.203 cal/molK and 108.210 cal/molK. These findings are a major asset in the application of such structures to biological systems.
FMO’s analysis
Within computational chemistry, the energies and allocations of FMO’s are imperative reactivity descriptors. The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) have significant roles in the electrical, electronic, optical properties, chemical reactivity and active sites of a given compound51. The HOMO's energy is proportional to the ionization potential (I) and describes the molecules sensitivity to electrophile attack52. The energy of LUMO is linked to the electron affinity (A) and defines the molecule's response to nucleophilic attack. A graphical illustration of the HOMO and LUMO for Pyz-1 and Pyz-2 is presented in Fig. 1. The positive phase is colored red, whereas the negative phase is colored blue. As shown in Fig. 1, with the exception of the methyl (CH3) groups, the highest HOMO and lowest LUMO orbitals of the Pyz-1 and Pyz-2 molecules are mainly localized almost throughout the structures.
The energy difference between the HOMO and LUMO orbitals determines the kinetic stability of the title compounds. Thus, molecules with a small energy gap are highly polarizable and are usually associated with high chemical reactivity as well as low kinetic stability53. According to the indications in the literature and the results reported in Table 2, a slight trend towards an increase in the energy gap can be observed when introducing the solvent effect for both titled molecules resulting in a lower reactivity in the aqueous phase. Furthermore, the energy gap reveals that Pyz-1 (ELUMO-HOMO = 5.118 eV) exhibits a lower value compared to Pyz-2 (ELUMO-HOMO = 5.166 eV), indicating easy molecular charge transfers, low kinetic stability and high chemical reactivity for Pyz-1.
Reactivity descriptors
Due to their wide range of applications in fields such as biology, chemistry and drug design, the development of chemical reactivity descriptors has gained considerable momentum54. Conceptual DFT theory is a developed branch of DFT which consists in deriving relevant concepts and principles from the electron density in order to make it possible to understand and predict the overall trends in chemical reactivity of a molecule55. With this aim, quantum chemical reactivity parameters have been calculated for the title compounds and their values are reported in Table 3.
Global hardness (η) is related to the resistance to electronic transfer between a molecule and its environment. It represents the gap between the EHOMO and ELUMO orbital energies and is associated with the stability of chemical systems. Soft molecules exhibit a small energy gap and are more reactive than hard ones because of their ability to easily donate electrons to an acceptor. The calculations show that Pyz-1 is less resistant to electronic transfer than Pyz-2 and therefore would be more reactive. It should be noted that whenever the ratio of energy change to maximum charge acceptance ∆Ec/∆Nmax = 0, this indicates that the molecule under consideration is electron-saturated and has no predisposition for charge transfer56. According to the tabulated values in Table 3, the ∆Ec/∆Nmax values are − 1.795 eV and − 1.761 eV for Pyz-1 and Pyz-2, respectively, with a maximum charge acceptance value of approximately 0.7 eV for both compounds. These values clarify the intramolecular charge transfer within the studied compounds and their ability to interact and bind to the active sites of α-glucosidase and α-amylase enzymes. Furthermore, the compounds' behavior suggests a tendency toward electrodonation rather than electroacceptance (ω− > ω+).
Molecular electrostatic potential (MEP) map
Molecular electrostatic potential is a three-dimensional representation allowing the visualization of charge distributions, the prediction of reactivity towards electrophilic and nucleophilic attacks and the relative polarity of a given molecule57. Moreover, it is used in biological recognition processes and to evaluate hydrogen bonding interactions. The visualization of the MEP relies on the mapping of the values onto the area corresponding to the boundaries of the molecule using the BWR (BlueWhite-Red) color transition: red, blue and white representing the regions of most positive, most negative and neutral electrostatic potential respectively58. The 3D maps of the MEP of the investigated molecules are displayed in Fig. 2.

Computed molecular electrostatic potential surface (MEP) of Pyz-1 and Pyz-2.
A high polarity is noted for Pyz-1 and Pyz-2 molecules. Indeed, we can notice that two distinct regions are formed: an electronically deficient region formed around the hydrogen atoms on the amine groups due to their high electronegativity, leading to a maximum value of 51.83 kcal/mol and 55.01 kcal/mol respectively for Pyz-1 and Pyz-2. These observations reveal a strong tendency to attract negatively charged atoms electrostatically and to act as a hydrogen bond donor group. Furthermore, For Pyz-1, the overall minimum values on the surface are − 40.99 kcal/mol and − 40.54 kcal/mol due to the isolated pairs of ketone groups. Whereas, for Pyz-2 the minimum values are − 46.38 kcal/mol and − 45.19 kcal/mol owing to isolated pairs of nitrogen atoms which are the most reactive sites for electrophilic attack during the interaction. From the MEP map, it can be derived that Pyz-1 and Pyz-2 molecules have significant biochemical activity and are key to drug recognition in biological systems.
NBO analysis
Natural bond orbital (NBO) analysis represents a computational tool for a more understandable and user-friendly interpretation of the computational solutions of the Schrödinger equation in chemical bonding concepts59. This approach allows studying the distribution of electron density on atoms and in bonds in an efficient way. It also provides a convenient way to examine charge transfer or conjugate interaction in molecular systems60. A useful aspect of the NBO method is that it provides information about the interactions of both filled (donor) orbital spaces (bonding orbitals or solitary pairs) and empty (anti-bonding and/or Rydberg orbitals) that could improve the analysis of intra-inter molecular interactions61. The strength of the interaction between electron donors and electron acceptors and thus the degree of conjugation of the system is measured by the value of the energy of hyperconjugal interactions, E(2). The larger this value, the stronger the interaction between electron donors and electron acceptors and the greater the degree of conjugation of the whole system.
To assess the donor–acceptor interactions, the second-order Fock matrix was performed. The stabilization energy E(2) associated with the i → j delocalization for each donor (i) and acceptor (j) is estimated as:
where qi is the donor orbital occupancy (2 for closed-shell, 1 for openshell), εi, εj are diagonal elements (orbital energies), and Fij is the off-diagonal NBO Fock matrix element.
For both compounds, the most important interaction analyses between the occupied Lewis-type NBO orbital (bonding) and the unoccupied non-Lewis-type NBO orbital (anti-bonding) have been calculated and donor–acceptor interactions having a stabilization energy above 20 kcal/mol are presented in Table S1 and S2. Inspection of different donors and acceptors reveals three types of donor’s LP, σ and π, and two types of acceptors σ* and π*. In Pyz-2, a remarkable stabilization energy contribution was noticed due to the interaction of filled molecular orbitals σ(N13-H21) and lone pairs LP(N13) with unfilled σ*(C7-N11) molecular orbital. The perturbative energy for these interactions are respectively 704.06 kcal/mol and 556.85 kcal/mol, which result in an important intramolecular charge transfer (ICT) causing a high stabilization of the investigated compound. The essential interaction energies related to SM3 molecule reveal that the most relevant donor–acceptor interactions are the interactions between the lone electron pair (LP) of the nitrogen atoms LP(N9) and LP(N1) to the anti-bonding acceptor π*(C7-O8), π*(C4-C5) and π*(N2-C3) orbitals. The interaction energies reported for the mentioned conjugative interactions are 61.25 kcal/mol, 37.87 kcal/mol and 31.35 kcal/mol respectively. The relatively strong electron donor donation from the π(C14-C16) → π*(C15-C17) (20.89 kcal/mol), π(C15-C17) → π*(C18-C19) (22.11 kcal/mol) and π(C18-C19) → π*(C14-C16) (21.34 kcal/mol) of the aromatic ring are identified. This phenomenon results from intramolecular hyperconjugative interactions between the π (C–C) and π*(C–C) orbitals, which lead to an ICT causing stabilization of the system.
Biological activities
Antidiabetic activity
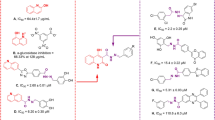
Compounds Pyz-1 and Pyz-2 were studied in vitro for their antidiabetic activities against α-glucosidase and α-amylase enzymes. Acarbose was used standard drug. The results of α-glucosidase and α-amylase inhibitory activity are summarized in Table 4. As shown in the Table 4, compound 1 showed the lowest activity against α-glucosidase and α-amylase with IC50 values of 279.0 and 500 µM, respectively. Pyz-1 and Pyz-2 exhibited good α-glucosidase and α-amylase inhibitory activities with IC50 values of 75.62 ± 0.56 and 95.85 ± 0.92 µM, and 120.2 ± 0.68, 119.3 ± 0.75 µM, respectively, compared to Acarbose (IC50(α-glucosidase) = 72.58 ± 0.68 and IC50(α-amylase) = 115.6 ± 0.574 µM). The obtained results showed a highly significant inhibition on α-amylase and α-glycosidase for the studied compounds. It was found that structural modification of carbohydrazide 1 resulted in increased enzymatic inhibition for compounds Pyz-1 and Pyz-2.
Antioxidant Activity
The antioxidant properties of the target compounds were determined by using DPPH, ABTS, H2O2 free radical scavenging, reducing power (FRAP), and Xanthine Oxidase (XO) assays. Ascorbic acid (A.A) and allopurinol were used as a standard. The results are summarized in Table 5.
For DPPH assay, the antioxidant activity of Pyz-2 was the highest as its SC50 value was the lowest (138.70 ± 1.45 µM), followed by Pyz-1 with SC50 value of 238.53 ± 1.12 µM. Pyz-1 and Pyz-2 exhibited moderate antioxidant capacities. For ABTS assay, compound Pyz-2 exhibited the potent scavenging activity with SC50 value of 20.54 ± 0.87 µM, which is 3.88-fold more than that Pyz-1 (SC50 = 79.77 ± 0.45 µM). Pyz-2 has an antioxidant activity comparable to that of A.A (SC50 = 22.49 µM). For the reducing power ability (FRAP), Pyz-2 displayed the maximum reducing power with IC50 value of 62.62 ± 0.49 µM. Statistically, the reducing power of Pyz-2 was stronger than A.A (IC50 = 88.12 µM). Moreover, in H2O2 method, Pyz-1 and Pyz-2 displayed a good antioxidant activity with IC50 values of 31.49 ± 0.92 and 24.82 ± 1.23 μM, respectively, compared to A.A (IC50 = 7.45 µM).
The results inhibitory effect on xanthine oxidase of title compounds, showed that Pyz-1 exhibited a considerable XO activity with IC50 value of 24.32 ± 0.78 µM. Furthermore, Pyz-2 manifested the highest XO inhibitory activity with IC50 value of 10.75 ± 0.54 µM, compared to Allopurinol (IC50 = 14.41 ± 1.01 µM).
Docking studies
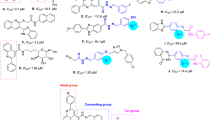
The binding interaction of the 1, Pyz-1 and Pyz-2 ligands with α-amylase and α-glucosidase was further examined by performing an in silico binding interaction analysis to understand their interactions at the enzymes active site. Similarly, Acarbose (ACA), a standard type 2 antidiabetic drug, was docked as a positive control. The most probable docking positions of ligands exhibiting the best binding affinity towards targeted enzyme α-amylase and α-glucosidase are grouped in Table 6 and illustrated in Figs. 3 and 4.

Binding between the docked compounds (1, Pyz-1, Pyz-2 and Acarbose) and α-amylase (PDB = 2GJP).

Binding between the docked compounds (1, Pyz-1, Pyz-2 and Acarbose) and α-glucosidase (PDB:3A4A).
As noted from Table 6, all values of binding energies are negative which stabilize the systems and favor interactions between the proteins and the ligands.
Against α-amylase enzyme target (PDB = 2GJP)
Among the newly suggested alternative compounds for treating diabetes, Pyz-1 and Pyz-2 demonstrate binding energies of − 6.7 kcal/mol and − 5.6 kcal/mol, respectively, both higher than the binding energy of ligand 1, which stands at − 5.3 kcal/mol.
Figure 3 presented the interacted amino acid residues of α-amylase with Pyz-1. The surrounding amino acid residues of the enzyme included Tyr198, Trp268, Ala237, Leu201 and Leu340. Two hydrogen bonds are formed in Pyz-1-α-amylase complex between hydrogen atom of Tyr198 residue and lone pairs of oxygen atom of keto group and between lone pairs of oxygen atom of keto group of Trp268 residue and hydrogen atom of amin group of Pyz-1 for a distance of 2.44 Å and 1.92 Å respectively. Furthermore, the π-sigma and π-Alkyl interactions between benzene ring and amino acids residues, consolidated the Pyz-1 scaffold in the Pyz-1-α-amylase complex. Pyz-2 molecule interacted α-amylase by establishing a conventional hydrogen bond with Glu266 (2.34 Å) and two hydrogen bonds with Trp268 (2.30 Å and 2.60 Å) (Fig. 3). This compound also exhibits π-cation, π-Sulfur, π-Alkyl and π-π interactions with Leu201, His240 and Tyr198. For acarbose, selected as a reference, the binding study revealed the involvement of fourteen amino acids in the molecular interactions, including five hydrogen bonds with the amino acids Lys269, Glu266 and Tyr198, two carbon hydrogen bonds with Asn270 and Tyr268 amino acids as well as eight Van der Waals type interactions with Asp271, Tyr295, Arg234, Ala237, His240, Leu340, Lys239 and Glu194 amino acids (Fig. 3). Unfavorable donor-donor and acceptor-acceptor interactions are also observed with the amino acid Asp333. Regarding compound 1, the affinity with α-amylase resulted in a binding energy of − 5.3 kcal/mol.
Against α-glucosidase enzyme target (PDB = 3A4A)
From the Table 6, the positive control ACA was found to be most effective α-glucosidase binder, thus forming the most stable complex with the lowest energy (− 5.9 kcal/mol) and interacting with Pro467, Trp468, Lys466, Asp34, Trp36 and Pro82 amino acids via strong hydrogen bonds (H-acceptor and H-donor). The second-most effective binder was Pyz-1 with an energy value of − 5.3 kcal/mol. Seven Van der Waals interactions, two π-π interactions and a single carbon hydrogen bond stabilized the Pyz-1-α-glucosidase complex (Fig. 4). Thus, the residues realizing Van der Waals interactions were Gln67, Met70, Arg413, Trp468, Glu408, Tyr407 and Lys406. Furthermore, amino acid residues such as Trp36 and Tyr470 also made a contribution to the stability of the complex through π-π interactions (Fig. 4). In the less stable Pyz-2-α-glucosidase complex (− 4.3 kcal/mol), numerous intermolecular interactions were established between the docked pyrazole and the active α-glucosidase amino acids, including classical hydrogen bond, Van der Waals interactions, π-Alkyl and π–π interactions (Fig. 4). Concerning compound 1, the binding energy with α-glucosidase was − 5.3 kcal/mol. In general, the binding energy analysis performed in this section for each coupling between the ligands (Acarbose, 1, Pyz-1 and Pyz-2) and the α-amylase and α-glucosidase macromolecule gives valuable information for predicting the affinity with the proteins. This analysis identifies which amino acid of proteins is in contact with the docked ligands in the active site.
ADME-T analysis
In this study, a computational study of Pyz-1 and Pyz-2 was conducted to determine the surface area and other physicochemical properties according to the directions of Lipinski’s rule62,63. Lipinski suggested that the absorption of a compound is more likely to be better if the molecule achieve at least three out of four of the following rules: (i) HB donor groups ≤ 5; (ii) HB acceptor groups ≤ 10; (iii) M. Wt less than 500; (iv) logP less than 5. As shown in Table 7, the two compounds (Pyz-1 and Pyz-2) obey all Lipinski's rules. Pyz-1 and Pyz-2 have a number of hydrogen‐bonding acceptor groups 3 and 2 and only 4 and 3 hydrogen‐bonding donors, respectively. Also, molecular weights are less than 500 and logP less than 5 and all these values agree with Lipinski's rules.
Also, ADMET profiles of Pyz-1 and Pyz-2 were preliminary assessed to analyze their potentials to build up as good medication candidates64,65,66. As shown in Table 7, the initial logD values of Pyz-1 and Pyz-2 are 1.447 and 0.245, respectively, thereby suggesting that the distribution of the title compounds remains constant up to pH 6.0, and then starts to vary with the formation of charged species. On the other hand, the logP values of Pyz-1 and Pyz-2 are 1.044 and 0.64, respectively, suggest that the title compounds may be slightly permeable to the biological barriers of physiological system, thus still having lipophilicity considered ideal in the intestinal absorption phase. The PSA value of Pyz-1 and Pyz-2 estimated at 98.91 and 88.31, respectively, shows a low permeability to the different biological barriers, with a slight affinity for plasma proteins evaluated at 29.32% and 30.68%.
Looking at the calculated parameter values, we find that these values are within the recommended standard. Thus, we can conclude that the molecules of the title had characteristics of drugability and their application as a theoretical drug is safe.
Conclusion
In conclusion, two novel pyrazole derivatives Pyz-1 and Pyz-2 were synthesized, characterized and evaluated for their antidiabetic and antioxidant activities. Pyz-1 and Pyz-2 were evaluated in vitro for their anti-diabetic, antioxidant activities. For anti-diabetic activity result, Pyz-1 and Pyz-2 showed a potent α-glucosidase and α-amylase inhibition. For antioxidant activity results, compound Pyz-2 showed excellent activity that the title compounds have considerable antioxidant and radical scavenger abilities. In xanthine oxidase assay, Pyz-1 and Pyz-2 exhibited remarkable inhibitory ability with IC50 values 24.32 ± 0.78 and 10.75 ± 0.54 µM, respectively. Also, molecular structure optimization, electronic and thermodynamic properties of Pyz-1 and Pyz-2 through DFT calculations supported the antidiabetic and antioxidant activities of the title compounds. However, as docking results it was found to have higher activity of compound Pyz-1 with a docking score of -5.7 kcal/mol against α-glucosidase and -6.7 kcal/mol against α-amylase. The predicted ADMET profiles of the title compounds were in line with Lipinski's rules and we can conclude that Pyz-1 and Pyz-2 had drugability characteristics. These promising results encourage further study of the antidiabetic and antioxidant activities of the title compounds by measuring their toxicity. Thus, advanced in vivo studies are in progress and will be published elsewhere.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
Chatterjee, S., Khunti, K. & Davies, M. J. Type 2 diabetes. The Lancet 389, 2239–2251 (2017).
Ademiluyi, A. O. & Oboh, G. Soybean phenolic-rich extracts inhibit key-enzymes linked to type 2 diabetes (α-amylase and α-glucosidase) and hypertension (angiotensin I converting enzyme) in vitro. Exp. Toxicol. Pathol. 65, 305–309 (2013).
Gao, Z. et al. Automatic interpretation and clinical evaluation for fundus fluorescein angiography images of diabetic retinopathy patients by deep learning. British J. Ophthalmol. 107, 1852–1858 (2023).
Yang, Y. Y., Shi, L. X., Li, J. H., Yao, L. Y. & Xiang, D. X. Piperazine ferulate ameliorates the development of diabetic nephropathy by regulating endothelial nitric oxide synthase. Mol. Med. Rep. 19, 2245–2253 (2019).
Chen, J. et al. Bone marrow stromal cell-derived exosomal circular RNA improves diabetic foot ulcer wound healing by activating the nuclear factor erythroid 2-related factor 2 pathway and inhibiting ferroptosis. Diabetes Med. 1, e15031 (2023).
Pai, Y. W. et al. Glycaemic control for painful diabetic peripheral neuropathy is more than fasting plasma glucose and glycated haemoglobin. Diabetes Metab. 47, 101158 (2021).
Chen, Y., Tan, S., Liu, M. & Li, J. LncRNA TINCR is downregulated in diabetic cardiomyopathy and relates to cardiomyocyte apoptosis. Scand. Cardiovasc. J. 52, 335–339 (2018).
Wang, H., Yang, T., Wu, J., Chen, D. & Wang, W. Unveiling the Mystery of SUMO-activating enzyme subunit 1: A groundbreaking biomarker in the early detection and advancement of hepatocellular carcinoma. Transplant. Proc. 1, 945–951 (2023).
Asmat, U., Abad, K. & Ismail, K. Diabetes mellitus and oxidative stress—A concise review. Saudi Pharm. J. 24, 547–553 (2016).
Araki, E. & Nishikawa, T. Oxidative stress: A cause and therapeutic target of diabetic complications. J. Diabetes Investig. 1, 90–96 (2010).
Matough, F. A., Budin, S. B., Hamid, Z. A., Alwahaibi, N. & Mohamed, J. The role of oxidative stress and antioxidants in diabetic complications. Sultan Qaboos Univ. Med. J. 12, 5 (2012).
Karrouchi, K. et al. Synthesis and pharmacological activities of pyrazole derivatives: A review. Molecules 23, 134 (2018).
Bennani, F. E. et al. Overview of recent developments of pyrazole derivatives as an anticancer agent in different cell line. Bioorg. Chem. 97, 103470 (2020).
Karrouchi, K. et al. Synthesis, X-ray structure, vibrational spectroscopy, DFT, biological evaluation and molecular docking studies of (E)-N’-(4-(dimethylamino) benzylidene)-5-methyl-1H-pyrazole-3-carbohydrazide. J. Mol. Struct. 1219, 128541 (2020).
Chaudhary, M. et al. Chloro and bromo-pyrazole curcumin Knoevenagel condensates augmented anticancer activity against human cervical cancer cells: design, synthesis, in silico docking and in vitro cytotoxicity analysis. J. Biomol. Struct. Dyn. 38, 200–218 (2020).
Tighadouini, S. et al. Synthesis, crystal structure, DFT studies and biological activity of (Z)-3-(3-bromophenyl)-1-(1, 5-dimethyl-1 H-pyrazol-3-yl)-3-hydroxyprop-2-en-1-one. Chem. Central J. 12, 1–11 (2018).
Karrouchi, K. et al. Synthesis, α-glucosidase inhibition, anticancer, DFT and molecular docking investigations of pyrazole hydrazone derivatives. Polycycl. Aromat. Compd. 1, 1–20 (2022).
Karrouchi, K. et al. Synthesis, characterization, free-radical scavenging capacity and antioxidant activity of novel series of hydrazone, 1, 3, 4-oxadiazole and 1, 2, 4-triazole derived from 3, 5-dimethyl-1H-pyrazole. Lett. Drug Des. Discov. 16, 712–720 (2019).
Abu-Melha, S. et al. Clean grinding technique: A facile synthesis and in silico antiviral activity of hydrazones, pyrazoles, and pyrazines bearing thiazole moiety against SARS-CoV-2 main protease (Mpro). Molecules 25, 4565 (2020).
Pogaku, V., Krishna, V. S., Sriram, D., Rangan, K. & Basavoju, S. Ultrasonication-ionic liquid synergy for the synthesis of new potent anti-tuberculosis 1, 2, 4-triazol-1-yl-pyrazole based spirooxindolopyrrolizidines. Bioorg. Med. Chem. Lett. 29, 1682–1687 (2019).
Karrouchi, K. et al. Synthesis, crystal structure, DFT, α-glucosidase and α-amylase inhibition and molecular docking studies of (E)-N’-(4-chlorobenzylidene)-5-phenyl-1H-pyrazole-3-carbohydrazide. J. Mol. Struct. 1245, 131067 (2021).
Mortada, S. et al. Synthesis, spectroscopic and DFT studies of 5-methyl-1H-pyrazole-3-carbohydrazide N-glycoside as potential anti-diabetic and antioxidant agent. J. Mol. Struct. 1267, 133652 (2022).
Karrouchi, K. et al. Synthesis, X-ray, spectroscopy, molecular docking and DFT calculations of (E)-N’-(2, 4-dichlorobenzylidene)-5-phenyl-1H-pyrazole-3-carbohydrazide. J. Mol. Struct. 1228, 129714 (2021).
Lee, C., Yang, W. & Parr, R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785 (1988).
Becke, A. D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 98, 1372–1377 (1993).
Krishnan, R. B. J. S., Binkley, J. S., Seeger, R. & Pople, J. A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 72, 650–654 (1980).
McLean, A. D. & Chandler, G. S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z= 11–18. J. Chem. Phys. 72, 5639–5648 (1980).
Kainat, S. et al. Theoretical modeling of B12N12 nanocage for the effective removal of paracetamol from drinking water. Computation 11, 183 (2023).
Shahab, M. et al. Structure based virtual screening and molecular simulation study of FDA-approved drugs to inhibit human HDAC6 and VISTA as dual cancer immunotherapy. Sci. Rep. 13, 14466 (2023).
Ali, Q. et al. Theoretical insight of ciprofloxacin removal from water using boron nitride (B12N12) nanocage. Surf. Interfaces. 31, 101982 (2022).
Cances, E., Mennucci, B. & Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 107, 3032–3041 (1997).
Cossi, M., Barone, V., Cammi, R. & Tomasi, J. Ab initio study of solvated molecules: a new implementation of the polarizable continuum model. Chem. Phys. Lett. 255, 327–335 (1996).
Barone, V., Cossi, M. & Tomasi, J. Geometry optimization of molecular structures in solution by the polarizable continuum model. J. Comput. Chem. 19, 404–417 (1998).
Lu, T. & Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012).
Humphrey, W., Dalke, A. & Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996).
Reed, A. E., Curtiss, L. A. & Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 88, 899–926 (1988).
Reed, A. E., Weinstock, R. B. & Weinhold, F. Natural population analysis. J. Chem. Phys. 83, 735–746 (1985).
F. Weinhold, J. E. Carpenter, The natural bond orbital Lewis structure concept for molecules, radicals, and radical ions. In The Structure of Small Molecules and Ions (pp. 227–236) (Springer, Boston, MA, 1988).
Parthasarathi, R., Subramanian, V., Roy, D. R. & Chattaraj, P. K. Electrophilicity index as a possible descriptor of biological activity. Bioorg. Med. Chem. 12, 5533–5543 (2004).
Chattaraj, P. K. & Giri, S. Electrophilicity index within a conceptual DFT framework. Phys. Chem. 105, 13–39 (2009).
Islam, N., Kaya, S. (Eds.). Conceptual Density Functional Theory and Its Application in the Chemical Domain (CRC Press, 2018).
Frisch, M. E., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R. et al. **Gaussian, Inc., Wallingford CT, 2016.
Dennington, R., Keith, T., & Millam, J. GaussView, version 5 (2009).
Morris, G. M. et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791 (2009).
Systemes, D. BIOVIA Discovery Studio Visualizer 2016. San Diego, CA: Dassault Systemes, 2016.
Schrodinger, L. L. C. The PyMOL molecular graphics system. Version, 1(5) (2010).
Ahmed, A. et al. Novel adamantyl clubbed iminothiazolidinones as promising elastase inhibitors: design, synthesis, molecular docking, ADMET and DFT studies. RSC Adv. 12, 11974–11991 (2022).
Karrouchi, K. et al. Experimental and computational interaction studies of (E)-N’-benzylidene-5-methyl-1H-pyrazole-3-carbohydrazide with α-glucosidase and α-amylase enzymes: A detailed structural, spectroscopic, and biophysical study. Polycycl. Aromat. Compd. 43, 1812–1832 (2023).
Pillai, R. R. et al. Synthesis, spectroscopic characterization, reactive properties by DFT calculations, molecular dynamics simulations and biological evaluation of Schiff bases tethered 1, 2, 4-triazole and pyrazole rings. J. Mol. Struct. 1177, 47–54 (2019).
Kostić, D. A. et al. Xanthine oxidase: Isolation, assays of activity, and inhibition. J. Chem. 2015, 1–9 (2015).
Arivazhagan, M., Manivel, S., Jeyavijayan, S. & Meenakshi, R. Vibrational spectroscopic (FTIR and FT-Raman), first-order hyperpolarizablity, HOMO, LUMO, NBO, Mulliken charge analyses of 2-ethylimidazole based on Hartree-Fock and DFT calculations. Spectrochim. Acta A Mol. Biomol. Spectrosc. 1, 493–501 (2015).
Karelson, M., Lobanov, V. S. & Katritzky, A. R. Quantum-chemical descriptors in QSAR/QSPR studies. Chem. Rev. 96, 1027–1044 (1996).
Streitwieser, A. & Salzberg, H. W. Molecular orbital theory for organic chemists. J. Electrochem. Soc. 109, 116 (1962).
Chattaraj, P. K., Nath, S., Maiti, B. Reactivity Descriptors 295–322. (Marcel Dekker, New York, 2003).
El-Hadki, H. et al. Theoretical Study of Reaction Between Nitrilimine and 1, 4 oxazine 2 Carboxylate by MP2 and DFT Methods. Oriental J. Chem. 34, 2992 (2018).
Parr, R. G., Szentpály, L. V. & Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1, 1922–1924 (1999).
Luque, F. J., López, J. M. & Orozco, M. Perspective on “Electrostatic interactions of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects”. Theor. Chem. Acc. 1, 343–345 (2000).
El Hadki, A. et al. Removal of oxytetracycline by graphene oxide and boron-doped reduced graphene oxide: A combined density function theory, molecular dynamics simulation and experimental study. FlatChem. 27, 100238 (2021).
Weinhold, F., Landis, C. R. & Glendening, E. D. What is NBO analysis and how is it useful?. Int. Rev. Phys. Chem. 1, 399–440 (2016).
Weinhold, F. & Landis, C. R. Natural bond orbitals and extensions of localized bonding concepts. Chem. Educ. Res. Pract. 1, 91–104 (2001).
Agwupuye, J. A. et al. Electronic structure investigation of the stability, reactivity, NBO analysis, thermodynamics, and the nature of the interactions in methyl-substituted imidazolium-based ionic liquids. J. Mol. Liq. 337, 116458 (2021).
Yu, Z. et al. Insights from molecular dynamics simulations and steered molecular dynamics simulations to exploit new trends of the interaction between HIF-1α and p300. J. Biomol. Struct. Dyn. 38, 1–12 (2020).
Lipinski, C. A., Lombardo, F., Dominy, B. W. & Feeney, P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 64, 4–17 (2012).
Xiong, G. et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucl. Acids Res. 49, W5–W14 (2021).
van Breemen, R. B. & Li, Y. Caco-2 cell permeability assays to measure drug absorption. Expert. Opin. Drug Metab. Toxicol. 1, 175–185 (2005).
Prabakaran, G., Manivarman, S. & Bharanidharan, M. Catalytic synthesis, ADMET, QSAR and molecular modeling studies of novel chalcone derivatives as highly potent antioxidant agents. Mater. Today: Proc. 48, 400–408 (2022).
Fettach, S. et al. Biological, toxicological and molecular docking evaluations of isoxazoline-thiazolidine-2, 4-dione analogues as new class of anti-hyperglycemic agents. J. Biomol. Struct. Dyn. 41, 1072–1084 (2021).
Fettach, S. et al. Synthesis, α-glucosidase and α-amylase inhibitory activities, acute toxicity and molecular docking studies of thiazolidine-2, 4-diones derivatives. J. Biomol. Struct. Dyn. 1, 1–12 (2021).
Acknowledgements
The authors are grateful to King Saud University, Riyadh, Saudi Arabia for funding the work through the Researchers Supporting Project number (RSPD2024R740).
Author information
Authors and Affiliations
Contributions
Conceptualization, S.M.; methodology, S.M. and K.K.; software, E.H.H.; resources, A.O.; funding acquisition, M.A.B.; formal analysis, H.M. and Y.A.; writing-original draft preparation, K.K., S.M., M.A.B and E.H.H.; Visualization, S.R. and A.M.; writing-review and editing, K.K.; supervision, M.A. and M.E.A.F. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mortada, S., Karrouchi, K., Hamza, E.H. et al. Synthesis, structural characterizations, in vitro biological evaluation and computational investigations of pyrazole derivatives as potential antidiabetic and antioxidant agents. Sci Rep 14, 1312 (2024). https://doi.org/10.1038/s41598-024-51290-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-51290-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
