
Abstract
This work is concerned with exploiting the power of chemometrics in the assay and purity determination of naphazoline HCl (NZ) and pheniramine maleate (PN) in their combined eye drops. Partial least squares (PLS) and artificial neural network (ANN) were the chosen models for that purpose where three selected official impurities, namely; NZ impurity B and PN impurities A and B, were successfully determined. The quantitative determinations of studied components were assessed by percentage recoveries, standard errors of prediction as well as root mean square errors of prediction. The developed models were constructed in the ranges of 5.0–13.0 μg mL−1 for NZ, 10.0–60.0 μg mL−1 for PN, 1.0–5.0 μg mL−1 for NZ impurity B and 2.0–14.0 μg mL−1 for two PN impurities. The proposed models could determine NZ and PN with respective detection limits of 0.447 and 1.750 μg mL−1 for PLS, and 0.494 and 2.093 μg mL−1 for ANN. The two established models were compared favorably with official methods where no significant difference observed.
Similar content being viewed by others


Introduction
Determining drug purity for ensuring its safety and quality is considered essential step in drug analysis. As a result, impurities detection and quantification take a great attention in pharmaceutical industries1. The demand to establish analytical methods which have the ability to determine and quantify the drug along with its impurities was consequently evoked2. On the other hand, as the number of the analyzed drugs and their impurities increased, the difficulty of their analysis using conventional methods was also amplified, taken in our consideration the simplicity and the availability of the spectrophotometric methods than the chromatographic ones3,4,5,6,7,8. In such case, multivariate data inspection is better in finding an answer to that complicated matrices9, 10. Therefore, chemometrics is commonly used to analyze such complex data obtained from spectrophotometric measurements to acquire valuable information. It is a useful tool when determining numerous drugs in their combined pharmaceutical formulations is required11, 12.
Naphazoline HCl (NZ), also known as 2-(naphthalen-1-ylmethyl)-4, 5-dihydro-1H-imidazole; HCl, is a drug that acts through decreasing pulmonary congestion. It reacts with α-adrenergic receptors located in the conjunctiva, producing a sympathomimetic effect leading to decrease swelling and edema of the eyes13. Reviewing its pharmacopeial monographs14, 15, shows that its quantification is achieved using two liquid chromatographic methods. Four reported impurities, namely; A, B, C and D, are also stated in its British pharmacopoeia (BP) monograph. Carefully reviewed spectrophotometric techniques reveal that NZ has been quantified in the existence of other substances16,17,18,19,20,21.
Pheniramine maleate (PN) is an alkylamine antihistaminic drug having anti-cholinergic properties through binding to H1 histaminic receptors. It is a first-generation drug which inhibits phospholipase-A2 and cyclic-GMP levels22. Its assay in the United States pharmacopoeia (USP) and BP is through high performance liquid chromatographic (HPLC) technique. In addition, two impurities, A and B, are reported in its BP monograph14, 15. A spectrophotometric method based on complexation with ferric ion has been published for its simultaneous determination with colorpheniramine maleate23.
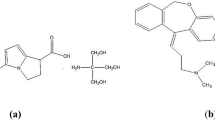
For the treatment of inflammatory eye disorders and allergic conjunctivitis, the approach of using NZ and PN together in combined eye drops is found to be more useful than using each drug alone24. Literature review reveals that NZ and PN have been quantified together using HPLC25,26,27,28, capillary electrophoresis29 and thin layer chromatography30. Two of these chromatographic methods have been published by our research group where their determination along with three reported official impurities, NZ impurity B, PN impurity A and PN impurity B, was achieved28, 30. In spite of the applicability of the chromatographic technique in analyzing such complex mixtures, it is still considered challenging. This is attributed to the requirements for the successive sample pretreatment steps as well as the selection of suitable mobile phase and stationary phase to attain the best separation and system suitability parameters. Besides all of this, the time needed for optimization, the expensive tools required, and the hazardous organic reagents utilized are considered potential obstacles to that technique31. On the other hand, spectrophotometric techniques can overcome these previously mentioned drawbacks as it is fast, cheap, simple and time saving32. To this end, our aim is to simultaneously analyze NZ, PN, and three official impurities, namely; NZ impurity B, PN impurity A and PN impurity B, using simple chemometrics-assisted spectrophotometric methods. Calibration mixtures are prepared based on five-level five-factor design. Partial least squares (PLS) and Artificial neural network (ANN) regressions are the chosen models for the determination of the five cited compounds (Fig. 1) in marketed dosage form and laboratory prepared mixtures.

Chemical structures of the five cited components.
Results and discussion
Among different analytical techniques used for drugs determination in their pharmaceutical formulations, spectrophotometry is the widely applicable one. This is attributed to simplicity and rapidity of spectro-analytical methods which has no need for neither sophisticated apparatus nor chemical pretreatment as other chromatographic methods9. Due to presence of sever overlapping between the spectra of the five studied components, Fig. 2. Multivariate spectrophotometric are the technique of choice to resolve this overlap. It was found that readings below 250.0 nm showed high noise which may affect the obtained result, otherwise readings above 300.0 nm gave almost zero absorption leading to invaluable information, thus the range between 250.0 and 300.0 nm was the range of choice in calculation. Brereton five-level calibration design was followed in order to prepare different mixtures of the five cited drugs33. 25 mixtures were prepared and divided into two groups. 15 mixtures for the calibration group whereas the remaining 10 mixtures were utilized for the validation one.

Normalized spectra of NZ (----), PN (\({\_}..{\_}\)), NZ impurity B (\({\_}.{\_}\)), PN impurity A (——) and PN impurity B (……) using methanol as solvent.
Partial least squares model
During construction of PLS model, mean centering of all spectral data was adopted. After applying the leave-one-out as a cross-validation tool, latent variables’ number was obtained6, 34. The plot relating this number to root mean square error of calibration (RMSEC) revealed that six was the optimum number to be utilized, Fig. 3. It is worth noting that numerous models for calibration were tried and built whereas obtaining least noise as well as satisfactory recovery results were the parameters for selecting the optimum one35.

Root mean square error of calibration (RMSEC) versus the number of latent variables used to construct the PLS calibration for the assay of NZ, PN, NZ impurity B, PN impurity A and B in their mixtures.
Artificial neural network model
Three transferable layers (input, hidden and output) are usually incorporated in ANN model8, 12 In our model, the 251 points of spectral data were used as input neurons11, 35. On the other hand, 5 neurons related to number of drugs determined by such model comprised the output layer. For the hidden layer, various numbers were tried whereas RMSEC was calculated in each time. It was found that the lowest error (< 0.1 for the five drugs) was gained upon using 4 neurons in this layer with no further enhancement upon increasing their number. It is worth noting that pure-line transfer function, 0.1-learning rate as well as 50-epochs were also utilized.
The prediction’s capability of the two proposed models was verified by the aid of the validation group mixtures. For each drug, average recovery percentages as well as relative standard deviations were tabulated, Table 1. It is worth noting that the concentration ranges of the two drugs were chosen to facilitate their direct determination in the challenging eye drops ratio. Linearity parameters, limits of detection and quantification were obtained, Table 2. As shown, our proposed models could detect NZ and PN impurities at limits of about ≈1% and ≈2%, respectively, of their parent drugs' highest calibration concentration. Moreover and in order to test the presence of possible over-fitting, root mean square error of prediction (RMSEP) and standard error of prediction (SEP) were computed as well. As shown in Table 2, their close values assured over-fitting absence in our models.
Eye drops application
Successful determination of NZ and PN in naphcon-A drops was attained by the proposed multivariate chemometrics models. As shown in Table 3, good recoveries were obtained and the models’ validity was further assured through recoveries of the added standards.
Statistical analysis
Student’s t-test as well as F- one were statistically applied to compare obtained results with that acquired by BP methods of NZ and PN analyses. Table 4 summarizes the outputs of this comparison along with average recoveries, standard deviations, variances, and number of observations utilized in those tests. It is worth noting that the lower t and F values obtained relative to theoretical ones assured the statistical absence of significant differences.
Comparison with reported chromatographic methods
Comparing our proposed models with the reported chromatographic methods for the determination of NZ, PN, and the three selected impurities was conducted. The solvents used, the linearity ranges as well as the obtained LOD values were the chosen items for this comparison, Table 5. The table proved the superiority of these proposed PLS and ANN spectrophotometric models in terms of the simplicity of solvents required. Lower LOD values were also obtained compared to the reported HPLC method.
Conclusion
In this work NZ, PN along with three selected official impurities are determined in their mixtures and eye drops pharmaceutical dosage form using accurate and simple chemometrics assisted spectro-analytical models. The proposed models are considered time-saving and cost-effective comparing with the reported chromatographic methods. The two proposed models show the advantage of the low values for prediction error. PLS supposes that the error is scattered equally between the two matrices; concentration and spectral response. As a result, robust results were given by this model through removing the absorbance as well as concentration data noises simultaneously. On the other hand, the ANN model’s predicting ability was exploited for obtaining more precise results during studied drugs’ quantification.
Methods
Instrument
A dual-beam Shimadzu, UV 1601 spectrophotometer, Kyoto, Japan. Spectra Scanning was conducted at 200.0–400.0 nm range with 0.2 nm intervals using 1.00-cm quartz cuvettes. The utilized Matlab® software (7.0.1) was integrated with a PLS Toolbox 2.1.
Materials
Standards
The studied drugs were supplied by Eva-pharma Co. (Egypt). Purities were checked as per BP methods to be 100.12% for NZ, and 99.58% for PN15. The impurities were purchased from Alfa Aesar Co. (Germany). Potencies were certified to be 99.00%, 100.30% and 99.70% for NZ impurity B, PN impurity A and PN impurity B, respectively.
Pharmaceutical dosage form
Naphcon-A drops, Alcon lab. INC. B. N. H13949-0615, containing 0.25 mg NZ and 3.0 mg PN in one mL.
Reagents
Methanol of analytical-grade was used (Alpha, Egypt).
Solutions
Five standard solutions, of 1.0 mg mL−1 concentration, were prepared separately using methanol.
Procedures
Construction of the calibration models
Spectral features of the five studied substances were determined through their normalized spectra using methanol as a blank. Twenty-five mixtures containing different concentrations of the five cited compounds were prepared following a five-levels five-factors experimental design to reach concentration ranges of 5.0–13.0 μg mL−1 for NZ, 10.0–60.0 μg mL−1 for PN, 1.0–5.0 μg mL−1 for NZ impurity B and 2.0–14.0 μg mL−1 for two PN impurities. Scanning of the prepared solutions were then conducted at 200.0–400.0 nm range. The range from 250.0 to 300.0 nm was used for further calculations. The data were analyzed using Matlab®, where multivariate calibration models were built by using fifteen mixtures from the previously prepared solutions.
Validation of the calibrated models
The remaining ten mixtures from the previously prepared solutions were used for the validation purpose. PLS and ANN obtained parameters were used for quantification of the five cited compounds.
Application to naphcon-A drops
A 1.0-mL solution was transferred from naphcon-A drops to a 50-mL flask, and 25.0 mL methanol was introduced. Sonication was then applied for 10.0 min. A final concentration of 5.0 µg mL−1 NZ and 60.0 µg mL−1 PN was obtained after completing the volume with methanol. The prepared solutions were scanned and concentrations were predicted by the developed models.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Rahman, N., Azmi, S. N. H. & Wu, H.-F. The importance of impurity analysis in pharmaceutical products: An integrated approach. Accred. Qual. Assur. 11, 69–74 (2006).
Gorog, S. Critical review of reports on impurity and degradation product profiling in the last decade. TrAC Trends Anal. Chem. https://doi.org/10.1016/j.trac.2017.09.012 (2018).
Ali, J., Tuzen, M. & Kazi, T. G. Evaluation of mercury in environmental samples by a Supramolecular solvent-based dispersive liquid–liquid microextraction method before analysis by a Cold vapor generation technique. J. AOAC Int. 100, 782–788 (2017).
Ali, J., Tuzen, M. & Kazi, T. G. Developed of a green water switchable liquid–liquid microextraction method for assessment of selenium in food and soft drink samples by using hydride generation atomic absorption spectrometry. Food Anal. Methods 12, 1298–1307 (2019).
Ali, J., Tuzen, M. & Kazi, T. G. Green and innovative technique develop for the determination of vanadium in different types of water and food samples by eutectic solvent extraction method. Food Chem 306, 125638 (2020).
Kelani, K. M., Hegazy, M. A., Hassan, A. M. & Tantawy, M. A. Univariate versus multivariate spectrophotometric methods for the simultaneous determination of omarigliptin and two of its degradation products. Spectrochim. Acta A Mol. Biomol. Spectrosc. 271, 120880 (2022).
Tantawy, M. A., Wahba, I. A., Saad, S. S. & Ramadan, N. K. Smart spectrophotometric methods for stability assessment of two co-formulated antigout drugs. Spectrochim. Acta A Mol. Biomol. Spectrosc. 273, 121062 (2022).
Tantawy, M. A., Wahba, I. A., Saad, S. S. & Ramadan, N. K. Classical versus chemometrics tools for spectrophotometric determination of fluocinolone acetonide, ciprofloxacin HCl and ciprofloxacin impurity-A in their ternary mixture. BMC Chem. 17, 1–11 (2023).
Hegazy, M. A., Bakr, M. A., Badawey, A. M. & Abbas, S. S. Univariate and multivariate assisted spectrophotometric methods for determination of rosuvastatin calcium and fenofibrate in bulk powders and tablets along with their degradation products. Spectrochim. Acta A Mol. Biomol. Spectrosc. 248, 1386–1425 (2020).
Ali, J., Tuzen, M., Feng, X. & Kazi, T. G. Determination of trace levels of selenium in natural water, agriculture soil and food samples by vortex assisted liquid-liquid microextraction method: Multivariate techniques. Food Chem. 344, 128706 (2021).
Yehia, A. M. & Mohamed, H. M. Chemometrics resolution and quantification power evaluation: Application on pharmaceutical quaternary mixture of Paracetamol, Guaifenesin, Phenylephrine and p-aminophenol. Spectrochim. Acta A Mol. Biomol. Spectrosc. 152, 491–500 (2016).
Tantawy, M. A. & Michael, A. M. Artificial neural networks versus partial least squares and multivariate resolution-alternating least squares approaches for the assay of ascorbic acid, rutin, and hesperidin in an antioxidant formulation. Spectrosc. Lett. 52, 339–345 (2019).
Meloun, M., Syrovy, T. & Vrana, A. The thermodynamic dissociation constants of ambroxol, antazoline, naphazoline, oxymetazoline and ranitidine by the regression analysis of spectrophotometric data. Talanta 62, 511–522 (2004).
USP, P. USP 39 - NF 34. in The United States Pharmacopeial (2016).
British pharmacopoeia. British Pharmacopoeia 2019 (2019).
el deen Sayed, N., Hegazy, M., Abdelkawy, M. & Abdelfatah, R. Spectrophotometric, chemometric and chromatographic determination of naphazoline hydrochloride and chlorpheniramine maleate in the presence of naphazoline hydrochloride alkaline degradation product. Bull. Facul. Pharm. 51, 57–68 (2013).
Goicoechea, H. C., Collado, M. S., Satuf, M. L. & Olivieri, A. C. Complementary use of partial least-squares and artificial neural networks for the non-linear spectrophotometric analysis of pharmaceutical samples. Anal. Bioanal. Chem. 374, 460–465 (2002).
Hemmateenejad, B., Ghavami, R., Miri, R. & Shamsipur, M. Net analyte signal-based simultaneous determination of antazoline and naphazoline using wavelength region selection by experimental design-neural networks. Talanta 68, 1222–1229 (2006).
Chocholous, P., Satinsky, D. & Solich, P. Fast simultaneous spectrophotometric determination of naphazoline nitrate and methylparaben by sequential injection chromatography. Talanta 70, 408–413 (2006).
Souri, E., Amanlou, M., Farsam, H. & Afshari, A. A rapid derivative spectrophotometric method for simultaneous determination of naphazoline and antazoline in eye drops. Chem. Pharm. Bull. (Tokyo) 54, 119–122 (2006).
Korany, M. A., Bedair, M. M. & El-Gindy, A. Analysis of diphenhydramine hydrochloride and naphazoline hydrochloride in presence of methylene blue in eye drops by second derivative spectrophotometry. Drug Dev. Ind. Pharm. 16, 1555–1564 (1990).
Parente, G., Pazzaglia, M., Vincenzi, C. & Tosti, A. Contact dermatitis from pheniramine maleate in eyedrops. Contact Dermat. 40, 338 (1999).
Abdel Fattah, S., Kelany, K. O., El-zeany, B. A. & El-tarras, M. F. Analysis of pheniramine maleate and colorpheniramine maleate via their Fe (III) Complexes. Anal. Lett. 20, 1667–1678 (1987).
Dockhorn, R. J. & Duckett, T. G. Comparison of naphcon-A and its components (naphazoline and pheniramine) in a provocative model of allergic conjunctivitis. Curr. Eye Res. 13, 319–324 (1994).
Huang, T., Chen, N., Wang, D., Lai, Y. & Cao, Z. A validated stability-indicating HPLC method for the simultaneous determination of pheniramine maleate and naphazoline hydrochloride in pharmaceutical formulations. Chem. Cent. J. 8, 7 (2014).
Oliveira, T. D. C., Freitas, J. M., Munoz, R. A. A. & Richter, E. M. Development of a novel versatile method for determination of two antihistamines in association with naphazoline using cathodically pretreated boron-doped diamond electrode. Electroanalysis 30, 868–876 (2018).
Sidhu, A. S., Kennedy, J. M. & Deeble, S. General method for the analysis of pharmaceutical dosage forms by high-performance liquid chromatography. J. Chromatogr. A 391, 233–242 (1987).
Kelani, K. M., Hegazy, M. A., Hassan, A. M. & Tantawy, M. A. Determination of naphazoline HCl, pheniramine maleate and their official impurities in eye drops and biological fluid rabbit aqueous humor by a validated LC-DAD method. RSC Adv. J. 11, 7051–7058 (2021).
Koziol, T. R., Jacob, J. T. & Achari, R. G. Ion-pair liquid chromatographic assay of decongestants and antihistamines. J. Pharm. Sci. 68, 1135–1138 (1979).
Kelani, K. M., Hegazy, M. A., Hassan, A. M. & Tantawy, M. A. A green TLC densitometric method for the simultaneous detection and quantification of naphazoline HCl, pheniramine maleate along with three official impurities. BMC Chem. 16, 1–11 (2022).
Elsonbaty, A., Serag, A., Abdulwahab, S., Hassan, W. S. & Eissa, M. S. Analysis of quinary therapy targeting multiple cardiovascular diseases using UV spectrophotometry and chemometric tools. Spectrochim. Acta A Mol. Biomol. Spectrosc. 238, 118415 (2020).
Attia, K. A. M., Nassar, M. W. I., El-Zeiny, M. B. & Serag, A. Zero order and signal processing spectrophotometric techniques applied for resolving interference of metronidazole with ciprofloxacin in their pharmaceutical dosage form. Spectrochim. Acta A Mol. Biomol. Spectrosc. https://doi.org/10.1016/j.saa.2015.10.040 (2016).
Saad, A. S., Elzanfaly, E. S., Halim, M. K. & Kelani, K. M. Comparing the predictability of different chemometric models over UV-spectral data of isoxsuprine and its toxic photothermal degradation products. Spectrochim. Acta A Mol. Biomol. Spectrosc. 219, 444–449 (2019).
El-Ragehy, N. A., Yehia, A. M., Hassan, N. Y., Tantawy, M. A. & Abdelkawy, M. Chemometrics tools in detection and quantitation of the main impurities present in aspirin/dipyridamole extended-release capsules. J. AOAC Int. 99, 948–956 (2016).
Yehia, A. M., Elbalkiny, H. T., Riad, S. M. & Elsaharty, Y. S. Chemometrics for resolving spectral data of cephalosporines and tracing their residue in waste water samples. Spectrochim. Acta A Mol. Biomol. Spectrosc. https://doi.org/10.1016/j.saa.2019.04.081 (2019).
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
K.M.K.: Conceptualization, Methodology, Software, Validation, Visualization, Supervision, Project administration, Funding acquisition, Writing—original draft. M.A.H.: Conceptualization, Methodology, Software, Formal analysis, Data curation, Visualization, Supervision, Project administration, Funding acquisition, Writing—review and editing. A.M.H.: Methodology, Software, Validation, Formal analysis, Investigation, Funding acquisition, Project administration, Writing—original draft, Writing—review and editing. M.A.T.: Methodology, Software, Validation, Formal analysis, Investigation, Funding acquisition, Project administration, Writing—original draft, Writing—review and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kelani, K.M., Hegazy, M.A., Hassan, A.M. et al. Application of multivariate chemometrics tools for spectrophotometric determination of naphazoline HCl, pheniramine maleate and three official impurities in their eye drops. Sci Rep 13, 19678 (2023). https://doi.org/10.1038/s41598-023-46940-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-46940-0
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
