
Abstract
The blotched nerite Nerita albicilla (Linnaeus 1758) is distributed in intertidal areas of the Indo-Pacific. In South Korea, it has been found only in the southernmost region of Jeju Island so far. Owing to its limited distribution, it can be a promising intertidal species helpful for monitoring global climate change effects in the Korean Peninsula. We performed population genetic analyses based on 393 COI haplotypes from 697 N. albicilla, including 167 from this study and 530 from public databases. The results showed that there are two distinct genetic lineages in N. albicilla: PAIO (Palearctic, Australasia, Indo-Malay, and Oceania) and Afrotropic lineages. DNA barcoding gap analyses indicated that the two lineages could be differentiated into two different species: N. albicilla (PAIO) and N. originalis sp. nov. (Afrotropic) (3.96%). Additionally, it was revealed that their divergence time was ca. 5.96 Ma and dramatic diversification of COI haplotypes occurred during the late Pliocene and Pleistocene. The results of MDA, BSP, and neutrality test implied recent population size expansion, which was estimated to be ca. 250 Ka. Finally, we discussed whether the observation of N. originalis sp. nov. in South Korea is due to the northward migration through ocean currents caused by global warming or due to artificial activity through marine transportation.
Similar content being viewed by others


Introduction
The blotched nerite, Nerita albicilla, an intertidal marine species, has long-lived planktotrophic larvae1, which can be characterized by its depressed spire, sculptured with broad, low spiral ribs, and an outer lip with one or two strong teeth posteriorly and sometimes anteriorly located2. Shells commonly reach 24 mm in height and primarily inhabit rubble and cobble fields with low wave energy, and are often associated with inner-reef flats3,4. Nerita albicilla is widely distributed extending to the shores of East Africa in the west and reaching its eastern boundary at the Cook Islands5. However, in South Korea, N. albicilla is restricted to the southernmost area of Jeju Island, located approximately 90 km south of the mainland6. Due to the interesting distribution pattern of N. albicilla, the National Institute for Biological Resources, Ministry of Environment, South Korea was designated as one of the 100 Climate Change Biological Indicator Species (CBIS) in 2010 for continuously monitoring and predicting climate change caused by global warming7.
The evolutionary rate of mitochondrial DNA is faster than that of nuclear DNA, enough to examine genetic diversity and differentiation of not only closely related sibling species but also phylogeographically diverged genetic groups within a single species8,9,10,11,12. These characteristics make mitochondrial DNA particularly appropriate for tracing recent evolutionary history, including colonization or translocation events and population bottlenecks13. In recent decades, many complete mitochondrial genomes mollusc consisting of 13 protein-coding genes, 22 tRNA genes, 2 rRNA genes, and one or two control region(s) have been sequenced and characterized14,15,16. Especially, a single mitochondrial gene, such as the cytochrome c oxidase subunit I gene (COI), can be used as an effective biological species identification system9,17 and can be informative in tracing the movement of a species18. This marker can also genetically distinguish closely related marine invertebrate species12, despite its short sequence (~ 543 bp). Crandall et al.5 analyzed the demographic history and phylogeographical patterns of N. albicilla and N. plicata. Using 529 COI sequences of N. albicilla, they found that N. albicilla exhibited a reciprocal monophyly of Indian and Pacific Ocean populations, indicating that N. albicilla comprises two distinct genetic lineages: clades A and B. Since then, no population genetic study of N. albicilla in the Indo-Pacific has been conducted.
In the present study, we attempted to characterize the genetic diversity and population genetic structure of Indo-Pacific N. albicilla based on 393 COI haplotypes extracted from 697 individuals, consisting of 530 individuals retrieved from public databases and 167 newly collected from five different sites located in the southernmost areas of Jeju Island, South Korea. The results confirmed that the presence of two distinct genetic lineages in Indo-Pacific N. albicilla, called Afrotropic and PAIO (Palearctic, Australasia, Indo-Malay, and Oceania) lineages, indicating probable N. albicilla species discrimination. In addition, we analyzed the DNA barcoding gap and its distribution patterns, which showed that the two genetic lineages may be reclassified into two independent Nerita species: N. albicilla (PAIO) and N. originalis sp. nov. (Afrotropic). In addition, their divergence times and demographic histories were explored. It is expected that the present results will be helpful for monitoring distribution patterns and population expansion of intertidal marine gastropods, as well as for understanding mechanisms of species discrimination of marine gastropods.
Results
Genetic diversity of N. albicilla based on COI
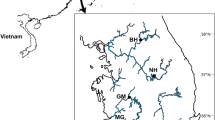
A total of 167 N. albicilla individuals were collected from five intertidal regions on Jeju Island, South Korea (Fig. 1; Table 1). From the collected samples, we amplified the COI barcoding region using PCR, and the resultant 575-bp PCR products were sequenced. In addition, we retrieved 530 previously published COI sequences of N. albicilla: 524 from Eric D. Crandall from https://geome-db.org/query and six from the NCBI GenBank database. As a result, a total of 393 COI haplotypes sequences (Data S1) were defined from 697 N. albicilla individuals which were collected from 26 localities: five from Jeju Island in South Korea and 21 from other locations including the Palearctic (Japan and Oman), Indo-Malay (China, China-Hong Kong, Indonesia, Japan-Okinawa, Malaysia, Philippines, Singapore, and Thailand), Australasia (Australia, New Caledonia, and Papua New Guinea), Oceania (American Samoa, Cook Island, Fiji, and Guam), and Afrotropic region (Kenya, Madagascar, Mauritius, South Africa, and Tanzania).

A map showing the collection sites of Nerita albicilla inhabiting intertidal areas of the Indo-Pacific including the Korean Peninsula (N = 697). Black dots represent the sites where the samples are directly collected in this study and white dots with black border are the collection sites where the samples are from references. Two individuals of Afrotropic in Jeju Island are directly collected from GJ and SM. The maps are provided from the d-maps site (https://d-maps.com) and the figure was edited by Adobe Illustrator v.22.2 (https://www.adobe.com).
The genetic diversity estimated with 393 COI haplotypes revealed that the average haplotype diversity (h) and nucleotide diversity (π) were 0.989 and 0.02001, respectively (Table 2). The highest haplotype diversity (h = 1) was observed in Nonjinmul (NJ), American Samoa (AMS), Guam (GUAM), Madagascar (MAD), and Singapore (SG), whereas the highest nucleotide diversity (π = 0.02490) was found in Thailand (TH). The most abundant haplotype was NAH37, which was found in 45 COI sequences from 697 N. albicilla individuals.
Population genetic analyses
We constructed a nucleotide sequence alignment set with 393 COI haplotypes of N. albicilla (Table S1), from which 186 polymorphic sites and 134 parsimony informative sites were identified. TCS network analysis (Fig. 2a) and principal coordinate analysis (PCoA) (Fig. 2b) showed the existence of two distinct genetic groups in N. albicilla. In the TCS network (Fig. 2a), 393 COI haplotypes were divided into two genetic lineages: one consisting of the 118 haplotypes detected primarily in the Afrotropic region, called the Afrotropic, and the other including 275 haplotypes found mainly in Palearctic, Australasia, Indo-Malay, and Oceania regions, called the PAIO. The lineages Afrotropic and PAIO have star-like topologies in common: NAH230 is a central haplotype in Afrotropic, and NAH03, NAH19, and NAH37 are central haplotypes in PAIO. As in the TCS network (Fig. 2a), the PCoA plot (Fig. 2b) also supported the existence of two genetic groups of N. albicilla. Furthermore, the TCS network (Fig. 2a) represents star-like topologies, which suggests a recent population demographic expansion for N. albicilla. When we calculated the pairwise FST values between the five different biogeographical groups of N. albicilla, the lowest value appeared between Palearctic and Oceania (-0.00027) and the highest between Oceania and Afrotropic (0.79859). The pairwise FST values between the Afrotropic and each of the other four PAIO members (0.72509–0.79859) were higher than those between the PAIO members (− 0.0027–0.01769) (Table S2). Analysis of molecular variance (AMOVA) based on COI were conducted to evaluate the degree of genetic differentiation statistically between PAIO and Afrotropic (Table 3). The results provide strong statistical support for the existence of two genetic lineages, PAIO and Afrotropic, with higher molecular variation between the two groups (69.88%) and lower variation within each group (8.08%). Assuming five different geographical groups, most of the variance (59.19%) was allocated to the level of individuals among the groups. The Mantel test did not show any statistically significant correlation between pairwise genetic distances and geographical distances (r = 0.166, P < 0.001; Fig. S2).

A TCS haplotype network, PCoA analyses, and a unrooted maximum likelihood tree based on the 393 COI haplotypes of Nerita albicilla in the Indo-Pacific. (a) A unrooted TCS haplotype network. Haplotype frequency is related to the size of the circle. Different colors within the nodes refer to different sampling sites divided into the five geographical regions depicted by the five different colors. (b) The PCoA result demonstrates the two genetic groups (PAIO and Afrotropic). The score of the two axes (Axis 1 = 67.61%, and Axis 2 = 2.07%) (c) A unrooted maximum likelihood tree. All the results consistently showed the existence of the two distinct genetic lineages (PAIO and Afrotropic) for N. albicilla in the Indo-Pacific. The figure was edited by Adobe Illustrator v.22.2 (https://www.adobe.com).
Examination of species discrimination of N. albicilla
Using Automatic Barcode Gap Discovery (ABGD), we explored the distribution of pairwise genetic divergences (Fig. 3a), ranks of pairwise genetic differences (Fig. 3b), and the results of automatic partition analyses (Fig. 3c) based on the COI haplotypes of N. albicilla, respectively. The results confirmed the existence of a distinct barcoding gap between intraspecific and interspecific variation (Fig. 3a), a distinct Kimura-2-parameter (K2P) distance gap at a specific rank (Fig. 3b), and two different groups, as shown in the automatic partition analysis (Fig. 3c). Based on the COI (575 bp) of Nerita albicilla, we identified the K2P intraspecific threshold with 393 haplotype sequences and the K2P interspecific distance ranges with 54 different species with the genus Nerita (Data S2, S3): NAH03 as a representative haplotype for Nerita albicilla (PAIO), NAH230 for Nerita originalis sp. nov., and the remaining 52 sequences of 52 different Nerita species retrieved from the GenBank database (Table S3). In Table S4, the intraspecific genetic distance ranges from 0 to 6.43% with 2.69% average (N = 67,575). The K2P distances between species varied from 1.80% to 26.73% (N = 1,431) with 16.62% mean. The pairwise K2P genetic distance between N. albicilla and N. originalis sp. nov. was 3.94%, which is higher than the mean value (= 2.69%) but lower than the maximum value (= 6.43%) in intraspecific genetic distances, whereas is lower than the mean value (= 16.62%) but higher than the minimum value (= 1.80%) in interspecific genetic distances. It may be interpreted as the two different species recently speciated (closely related with lower intraspecific distance) of N. albicilla and N. originalis sp. nov., or an identical species genetically differentiated (above the average value of intraspecific distance). Considering the existence of an apparent barcoding gap between the two species (Fig. 3), the former is likely to be more reliable than the latter. We also found the three pairwise K2P genetic distance lower than 3.94% between Nerita species: N. reticulata and N. signata (= 2.54%), N. melanotragus and N. morio (= 1.80%), and N. planospira and N. sanguinolenta (= 2.56%). These analyses strongly support the differentiation of N. albicilla into two independent species: N. albicilla (PAIO) and N. originalis sp. nov. (Afrotropic).

Distribution of pairwise genetic divergence, ranked pairwise difference, and automatic partition based on the 393 COI haplotypes of Nerita albicilla, indicating that it may be divided into two different species. (a) Distribution patterns of pairwise genetic divergences. The horizontal axis represents intervals of pairwise Kimura-2-parameter (K2P) genetic distance in percentage, and the vertical axis represents the number of individuals associated with each distance interval. (b) The result of ranked pairwise differences. The results are similar to the distribution of pairwise genetic divergence shown in (a). The horizontal axis indicates a ranked ordered value based on K2P genetic distance, and the vertical axis represents the K2P genetic distance in percentage. (c) The result of automatic partition analyses. The horizontal axis represents the prior maximum intraspecific divergence (P), and the vertical axis represents the number of groups inside the partitions (primary and recursive). The figure was edited by Adobe Illustrator v.22.2 (https://www.adobe.com).
Phylogenetic analysis and divergence time estimation
Based on the haplotype sequence alignment (Table S1), we reconstructed the maximum likelihood (ML) tree based on the 393 COI haplotypes with two outgroups, N. adenensis and N. planospira. The resultant ML tree revealed monophyly of the 393 COI haplotypes of N. albicilla (BP 100) and strongly supported the existence of the two distinct genetic lineages shown in population genetic analyses (Fig. S1), and strongly supported monophyly of Afrotropic (BP 100) and PAIO (BP 100). Likewise, an unrooted ML tree (without outgroups) showed two distinctive phylogenetic groups: N. albicilla (PAIO) and N. originalis sp. nov. (Afrotropic) (Fig. 2c). In addition, to elucidate phylogenetic relationships of N. albicilla and N. originalis sp. nov. within the genus Nerita, we reconstructed ML and BI trees with 133 COI haplotypes, which include N. albicilla (54 haplotypes), N. originalis (24 haplotypes), and 52 Nerita species (one haplotype per species) (Table S3). According to the well-resolved ML and BI trees (Fig. S3), N. albicilla (BP 100 and BPP 1.00) and N. originalis sp. nov. (BP 100 and BPP 0.99) formed independent monophyletic groups with high node confidence values, respectively, which are grouped together.
As shown in Fig. 4a, the molecular clock analysis performed by using BEAST 2.6.619,20 indicated that the N. albicilla (PAIO) and N. originalis sp. nov. (Afrotropic) shared their most recent common ancestor at about 5.96 Ma (4.69–7.04 Ma), corresponding to the late-Miocene. The N. albicilla lineage apparently derived from an Afrotropical ancestral haplotype (NAH324) around 4.81 Ma. In addition, dramatic diversification of COI haplotypes occurred mainly during the late Pliocene and Pleistocene.

The results of time-calibrated Bayesian tree, hypothetical common ancestor using S-DIVA, mismatch distribution analyses (MDA), and Bayesian skyline plots of Nerita albicilla and Nerita originalis sp. nov. (BSPs). (a) Highlighted time-calibrated Bayesian tree and hypothetical common ancestor. The tree was reconstructed with 133 COI haplotypes, which include N. albicilla (54 haplotypes), N. originalis (24 haplotypes), and 52 Nerita species (one haplotype per species) (Table S3) using the BEAST (refer to Fig. S4). Additionally, a S-DIVA analysis was done with 393 COI haplotypes of N. albicilla and N. originalis (Table S1) to seek for the inference of ancestral areas under the Bayesian binary MCMC (BBM) model implemented in RASP ver 3.2. Ancestral areas were hypothesized based on the distribution range of the fossil records of Nerita and the contemporary distribution of N. albicilla and N. originalis sp. nov. Pie charts at nodes represent the probabilities of the ancestral distributions. (b) MDA graphs. The X-axis represents the number of pairwise differences, and the Y-axis represents the frequency of the pairwise comparisons. The observed frequencies were represented by the line marked with an x. The frequency expected under the hypothesis of the population expansion model was depicted by a continuous line. (c) Bayesian skyline plots. The X-axis represents time (Ka) and the Y-axis represents effective population size × mutation rate per site per generation. Medians are shown as solid lines and blue areas represent 95% HPD limits. The map provided by the d-maps site (https://d- maps.com) was edited to refer to biogeographic realms (https://ecoregions.appspot.com) by Adobe Illustrator v.22.2 (https://www.adobe.com).
Demographic history with population expansion and dispersal
To reconstruct historical demography in terms of population expansion and dispersal, with the 393 COI haplotypes including 275 for N. albicilla (PAIO) and 118 for N. originalis sp. nov. (Afrotropic), we performed neutrality tests, mismatch distribution analysis (MDA), and Bayesian skyline plot (BSP) analysis. Neutrality tests (Tajima’s D and Fu’s FS) showed statistically significant negative values, except for the weak positive value of Tajima’s D observed in the TH population (Table 2). The negative values of Tajima’s D and Fu’s FS tests (Table 2) provide data that support a recent expansion. The MDA results (Fig. 4b, S5a) showed that each of the five different geographical groups, as well as N. albicilla and N. originalis sp. nov. form only one unimodal curve. The unimodal characteristics also indicate that N. albicilla and N. originalis sp. nov. have undergone recent demographic expansion. However, the total that merges all the data of N. albicilla (PAIO) and N. originalis sp. nov. (Afrotropic) forms a bimodal curve, indicating the existence of two different population expansion histories. The bimodal peaks may indicate the presence of two distinct genetic lineages, representing N. albicilla (PAIO) and N. originalis sp. nov. (Afrotropic). The BSP results also showed that the effective population sizes of N. albicilla and N. originalis sp. nov. had begun to gradually increase around 250 Ka (Fig. 4c, S5b).
Discussion
In this study, 697 COI sequences of N. albicilla, which consisted of 167 from Jeju Island, South Korea, and the remaining 530 from published data, were employed to examine the population genetic structure and taxonomy of N. albicilla inhabiting the Indo-Pacific region. The TCS network (Fig. 2a), PCoA (Fig. 2b), phylogenetic trees (Fig. 2c; Figs. S1 and S3), and DNA barcoding gap (Fig. 3) clearly showed that there are two different genetic lineages. The two genetic lineages were statistically supported by AMOVA (Table 3). The PAIO lineage is predominantly distributed in the Palearctic, Australasia, Indo-Malay, and Oceania regions, and the Afrotropic lineage is dominantly distributed in the Afrotropic region. This result supports the perspective of Crandall et al.5: Clade A corresponds to Afrotropic and Clade B to PAIO.
Our results indicate that the two genetic lineages of N. albicilla could be discriminated into two different species: N. albicilla (PAIO) and N. originalis sp. nov. (Afrotropic). We decided that the PAIO type continuously retains the original name of N. albicilla Linnaeus 1758, and the Afrotropic type is newly named N. originalis sp. nov.: “originalis” is a Latin word that means an English word “origin”, indicating that N. albicilla may have originated from N. originalis sp. nov. The original description of N. albicilla Linnaeus, 1758 was conducted with the sample from Hitoe, Tsushima, Japan which is the closest Japanese island to the Korean peninsula. Although the original description by Linnaeus21 was not sufficiently detailed to distinguish one from the other species, we could conclude that the PAIO type corresponds to N. albicilla because all individuals collected from five different Japanese sites including Okinawa I. (1), Shiova-wan(11), Cape Hedo (9), Oku (1), and Misaki (20) (Table 1) are appeared in N. albicilla (Clade B) without any exception (Figs. 2, S1, and S3). It implies that the Afrotropic type has never been reported from Japan so far.
The Nerita species, including N. albicilla and N. originalis sp. nov., display extensive dispersal potential because of their pelagic larval stage (up to 6 months)22. N. albicilla and N. originalis sp. nov. go through veliger larvae that remain in the plankton stage for weeks to months, floating along the flow of ocean currents in the Indo-Pacific, and are less affected by geographical barriers for dispersal. Nevertheless, in this study, it was observed that the distributional ranges of N. albicilla and N. originalis sp. nov. overlap in many locations in the Indo-Pacific, such as Jeju Island, Cook Islands, Indonesia, New Caledonia, Singapore, and South Africa, indicating broadly sympatric distributions. The sympatric distribution with their genetic differences could provide evidence for the possible species discrimination of N. albicilla. Through further studies including comparative morphological analyses, it may be possible to obtain additional evidence for the species discrimination of N. albicilla into N. albicilla and N. originalis sp. nov. The ancestral Afrotropic origin of N. albicilla was shown in S-DIVA and the basal-branch positioning of a South African COI haplotype (NAH 324) in N. albicilla of the ML trees (Fig. 4a; Fig. S1), which suggests that it is most likely that N. albicilla may have originated from N. originalis sp. nov. inhabiting the Afrotropic region. It is conceivable that the N. albicilla might have first appeared in South Africa at about 5.96 Ma and later they had spread to the Palearctic region through Indo-Malay, Australasia, and Oceania. Along the dispersal of N. albicilla east and north, possibly out of Africa, its COI haplotype diversity dramatically increased during the late Pliocene and mainly Pleistocene (ca. 5.0–1.0 Ma; Fig. 4a) with their adaptation and positive selection process to a variety of new environments11,23. Changes in shorelines, oceanic circulation, upwelling zones, temperatures, salinities, nutrient availability, and sea level fluctuations affect coastal habitats, causing habitat shifts in rocky habitats24. Interglacial transgressions may have provided opportunities for some species that survived glacial periods to expand and move into newly flooded coastal habitats25. During the warm periods of deglaciation, species that were able to adapt to warming conditions experienced population expansion and shifted their range northward26. Due to these environmental conditions, N. albicilla and N. originalis sp. nov. may have experienced dramatic population expansions. With the dispersal out of Africa and the late Pliocene and Pleistocene explosions of COI haplotype diversity of N. albicilla and N. originalis sp. nov., its effective population size expansions simultaneously occurred in all five regions of the Indo-Pacific area at approximately 250 Ka before the Last Glacial Maximum (LGM: ca. 0.027–0.021 Ma) (Fig. 4c, S5b).
In general, the genus Nerita has a long-lived planktotrophic larval period1,22, which makes it possible to increase the time to dispersal, allowing a wide range of habitats to be reached27. The molluscan fauna of eastern Asia has been created by the repeated migration of fauna from the Indo-West Pacific Ocean28,29. Strong oceanic surface currents can transport planktonic larvae to distant oceanic islands, and genetic variations may accumulate, causing speciation on these islands30,31,32,33. Therefore, the possibility of moving to the east and north along sea currents (Fig. 5a)34,35 is considered the primary reason for the observed distribution. A summer monsoonal wind (southwest) can drive seasonal reversals in surface circulation patterns and water exchange, and these seasonal currents and the long planktotrophic larval period could be responsible for the larval dispersal of N. albicilla. Interestingly, we found two N. originalis sp. nov. individuals on Jeju Island, South Korea. As previously mentioned, they likely moved from the Afrotropic region through sea currents. However, recent anthropogenic introductions should not be overlooked because of the globalization of maritime trade through commercial shipping (Fig. 5b)36,37,38. Further studies are necessary to examine the major reasons and routes of their historical dispersal in Africa.

The maps of sampling sites and possible migration routes by sea currents and core and secondary maritime shipping routes that might be related to artificial migration for Nerita albicilla and Nerita originalis sp. nov. (a) A map of sampling sites and possible migration routes by sea currents for Nerita albicilla and Nerita originalis sp. nov. The blue dots represent the sites where only N. albicilla are found and the red dots represent the sites where only N. originalis sp. nov. are found. Mixed colored dots mean that both N. albicilla and N. originalis sp. nov. are found on the sample site. Pink lines are demonstrated as sea currents. The abbreviations of sea currents are as follows: EACC, East African Coastal Current; EICC, East India Coastal Current; EKWE, East Korean Warm Current; GW, Great Whirl; JWC, Jeju Warm Current; KC, Kuroshio Current; SC, Somali Current; SMC, Summer Monsoon Current; TWC, Tsushima Warm Current; WKCC, West Korea Coastal Current; WICC, West India Coastal Current, and YSWC, Yellow Sea Warm Current. Schematic representation of the circulation of sea current during July (summer monsoon). (b) A map of core and secondary maritime shipping routes in the Indo-Pacific and the pie charts representing the ratio of the number of N. albicilla and N. originalis sp. nov. In the Afrotropic region, there was only one N. albicilla individual and 173 N. originalis sp. nov. individuals. In the Palearctic, Australasia, Indo-Malay, and Oceania regions, there were 504 N. albicilla individuals and 19 N. originalis sp. nov. individuals. We found two N. originalis sp. nov. individuals in Jeju Island, South Korea that might have recently come north from the Afrotropic region. The basic map is from a free map providing site (https://d- maps. com) and the figure was edited refer to Soegiarto, A. & Birowo (1975) and Main Maritime Shipping Routes and Checkpoints (2011) by Adobe Illustrator v.22.2.
Molecular markers can be used to track the demographic responses of populations to past climatic shifts and other historical processes39,40, and to analyze the current intraspecific genetic variation to predict the impact of future climate change on certain populations41. As mentioned before, the National Institute for Biological Resources (NIBR), Ministry of Environment, South Korea selected the 100-representative organism of CBIS in 2010 which are sensitively affected by global warming inhabiting the Korean Peninsula. N. albicilla is included in the list of CBIS and this study is the first report with respect to this species. Through continuous monitoring of distributional changes and intraspecific genetic variations of N. albicilla and N. originalis sp. nov. could be a cornerstone for exploring and predicting the global warming effect in the Indo-Pacific with the northward migration of marine gastropods.
Materials and methods
Sample collection
In total, 167 N. albicilla individuals were collected from five intertidal regions (GJ, NJ, SC, TS, and SM) of Jeju Island on the Korean Peninsula in 2021 (Fig. 1). Collected individuals were immediately fixed with 100% alcohol. They were brought to the laboratory and stored at -20 °C. Species identification was performed using shell morphology and COI sequence.
DNA extraction, PCR amplification, and sequencing
Genomic DNA was isolated from the muscle tissue (foot) using a DNeasy Blood and Tissue Kit (QIAGEN, Valencia, California, USA) following the manufacturer’s protocol. The concentration of extracted DNA was evaluated using NanoDrop 2000 (Thermo Fisher Scientific Co, USA) and 1% agarose gel electrophoresis.
To amplify partial fragments of the mitochondrial COI gene, PCR was performed using universal primers (LCO1490/HCO2198)42. The primer sequences are shown in Table S5. The thermal cycling profile consisted of denaturation at 94 °C for 2 min, 35 cycles of 94 °C for 1 min, 48 °C for 1 min, and 72 °C for 1 min; a final extension at 72 °C for 5 min; and a cooling down step was performed at 4 °C. The PCR mixtures were prepared to a total volume of 50 μl containing 1 μl of 10 pM of each primer, 1 μl of 10 mM dNTP mix, 5 μl of 10 × Taq DNA polymerase reaction buffer, 0.25 μl of 5 U/μl DiaSter Taq DNA polymerase, and 3 μl of total genomic DNA. One microliter of each PCR product was electrophoresed on a 1% agarose gel containing the eco-dye which is fluorescent dye as DNA staining solution and observed under UV light. When the PCR bands were detected, the PCR products were purified using a QIAquick PCR Purification Kit (QIAGEN Co., USA) and directly sequenced by the commercial sequencing service company SolGent Co., Ltd. (Daejeon, South Korea) with an ABI Prism 3730 DNA sequencer (PerkinElmer Inc., USA) using a Big Dye Termination Sequencing Kit (PerkinElmer Inc., USA).
Population genetic analyses
We used COI sequences from 524 N. albicilla individuals, provided by Eric D. Crandall (https://geome-db.org/query), and six N. albicilla individuals which were previously reported in the NCBI GenBank database. The nucleotide sequences of mitochondrial COI obtained from N. albicilla were aligned using BioEdit 7.0.5.343 and Clustal X244. Pairwise genetic distances were calculated with the Kimura-2-Parameter (K2P) in Mega 11.0.1345. The identification of variable and parsimony informative sites and the number of haplotypes (h) were estimated using DnaSP 6.12.0346. Based on the haplotype list generated by DnaSP, the number of private haplotypes unique to each population was determined (Table S1). Descriptive statistics including the number of polymorphic sites and haplotypes, haplotype diversity, and nucleotide diversity were estimated for each population using the program DnaSP 6.12.0346. A haplotype network was constructed using a statistical parsimony approach at the population level using PopART47. To further evaluate and visualize the geographic genetic structure among the populations, PCoA was performed to visualize the geographic genetic structure among species using the DARwin 6.0.2148 program which ordinates genetic distance estimates calculated with the haplotype data used in this study. FST values between N. albicilla populations using COI data were calculated in Arlequin 3.5.2.249 to test for significant differentiation among each population from sampled sites. Hierarchical analysis of molecular variance (AMOVA) was performed to examine the amount of genetic variability partitioned within and among the five groups of N. albicilla according to biogeographic regions (Afrotropic, Australasia, Indo-Malay, Oceania, and Palearctic) and Afrotropic and PAIO in Arlequin 3.5.2.249. The correlation between pairwise genetic distances and geographic distances between individuals was established using the Mantel test (Alleles in Space 1.050).
DNA barcoding gap analyses
Analyses of barcoding gaps based on COI were conducted using an online version of ABGD (https://bioinfo.mnhn.fr/abi/public/abgd/abgdweb.html) to generate distance histograms, distance ranks, and automatic partitions. These analyses were conducted using the Kimura-2-parameter (K2P) distance matrix and two different parameters: the range of prior intraspecific divergence from Pmin (0.001) to Pmax (0.1) and relative gap width (X = 1.5).
Phylogenetic analyses and divergence time estimation based on COI
Phylogenetic analyses based on the 393 COI haplotype sequences obtained in this study were performed using the ML method. In the ML tree, model selection in the IQ-Tree software package (http://www.iqtree.org) was tested and the substitution model HKY + F + I was chosen as the best-fit model under the Bayesian information criterion. The phylogenetic tree was analyzed from 1,000 ultrafast bootstrap replicates using the IQ-Tree web server (http://iqtree.cibiv.univie.ac.at). To elucidate phylogenetic relationships of N. albicilla and N. originalis sp. nov. within the genus Nerita, we additionally reconstructed ML and BI trees with 133 COI haplotypes (Fig. S3), which include N. albicilla (54 haplotypes), N. originalis (24 haplotypes), and 52 Nerita species (one haplotype per species) (Table S3). The maximum likelihood tree was reconstructed under the TIM F + I + G4 in IQtree online site and the Bayesian topology was inferred under the GTR + F + I + G4 model using MrBayes 3.2.251. Bathynerita naticoidea, Clypeolum owenianum, and Puperita pupa were used as outgroup species.
Divergence time estimation for the two phylogenetic lineages of N. albicilla was conducted based on COI haplotype sequence matrix used in Fig. S3 by BEAST 2.6.619,20. The analysis was estimated using the strict molecular clock algorithm under the calibrated-Yule model52, which was calibrated using the earliest fossil record data of Nerita (ca. 90 Ma)53 for the basal node (90 Ma; normal distribution) and a “monophyly” option was chosen in the BEAUti 2 program (the genus Nerita). Posterior distributions of the parameter were estimated using 10,000,000 MCMC iterations and sampled every 1,000 iterations after discarding the initial 20% of the iterations as burn-in. The best-fit substitution model was selected using jModelTest 2.1.754, as GTR + F + I + G4. TreeAnnotator 2.6.655 was used to produce a tree with maximum clade credibility and median height after removing the initial 20% of iterations as burn-in. FigTree 1.4.456 was used to visualize the topology of the resultant consensus tree. To estimate the distribution of a hypothetical common ancestor, S-DIVA analysis based on the Bayesian binary MCMC method57 based on the BEAST tree of COI using the RASP 3.2 program58. When running the program, three possible distributed areas were coded for the ingroup taxa as follows: (A) Palearctic; (B) Afrotropic; (C) Australasia, Indo-Malay, and Oceania.
Demographic history analyses
To investigate the demographic history of populations of N. albicilla, we used three approaches: neutrality tests, MDA59, and BSP60. Neutrality tests such as Tajima’s D61 and Fu’s Fs62 were conducted to examine the population demographic history and evolutionary neutrality of N. albicilla using the Arlequin 3.5.2.249 program based on COI haplotypes. MDA59 was also conducted to investigate the demographic stability of the phylogenetic lineages and species using DnaSP 6.12.0346. BSP60 was computed to examine historical demographic fluctuations since the most recent common ancestor. For the analyses, the HKY model was selected, mutation rates of 2.0 × 10–8 under a strict molecular clock were used with BEAST 2.6.619,20, and MCMC was run for 10 million steps. TRACER 1.7.263 was used to calculate the ESS value and construct the BSP.
Data availability
The COI haplotype sequences of Nerita albicilla have been deposited in the GenBank database under accession numbers ON982333–ON982444 (NAH001–NAH112). The pre-processed dataset can be found in the Supplementar Data as follows: Data S1, nucleotide sequence alignment of 393 COI haplotypes of N. albicilla, Data S2, the K2P intraspecific genetic distance with 393 haplotype sequences of Nerita albicilla, and Data S3, the K2P interspecific distance genetic ranges with 54 different species with the genus Nerita.
References
Kano, Y. Usefulness of the opercular nucleus for inferring early development in neritimorph gastropods. J. Morphol. 267, 1120–1136 (2006).
Echem, R. T. Morphometric relations of gastropod species: Nerita albicilla and Patella nigra. WNOFNS 7, 30–36 (2017).
Hughes, R. N. Notes on the Nerita (Archaeogastropoda) populations of Aldabra Atoll, Indian Ocean. Mar. Biol. 9, 290–299 (1971).
Vermeij, G. J. Substratum relationships of some tropical Pacific intertidal gastropods. Mar. Biol. 10, 315–320 (1971).
Crandall, E. D., Frey, M. A., Grosberg, R. K. & Barber, P. H. Contrasting demographic history and phylogeographical patterns in two Indo-Pacific gastropods. Mol. Ecol. 17, 611–626 (2008).
Choe, B. L. Ilustrated encyclopedia of fauna and flora of Korea. Mollusca (II) (ed. Choe, B. L.) 281 (Ministry of Education, 1992).
National Institute of Biological Resources. CBIS; Climate-sensitive Biological Indicator Species. https://species.nibr.go.kr/home/mainHome.do?cont_link=011&subMenu=011017&contCd=011017 (2010).
Hwang, U. W. & Kim, W. General properties and phylogenetic utilities of nuclear ribosomal DNA and mitochondrial DNA commonly used in molecular systematics. Korean J. Parasitol. 37, 215 (1999).
Baek, S. Y. et al. DNA barcoding of metazoan zooplankton copepods from South Korea. PLoS ONE 11, e0157307 (2016).
Choi, E. H. et al. Molecular phylogenetic, population genetic and demographic studies of Nodularia douglasiae and Nodularia breviconcha based on COI and 16S rRNA. Sci. Rep. 10, 1–14 (2020).
Shin, C. R. et al. Characterization of metapopulation of Ellobium chinense through Pleistocene expansions and four covariate COI guanine-hotspots linked to G-quadruplex conformation. Sci. Rep. 11, 1–13 (2021).
Choi, E. H. et al. Liolophura species discrimination with geographical distribution patterns and their divergence and expansion history on the northwestern Pacific coast. Sci. Rep. 11, 1–17 (2021).
Wilson, A. C. et al. Mitochondrial DNA and two perspectives on evolutionary genetics. Biol. J. Linn. Soc. 26, 375–400 (1985).
Lee, J. H., Kim, S. K. & Hwang, U. W. Complete mitochondrial genome of Lamprotula coreana (Unionidae, Unionoida, Bivalvia). Mitochondrial DNA 27, 1026–1027 (2016).
Cha, S. H. et al. The complete mitochondrial genome of a freshwater mussel Nodularia douglasiae (Bivalvia: Unionidae). Mitochondrial DNA 3, 947–948 (2018).
Choi, E. H. & Hwang, U. W. The complete mitochondrial genome of an endangered triton snail Charonia lampas (Littorinimorpha: Charoniidae) from South Korea. Mitochondrial DNA 6, 956–958 (2021).
Gunyakti, K. S., Celik, F., Kesik, H. K. & Simsek, S. In silico analysis of the biodiversity and conservation status of mitochondrial cytochrome C oxidase subunit 1 (COI) gene of Taenia multiceps. Acta Parasitol. 65, 852–858 (2020).
Baek, S. Y. et al. COI-based genetic structure of an exotic snapping turtle Chelydra serpentina imported to South Korea. ASED 36, 354–362 (2020).
Drummond, A. J. & Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 1–8 (2007).
Bouckaert, R. et al. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 10, e1003537 (2014).
Linnaeus, C. V. Systema Naturae per regna tria naturae. Secundum classes, ordines, genera, species, cum characteribus, differentiis, synonymis, locis. Editio, 1, 823 (1758).
Underwood, A. J. Comparative studies on the biology of Nerita atramentosa Reeve, Bembicium nanum (Lamarck) and Cellana tramoserica (Sowerby) (Gastropoda: Prosobranchia) in SE Australia. J. Exp. Mar. Biol. Ecol. 18, 153–172 (1975).
Mirabello, L. & Conn, J. E. Population analysis using the nuclear white gene detects Pliocene/Pleistocene lineage divergence within Anopheles nuneztovari in South America. Med. Vet. Entomol. 22, 109–119 (2008).
Graham, M. H., Dayton, P. K. & Erlandson, J. M. Ice ages and ecological transitions on temperate coasts. Trends Ecol. Evol. 18, 33–40 (2003).
Hewitt, G. M. Post-glacial re-colonization of European biota. Biol. J. Linn. Soc. 68, 87–112 (1999).
He, L. et al. Late Pleistocene population expansion of Scylla paramamosain along the coast of China: A population dynamic response to the last interglacial sea level highstand. J. Exp. Mar. Biol. Ecol. 385, 20–28 (2010).
Paulay, G. & Meyer, C. Dispersal and divergence across the greatest ocean region: do larvae matter?. Integ. Comput. Biol. 46, 269–281 (2006).
Jablonski, D., Roy, K. & Valentine, J. W. Out of the tropics: evolutionary dynamics of the latitudinal diversity gradient. Science 314, 102–106 (2006).
Jablonski, D. et al. Out of the tropics, but how? Fossils, bridge species, and thermal ranges in the dynamics of the marine latitudinal diversity gradient. Proc. Natl. Acad. of Sci. U.S.A. 110, 10487–10494 (2013).
Paulay, G. & Meyer, C. Diversification in the tropical Pacific: comparisons between marine and terrestrial systems and the importance of founder speciation. Integr. Comp. Biol. 42, 922–934 (2002).
Nakano, T., Yazaki, I., Kurokawa, M., Yamaguchi, K. & Kuwasawa, K. The origin of the endemic patellogastropod limpets of the Ogasawara Islands in the northwestern Pacific. J. Molluscan Stud. 75, 87–90 (2009).
Bird, C. E., Holland, B. S., Bowen, B. W. & Toonen, R. J. Diversification of sympatric broadcast-spawning limpets (Cellana spp.) within the Hawaiian archipelago. Mol. Ecol. 20, 2128–2141 (2011).
Williams, S., Apte, D., Ozawa, T., Kaligis, F. & Nakano, T. Speciation and dispersal along continental coastlines and island arcs in the Indo-West Pacific turbinid gastropod genus Lunella. Evolution 65, 1752–1771 (2011).
Soegiarto, A. & Birowo, S. Atlas oceanology of the Indonesian and the adjacent waters. Book 1. The present state of knowledge of oceanology in Indonesia. Nat. Inst. Oceanol. Jakarta-Indonesia (Indonesian) (1975).
Shankar, D., Vinayachandran, P. N. & Unnikrishnan, A. S. The monsoon currents in the north Indian Ocean. Prog. Oceanogr. 52, 63–120 (2002).
Jean-Paul, R. Department of Global Studies & Geography. Hofstra University. Main Maritime Shipping Routes and Checkpoints. https://porteconomicsmanagement.org/pemp/contents/part1/interoceanic-passages/main-maritime-shipping-routes/ (2011).
Kolar, C. S. & Lodge, D. M. Ecological predictions and risk assessment for alien fishes in North America. Science 298, 1233–1236 (2002).
Hulme, P. E. Trade, transport and trouble: Managing invasive species pathways in an era of globalization. J. Appl. Ecol. 46, 10–18 (2009).
Templeton, A. R., Routman, E. & Phillips, C. A. Separating population structure from population history: A cladistic analysis of the geographical distribution of mitochondrial DNA haplotypes in the tiger salamander, Ambystoma tigrinum. Genetics 140, 767–782 (1995).
Lessa, E. P., Cook, J. A. & Patton, J. L. Genetic footprints of demographic expansion in North America, but not Amazonia, during the Late Quaternary. Proc. Natl. Acad. of Sci. U.S.A. 100, 10331–10334 (2003).
Davis, M. B. & Shaw, R. G. Range shifts and adaptive responses to Quaternary climate change. Science 292, 673–679 (2001).
Folmer, O., Black, M., Hoeh, W., Lutz, R. & Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 3, 294–299 (1994).
Hall, T. A. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41, 95–98 (1999).
Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F. & Higgins, D. G. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882 (1997).
Tamura, K., Glen, S. & Sudhir, K. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027 (2021).
Rozas, J. et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302 (2017).
Leigh, J. W. & Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 6, 1110–1116 (2015).
Perrier, X. DARwin software, available at, http://darwin.cirad. fr/darwin (2006).
Excoffier, L. & Lischer, H. E. L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567 (2010).
Miller, M. P. Alleles In Space (AIS): Computer software for the joint analysis of interindividual spatial and genetic information. J. Hered. 96, 722–724 (2005).
Ronquist, F. et al. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542 (2012).
Drummond, A. J., Ho, S. Y. W., Phillips, M. J. & Rambaut, A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 4, e88 (2006).
Frey, M. A. & Vermeij, G. J. Molecular phylogenies and historical biogeography of a circumtropical group of gastropods (Genus: Nerita): implications for regional diversity patterns in the marine tropics. Mol. Phylogenet. Evol. 48, 1067–1086 (2008).
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 9, 772 (2012).
Rambaut, A. & Drummond, A. J. TreeAnnotator v. 2.3. 0., Available at, http://beast2.orgpackage (2014).
Rambaut, A. FigTree v. 1.4. 4., Available at, http://tree.bio.ed.ac.uk/sofware/fgtree/ (2012).
Nylander, J. A. A., Olsson, U., Alström, P. & Sanmartin, I. Accounting for phylogenetic uncertainty in biogeography: A Bayesian approach to dispersal-vicariance analysis of the thrushes (Aves: Turdus). Syst. Biol. 57, 257–268 (2008).
Yu, Y., Harris, A. J., Blair, C. & He, X. RASP (Reconstruct Ancestral State in Phylogenies): a tool for historical biogeography. Mol. Phylogenet. Evol. 87, 46–49 (2015).
Rogers, A. R. & Harpending, H. Population growth makes waves in the distribution of pairwise genetic differences. Mol. Biol. Evol. 9, 552–569 (1992).
Heled, J. & Drummond, A. J. Bayesian inference of population size history from multiple loci. BMC Evol. Biol. 8, 1–15 (2008).
Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595 (1989).
Fu, Y.-X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147, 915–925 (1997).
Rambaut, A., Suchard, M. A., Xie, D. & Drummond, A. J. Tracer v1. 6. 2014. Preprint at (2015).
Acknowledgements
This work was supported by a grant from the National Institute of Biological Resources (NIBR), funded by the Ministry of Environment (MOE) of the Republic of Korea (NIBR202206203, NIBR202231206) and the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (2020R1A6A3A01095736 to BP and 2021R1C1C2012882 to EHC).
Author information
Authors and Affiliations
Contributions
U.W.H. conceived the study. U.W.H. and E.H.C. designed and set up the experiment. S.H., B.P., G.K., and U.W.H. performed the sample collections. S.H., G.K., and E.H.C. performed the experiment and analyzed the data. U.W.H., S.H., and B.P. wrote the manuscript. All authors thoroughly reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hong, S., Park, B., Kim, G. et al. Possible species discrimination of a blotched nerite Nerita albicilla with their distribution pattern and demographic history in the Indo-Pacific. Sci Rep 13, 4545 (2023). https://doi.org/10.1038/s41598-023-31004-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-31004-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
