
Abstract
A part of the radiocaesium from the Fukushima Daiichi Nuclear Power Plant (FDNPP) accident was emitted as glassy, water-resistant caesium-bearing microparticles (CsMPs). Here, we isolated and investigated seven CsMPs from marine particulate matter and sediment. From the elemental composition, the 134Cs/137Cs activity ratio, and the 137Cs activity per unit volume results, we inferred that the five CsMPs collected from particulate matter were emitted from Unit 2 of the FDNPP, whereas the two CsMPs collected from marine sediment were possibly emitted from Unit 3, as suggested by (i) the presence of calcium and absence of zinc and (ii) the direction of the atmospheric plume during the radionuclide emission event from Unit 3. The presence of CsMPs can cause overestimation of the solid–water distribution coefficient of Cs in marine sediments and particulate matter and a high apparent radiocaesium concentration factor for marine biota. CsMPs emitted from Unit 2, which were collected from the estuary of a river that flowed through a highly contaminated area, may have been deposited on land and then transported by the river. By contrast, CsMPs emitted from Unit 3 were possibly transported eastward by the wind and deposited directly onto the ocean surface.
Similar content being viewed by others


Introduction
Large amounts of radionuclides were emitted into the environment by the Fukushima Daiichi Nuclear Power Plant (FDNPP) accident. Among the many radionuclides emitted, radioactive caesium (RCs) has been intensively investigated because of the large amount of emission and its relatively long half-life (T1/2 of 137Cs = 30 years). Previous studies have shown that 3–6 PBq of 137Cs was deposited on the land, 12–15 PBq was deposited from the atmosphere onto the Pacific Ocean surface, and 3.6 ± 0.7 PBq was released directly from the accident site into the ocean1,2. RCs concentrations in surface seawater in the coastal area near the FDNPP decreased rapidly during the several years following the accident, partly because of the physical decay of RCs, but mostly because of dispersion by seawater circulation1,3,4. However, particulate RCs concentrations in coastal sediments showed large variations5 and decreased more slowly than the dissolved RCs concentrations6. We hypothesized that the large variations and slow decrease of particulate RCs concentrations might be due to the presence of water-resistant Cs-bearing microparticles (CsMPs); this possibility was also suggested by Kusakabe et al.7 and Otosaka8.
Adachi et al.9 first reported CsMPs from the FDNPP accident on filters used to collect aerosols in Tsukuba, Ibaraki Prefecture (this type of CsMPs are known as Type-A particles following Igarashi et al.10). Subsequent studies have shown that the matrix of Type-A particles is silicon dioxide (SiO2) glass, and Cs, iron (Fe), zinc (Zn), and other various elements are included in the matrix11,12,13,14. Type-A particles are ~ 0.1–10 µm in diameter, and their 137Cs activity is ~ 10–2 to 102 Bq/particle10,15. Uranium (U) and Cs isotopic ratios (U isotopes, 235U and 238U; Cs isotopes, 133Cs, 134Cs, 135Cs, and 137Cs)16, and 134Cs/137Cs activity ratios17 of Type-A particles are consistent with those of Unit 2 or 3 (Unit 2/3 hereinafter) of the FDNPP calculated theoretically based on the nuclear fuel burnup18. Another type of CsMPs, Type-B particles10, emitted from Unit 1 of the FDNPP was collected at the vicinity of the FDNPP19. Type-B particles are also composed mainly of SiO2, but they are variable in shape and range in diameter from 50 to 400 μm. Their 137Cs activity is 101–104 Bq/particle, and they include refractory elements such as calcium (Ca)17,20.
Type-A particles were discovered from suspended particles in the downstream of the Kuchibuto River in Fukushima Prefecture21. Previous studies did not succeed in isolating CsMPs from suspended particles in seawater and marine sediments, although they reported highly radioactive spots on autoradiographic images of marine samples22,23. The presence of CsMPs in river water and the radioactive spots on autoradiographic survey images of the marine samples suggest that CsMPs, which deposited on the land, eventually flow into the ocean via rivers. To confirm this possibility, the particles must be isolated and directly observed, for example by conducting a scanning electron microscopy (SEM) with energy-dispersive spectrometer (EDS) analysis, because CsMPs and RCs-enriched clay minerals are hard to distinguish on autoradiographic images15. Despite the importance of CsMPs in evaluating the behaviour of RCs in the ocean and its influence on marine biota, to the best of our knowledge CsMPs have not yet been isolated from marine samples. In this study, we efficiently isolated CsMPs from marine samples collected in coastal areas of the Pacific Ocean by the wet separation method16,17,21. After isolating CsMPs from the marine samples, we conducted an SEM–EDS analysis of the isolated CsMPs and measured their radioactivity with a high-purity germanium semiconductor (HPGe) detector. We also measured their size, elemental composition, 137Cs activity, and 134Cs/137Cs activity ratio, and we calculated the ratio of 137Cs in CsMPs to the total 137Cs in the sample. That ratio affects evaluations of (i) the solid–water distribution coefficient (Kd) value between seawater and particulate matter in the water column or marine sediments, and (ii) the RCs concentration factor (CF) for marine organisms. Moreover, we discussed the sources of CsMPs in the marine samples and their migration behaviour in the ocean.
Results and discussion
We isolated seven radioactive particles (RPs) from the marine samples collected at points A–F (Fig. 1). We refer to the five RPs isolated from samples of particulate matter (suspended particles, sinking particles, and zooplankton) as PM particles; those from points A, B, C, D, and F are referred to as PM-A, -B, -C, -D, and -F, respectively. We refer to the two RPs isolated from the marine sediment sample collected at point E as MS particles (MS-E1 and -E2).

Sampling points A–F and rivers near the FDNPP, shown on a Cs inventory map from MEXT40.
RCs activity in RPs isolated from bulk marine samples
The analytical data for the bulk samples and isolated RPs are summarized in Table 1 (all RCs data have been decay-corrected to 11 March 2011). The 134Cs and 137Cs activities of the PM particles were 0.250–1.83 Bq and 0.229–1.66 Bq, respectively, whereas the 134Cs and 137Cs activities of the MS particles were 20.4–42.0 Bq and 18.6–38.6 Bq, respectively. These differences are attributable to the difference in the particle sizes (MS particles > PM particles). The 134Cs/137Cs activity ratio of all RPs was approximately 1 (average = 1.09); therefore, the RPs isolated from the marine samples were derived from the FDNPP accident18. In addition, the 134Cs/137Cs activity ratios of the RPs (~ 1.07 to 1.11; Table 1) indicated that they were emitted from Unit 2/3 of the FDNPP because the ratios for RPs were around 1.05 (Unit 3) and 1.08 (Unit 2) calculated by Nishihara et al.18.
Characterisation of the isolated RPs
PM-A, -B, and -F were similar to terrestrial Type-A particles17 with regard to their size and shape (Fig. 2), elemental composition (Fig. 3, Table 2), and RCs activity (Table 1). PM-C and -D were attached on a fiber-reinforced plastic particle (~ 2 mm) and Fe-rich particle (~ 20 µm), respectively. Although Cs was detected only in an area with a radius of about 1 µm on these larger particles (within the white dashed circles in Fig. 2), Si, chlorine, potassium, Fe, and Zn peaks were detected on the EDS spectra of these areas; thus, they were compositionally similar to Type-A particles from terrestrial samples17. It was necessary to obtain elemental maps of PM-D by EDS (Fig. S1) because the area where Cs was detected could not be distinguished on backscattered electron images. The EDS spectrum of PM-D showed a weak but clear U peak (Fig. 3). Abe et al.11 showed by a synchrotron radiation analysis that Type-A particles from a terrestrial sample contained U. The aluminium (Al) detected in some PM particles is probably attributable to secondary adhesion of Al to the particles surface because previous studies scarcely detected Al inside CsMPs12,13,14.

SEM images of the seven radioactive particles (RPs) isolated from marine samples. PM and MS indicate RPs isolated from particulate matter and marine sediment, respectively. RPs were placed on Kapton tape for the SEM–EDS analyses. Cs was detected only in areas within the white dashed circles of each large particles incorporating PM-C and -D.

EDS spectra of the seven RPs. The carbon was from the carbon coating done during sample preparation.
The volume of each RP was calculated from its apparent diameter and compared with CsMP volume data reported by Miura et al.17 (Fig. 4); the diameters of the white dashed circles on the SEM images (Fig. 2) were used to calculate the volumes of PM-C and -D. In terrestrial samples, the 137Cs activity per unit volume of Type-A particles is ~ 10,000 times higher than that of Type-B particles emitted from Unit 1 of the FDNPP17 (Fig. 4). The RPs isolated from the marine samples in this study plotted approximately along the line corresponding to the relationship in Type-A particles from terrestrial samples. Thus, the PM particles in this study were classified as Type-A particles on the basis of their elemental composition (Table 2), 134Cs/137Cs activity ratio (Table 1), and 137Cs activity per unit volume (Table 2, Fig. 4).

Relationships between 137Cs activity and particle volume (137Cs activity per unit volume) of Type-A and Type-B particles, reported by Miura et al.17, compared with that of the marine RPs in this study. 137Cs activity per volume of RPs from marine samples is similar to that of terrestrial Type-A particles. The relationship in sample SP reported by Zhang et al.24 is also plotted.
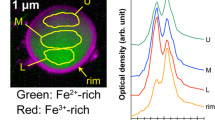
The MS particles were found to be aggregates of Type-A particles. MS-E1 and -E2 were ~ 10 µm and ~ 15 µm in size, respectively. To calculate the volumes (Fig. 4), we approximated their volume by summing the volumes of the spheres composing each aggregate (Fig. S2). Because the two MS particles were not wholly visible in the SEM images, MS particle volume was underestimated. The 134Cs/137Cs activity ratio (Table 1) and 137Cs activity per unit volume of the MS particles were similar to those of Type-A particles. However, although the Type-A particles reported so far contain Zn10, no Zn was detected in the MS particles (Fig. 3, Table 2). Moreover, the MS particles included a Ca-rich area that overlapped the Cs-rich area (Figs. 3, S3, and S4). This characteristic is not found in the previously reported CsMPs (i.e., Type-A and Type-B particles)17,19. Zhang et al.24 considered a RP that they called SP, which had a 134Cs/137Cs activity ratio of more than 1 and lacked Zn, to be a new type of CsMPs (Table 2). However, SP differed from the MS particles with regard to 137Cs activity per unit volume (SP < MS-E1 and -E2; Fig. 4, Table 2) and elemental composition (e.g., Ca; Table 2). Therefore, the MS particles represent a new type of RP that is different from the previously reported CsMPs17,19 and SP24.
Influence of CsMPs on K d and CF values
We calculated the ratio of 137Cs activity in RPs to the total 137Cs in the bulk sample (R(RPs)) (Table 1). In the bulk samples collected from points A, B, C, E, and F, no radioactive spots were found by autoradiography with imaging plate (IP) apart from the isolated RPs. By contrast, most of the time-series samples collected at point D (GST#1–7) had radioactive spots, although a RP was isolated from only GST#5 (Table 1). In the current study, we assumed that radioactive spots with 137Cs activity of > 0.1 Bq on the IP image were CsMPs and calculated R(RPs) of GST#1–7 by using the photo-stimulated luminescence (PSL) values of the IP images (Table 1)21. The R(RPs) values of the zooplankton sample from point C and the sediment sample from point E were high, possibly because the concentration of non-RP 137Cs present as adsorbed species on the bulk samples was low. Both of these samples were collected in 2011, but the mesh size of the plankton net used to collect the sample from point C was 330 µm, and the < 2 mm fraction of sediment was recovered from the sample at point E by sieving. Consequently, the total surface area of the samples onto which Cs could be adsorbed was small and the 137Cs concentration of the bulk samples was low. In addition, the higher 137Cs activity of MS particles (~ 57 Bq), compared with the other RPs, caused R(RPs) of the sample from point E to be relatively high.
The large variation of RCs activity in marine sediments reported by Kusakabe et al.5 might be explained by the presence of CsMPs in the ocean. In a sample that included CsMPs, the Kd value of Cs would be overestimated because the Cs in CsMPs would be considered to be adsorbed on clay minerals. Similarly, the CF value would be overestimated because the Cs would be considered to have been absorbed by plankton. In actuality, however, Cs in CsMPs does not affect Kd or CF values because of the water-resistant character of CsMPs. CsMPs in rivers have also led to variation of Cs Kd values21. When the amount of Cs adsorbed to clay minerals (absorbed by plankton) is assessed on the basis of its Kd (CF) value, the Cs activity of CsMPs should be excluded. In the current study, radioactive spots with > 0.1 Bq of 137Cs activity were assumed to be CsMPs. However, Okumura et al.15 have reported that some CsMPs have 137Cs activity of less than 0.1 Bq, and weathered biotite may have 137Cs activity of more than 0.1 Bq; thus, distinguishing between CsMPs and weathered biotite by using IP images is difficult. In the future, a non-destructive method should be developed to allow them to be distinguished.
Source of CsMPs in marine samples
A key question of this study was where did the CsMPs in the marine samples come from? Widespread transport of CsMPs has been linked to flows of radioactive plumes in the atmosphere9. Nine major RCs-bearing plumes (P1–P9) from the FDNPP have been reported, based on atmospheric diffusion model calculations25 and the measurement of Cs activity concentrations in the atmosphere around eastern Japan26. These plumes were released between 12 March 2011 (Plume 1: P1) and 21 March 2011 (Plume 9: P9)25. In this study, we used the plume numbers defined by Nakajima et al.25 and Tsuruta et al.26.
P1 (emitted 12–13 March 2011), which included Type-B particles, was emitted from Unit 1 of the FDNPP at 15:36 JST on 12 March 2011 as a result of a hydrogen (H) explosion, and it flowed north-westward27,28 (Fig. 5). Type-B particles derived from this explosion were found to be distributed in a very narrow area north-northwest of the FDNPP17,19,20.

According to the International Research Institute for Nuclear Decommissioning (IRID)29, the Zn in Type-A particles was derived from coating materials in the FDNPP suppression chamber (S/C) because the water source for reactor Unit 2 isolation cooling was switched from a condensate storage tank to the S/C. Thus, the IRID29 concluded that Type-A particles were from Unit 2 of the FDNPP. In support of the IRID conclusion29, Kurihara et al.16 inferred that Unit 2 was the emission source of Type-A particles (particularly spherical ones) on the basis of (i) U and Cs isotope ratios, (ii) the sequence of events during the accident30,31, and (iii) reported temporal variations of polluted air25,26,27,28,32. Type-A particles were collected on an air filter on 14–15 March 2011 in Tsukuba9; therefore, P2 (emitted from the night of 14 March until the morning of 15 March 2011) contained Type-A particles. P2 flowed southwest toward the Tokyo Metropolitan Area (TMA). Ikehara et al.33 showed by an autoradiography analysis of IP of contaminated soil samples that both P2 and P3 (emitted on the afternoon of 15 March 2011) contained Type-A particles. P3 flowed first toward the central part of Fukushima Prefecture, and later northward and northwestward. Therefore, in this study we considered that both P2 and P3 included Type-A particles emitted from Unit 2 (Fig. 5).
Considering that wet venting reduced particulate emissions, CsMPs may have been released from Unit 3 during two events. The first was a H explosion that occurred at 11:01 JST on 14 March 2011, and the other occurred on the morning of 16 March 2011 and produced P4. The decreasing dry well pressure31 and white smoke from the Unit 3 building32 observed on 16 March indicated that RCs in P4 were derived mainly from Unit 327. At the time of the H explosion, the wind was blowing towards the east. P4 flowed towards the south-southwest direction and was transported to offshore of the TMA25 (Fig. 5).
P5 (18 March 2011) and P6 (19 March 2011) were transported northward from the FDNPP; then, after reaching the northern coastal area about 50 km from the plant, they flowed northeast. It is not clear whether P5 and P6 contained CsMPs, but their transport pathways did not overlap any of the sampling points of this study. P7 (20 March 2011) flowed first toward the Pacific Ocean and subsequently the northern part of the TMA. P8 (20–21 March 2011) was transported to the northwest about 50 km from the plant and then gradually moved southward. P9 (21 March 2011) flowed southeast and spread over a wide area of the TMA. Radioactive materials were released continuously during the origin periods of the P7–P925, and no CsMPs were collected on air filters during 20–21 March in Tsukuba9; thus, P7–P9 probably contained no CsMPs. Therefore, we focused on P1–P4 and suggested three possible sources of Type-A particles in the marine samples: rivers, direct atmospheric deposition, and direct release to the ocean.
(1) River transport
P2 and P3 deposited Type-A particles mainly on land areas (Fig. 5). In the five years after the FDNPP accident, 12 TBq of 137Cs was transported to the ocean by the Abukuma River and almost all of the RCs (96.5%) were transported in particulate form34. Miura et al.21 has reported Type-A particles in suspended particles from the Kuchibuto River (Fig. 1), a tributary of the Abukuma River. PM-F, a Type-A particle, was found in the sample collected at point F from the Abukuma River estuary. Several other rivers also flow through highly contaminated areas (Fig. 1), and Kubo et al.22 collected suspended particles with highly radioactive spots from the estuary of the Kuma River (4 km to the south of the FDNPP). These findings indicate that Type-A particles deposited on land after the accident subsequently entered various rivers as a result of surface soil erosion and were transported to the ocean.
(2) Direct atmospheric deposition
Few Type-A and Type-B particles were deposited from atmospheric plumes directly onto the ocean surface (Fig. 5). Okumura et al.35 have suggested on the basis of a dissolution experiment that Type-A particles (1 µm) might dissolve completely in seawater within about 10 years because of the high ionic strength of seawater. Therefore, Type-A particles directly deposited in the ocean might already have dissolved. CsMPs emitted from Unit 3, however, might have been directly deposited on the ocean because at the time of the H explosion, the wind was blowing toward the ocean, and the P4 pathway was also partly over the ocean (Fig. 5).
(3) Direct release to the ocean
Direct release of highly radioactive liquid wastes from the FDNPP to the ocean was also a possible source, which was discussed by Tsumune et al.2. However, whether particulate RCs was released directly from the FDNPP to the ocean remains unknown. Therefore, we cannnot evaluate whether direct release was a source of CsMPs. We therefore inferred that the PM particles in this study were Type-A particles that were deposited on land from P2 and P3, transported to rivers by surface soil erosion and runoff, and then transported via the rivers to the ocean.
In contrast, the MS particles were possibly derived from P4, because sampling point E is along the transport pathway of P4. In addition, the characteristics of the MS particles differed from those of previously reported CsMPs17. Furthermore, the molten core concrete interaction was more limited in Unit 2 than in Unit 329, which supported that the MS particles were emitted from Unit 3 because of containing Ca probably derived from concrete. These results and reports suggest that the MS particles were emitted from Unit 3, directly deposited onto the ocean surface, and possibly transported by ocean currents to point E. By contrast, Zhang et al.24 suggested that SP, which they isolated from a terrestrial soil sample collected from Okuma Town (3 km south–southwest of the FDNPP), was emitted from Unit 3 by H explosion at 11:01 JST on 14 March 2011. They suggested that the force of the explosion transported SP against the wind (Fig. 5) and deposited it on the land because of its large size and high density. We consider it likely that the MS particles in this study and SP came from Unit 3, but more particles need to be analysed to confirm this inference.
Migration of Type-A particles in the ocean
To understand the migration of Type-A particles in the ocean, we conducted the following calculation to estimate the horizontal transport distance of Type-A particles in the ocean until their deposition on the seafloor.
The reported diameter of Type-A particles (Dp) ranges from ~ 0.1 to 10 µm, and their density (ρp) is assumed to be around 2.5 g cm–3, which is the density of soda-lime glass. Thus, in the calculation, we used 2.0–3.0 g cm–3 as the range of ρp. The density (ρf) and viscosity (η) of seawater are 1.024 g cm–3 and 1.01 × 10–2 g cm–1 s–1, respectively36,37. In accordance with Stokes’ Law, the sinking velocity (vs) of Type-A particles with Dp from 0.1 µm to 10 µm can be calculated as follows:
where g is gravitational acceleration. Then, the horizontal transport distance of Type-A particles until they are deposited on the seafloor (dH) can be calculated with the following equation:
where dw and vH are water depth and horizontal flow velocity, respectively. If dw = 100 m and vH = 0.1 m s–1 (the order of vH was estimated from Tsumune et al.38), the dH of Type-A particles with Dp of 1 µm would be ~ 10,000 km (Fig. 6). Such particles would likely not be found in coastal sediments because they would be transported further offshore. By contrast, dH of Type-A particles with Dp of 10 µm would be ~ 100 km, and such particles might therefore be found in coastal sediments.

Relationship between horizontal transport distance and particle size by calculation.
The result of this calculation is consistent with the characteristics of the Type-A particles collected in the current study. PM-A, -B, and -F (diameter ~ 1 µm) were collected from suspended particles, whereas PM-D, MS-E1, and MS-E2 (diameter > 10 µm) were collected from sinking particles and sediment. Moreover, it is important to note that PM-D was attached to an Fe-rich particle, the density of which was likely high, whereas the matrix of PM-C, which was isolated from the plankton net sample, was a low-density plastic (mostly < 1.5 g cm–3)39.
PM-A and -B, which were collected in July 2015 and December 2013, respectively, from suspended particles in the coastal area, were Type-A particles with a diameter of ~ 1 µm. Because their shape indicated that they had not been altered by dissolution35, these CsMPs were likely collected relatively soon after their transport via rivers to the ocean.
Materials and methods
Bulk sample collection and measurement of 134Cs and 137Cs in the samples
Sampling points A–F and rivers near the FDNPP are shown in Fig. 1 on a Cs inventory map from MEXT40. Bulk samples were collected by several methods as described below; sampling information is summarized in Table S1.
(1) Suspended particles collected by water filtration using large-volume pumps (points A and B)
Large-volume pumps (WTS-LV, McLane Research Laboratories, Inc., Falmouth, MA) were used to collect suspended particles in July 2015 (point A) and December 2013 (point B) by the T/S Oshoro-maru of Hokkaido University and the fishing vessel Koumei-maru, respectively. The seawater was filtered through a PVDF membrane filter (Durapore, diameter 142 mm, pore size 0.45 μm, Merck Ltd., Tokyo, Japan). Then, the samples of suspended matter were removed from the filters with a plastic spatula, dried at 60 °C, and transferred to Teflon tubes. RCs concentrations of the suspended matter samples were measured by using a gamma-ray spectrometer with a well-type Ge detector (Canberra EGPC 250-P21, Meriden, CT). Samples were prepared for this measurement by the method of Kubo et al.22.
(2) Zooplankton collected by plankton net (point C)
A plankton net with a 330 µm mesh was used to collect zooplankton at point C in July 2011 by the T/V Umitaka-maru of the Tokyo University of Marine Science and Technology. The sample was dried at 60 °C and transferred to Teflon tubes. The RCs concentration of the plankton net sample was measured by using a gamma-ray spectrometer with a well-type Ge detector, as described above for the suspended particle samples.
(3) Sinking particles collected in a sediment trap (point D)
Sinking particles were collected by a time-series sediment trap (SMC7S-500ex, NiGK Corp., Japan) at point D between October and December 2014. The sinking particle samples were dried at 60 °C and transferred to Teflon tubes. RCs concentrations in the sinking particle samples were also measured by using a gamma-ray spectrometer with a well-type Ge detector.
(4) Marine sediment collected by a grab sampler (point E)
During a cruise of R/V Seikai (Japan Atomic Energy Agency) in July 2011 in the shallow region (bottom depth < 50 m) of the coastal area of Ibaraki Prefecture, a sediment sample was collected with a Smith–McIntyre sampler at point E, and the upper layer (0–3 cm) was separated from the rest of the grab sample on board. The sample consisted of very fine sand/fine sand (0.0625–0.25 mm; Wentworth scale). After transfer to a laboratory on land, the sample was dried at 105 °C and crushed, and the coarse fraction was removed by using a 2-mm sieve. The sieved sample was transferred to a plastic container, and gamma-rays specific to 134Cs (604.7 and 795.9 keV) and 137Cs (661.7 keV) were measured by using a coaxial Ge detector (ORTEC GEM20P4, resolution 1.7 keV/1.33 meV, relative efficiency 29%–31%). This measurement has been described in detail by Otosaka and Kobayashi41 and Otosaka and Kato6.
(5) Suspended particles collected by water filtration (point F)
At point F, 60 L of estuarine water was filtered through a sieve with 64-µm pore size in November 2012. The water was then filtered in situ through 3-µm and 0.45-µm pore size membrane filters by using an ADVANTEC pressurised filtering system (DV-10 and KS-142-UH, Advantec, Tokyo, Japan). The filters were dried at 60 °C for 12 h, and then the suspended particles (3–63 µm) on the filters were placed into a custom plastic bag and sealed for later gamma-ray measurement. The sample was measured for 137Cs (661.7 keV) by using a planar-type Ge detector (GC4018/7915-30/ULB-GC, CANBERRA). This measurement is explained in detail by Sakaguchi et al.42,43.
Isolation of RPs from bulk samples
The spatial distribution of radioactivity, particularly RCs, in each bulk sample was measured by autoradiography with an IP (BAS-MS 2040, 130 Fujifilm Corp., Japan) and an IP reader (FLA-9000, Fujifilm Corp., Japan). Three CsMPs isolated from terrestrial samples17 with known activity (~ 1, ~ 4, and ~ 20 Bq of 137Cs) were measured along with the samples as calibration standards for calculating 137Cs activity at each hot spot. High radioactive spots in the samples identified on the images by the IP reader were considered to be CsMPs. Particles with relatively high radioactivity (RPs) were isolated from the samples by the wet separation method, which is an efficient method of isolating RPs16,17,21. After isolation, each RP in the separation water was carefully dropped onto a Kapton tape and air-dried for subsequent analysis.
Measurement of 134Cs and 137Cs activities in the isolated RPs
The 134Cs (604.7 keV) and 137Cs (661.7 keV) activities of the isolated RPs were determined by gamma-ray spectrometry with a HPGe (GX4018, CANBERRA Industries 142 Inc., USA) to determine the FDNPP unit from which the CsMPs originated. Radioactivity standard solutions for 134Cs (0.182 Bq as of 25 November, 2016, Japan Radioisotope Association, CZ-010) and 137Cs (1.40 Bq as of 25 November, 2016, Japan Radioisotope Association, CS-005) dispersed on a filter over an area of 1 mm2 were used for calibration of the gamma-ray spectrometer. These standard radioactivity solutions were calibrated by the Japan Calibration Service System (JCSS; http://www.nite.go.jp/en/iajapan/jcss/index.html).
SEM-EDS analysis of the isolated RPs
SEM (TM3030Plus, Hitachi, Japan) with EDS (AZtecOne, Oxford Instruments, UK) analysis was performed to observe the shape and elemental composition of RPs. Elemental maps of some RPs were also obtained by EDS measurement.
Calculation of the ratio of 137Cs activity in RPs to total 137Cs in the bulk sample
To assess the influence of RPs on Kd and CF values, we calculated the ratio of 137Cs activity in RPs to the total 137Cs in the bulk samples, R(RPs):
where A(RPs) is the 137Cs activity in RPs and A(bulk) is the total 137Cs activity in the bulk sample containing the RP. A(RPs) is the sum of 137Cs activity of the isolated RPs measured by HPGe detector and the 137Cs activity of radioactive spots (> 0.1 Bq of 137Cs) calculated from the PSL of IP images. A(bulk) was measured by a HPGe detector.
References
Aoyama, M. et al. 134Cs and 137Cs in the North Pacific Ocean derived from the March 2011 TEPCO Fukushima Dai-ichi Nuclear Power Plant accident, Japan. Part two: Estimation of 134Cs and 137Cs inventories in the North Pacific Ocean. J. Oceanogr. 72, 67–76 (2016).
Tsumune, D. et al. One-year, regional-scale simulation of 137Cs radioactivity in the ocean following the Fukushima Dai-ichi Nuclear Power Plant accident. Biogeosciences 10, 6259–6314 (2013).
Buesseler, K. et al. Fukushima Daiichi-derived radionuclides in the ocean: Transport, fate, and impacts. Annu. Rev. Mar. Sci. 9, 173–203 (2017).
Kaeriyama, H. Oceanic dispersion of Fukushima-derived radioactive cesium: A review. Fish. Oceanogr. 26, 99–113 (2017).
Kusakabe, M., Oikawa, S., Takata, H. & Misonoo, J. Spatiotemporal distributions of Fukushima-derived radionuclides in nearby marine surface sediments. Biogeosciences 10, 5019–5030 (2013).
Otosaka, S. & Kato, Y. Radiocesium derived from the Fukushima Daiichi Nuclear Power Plant accident in seabed sediments: Initial deposition and inventories. Environ. Sci. Process. Impacts 16, 978–990 (2014).
Kusakabe, M., Inatomi, N., Takata, H. & Ikenoue, T. Decline in radiocesium in seafloor sediments off Fukushima and nearby prefectures. J. Oceanogr. 73, 529–545 (2017).
Otosaka, S. Processes affecting long-term changes in 137Cs concentration in surface sediments off Fukushima. J. Oceanogr. 73, 559–570 (2017).
Adachi, K., Kajino, M., Zaizen, Y. & Igarashi, Y. Emission of spherical cesium-bearing particles from an early stage of the Fukushima nuclear accident. Sci. Rep. 3, 2554 (2013).
Igarashi, Y. et al. A review of Cs-bearing microparticles in the environment emitted by the Fukushima Dai-ichi Nuclear Power Plant accident. J. Environ. Radioact. 205–206, 101–118 (2019).
Abe, Y. et al. Detection of uranium and chemical state analysis of individual radioactive microparticles emitted from the Fukushima nuclear accident using multiple synchrotron radiation X-ray analyses. Anal. Chem. 86, 8521–8525 (2014).
Furuki, G. et al. Caesium-rich micro-particles: A window into the meltdown events at the Fukushima Daiichi Nuclear Power Plant. Sci. Rep. 7, 42731 (2017).
Kogure, T. et al. Constituent elements and their distribution in the radioactive Cs-bearing silicate glass microparticles released from Fukushima nuclear plant. Microscopy 65, 451–459 (2016).
Yamaguchi, N. et al. Internal structure of cesium-bearing radioactive microparticles released from Fukushima nuclear power plant. Sci. Rep. 6, 20548 (2016).
Okumura, T., Yamaguchi, N. & Kogure, T. Finding radiocesium-bearing microparticles more minute than previously reported, emitted by the Fukushima nuclear accident. Chem. Lett. 48, 1336–1338 (2019).
Kurihara, Y. et al. Isotopic ratios of uranium and caesium in spherical radioactive caesium-bearing microparticles derived from the Fukushima Dai-ichi Nuclear Power Plant. Sci. Rep. 10, 3281 (2020).
Miura, H. et al. Characterization of two types of cesium-bearing microparticles emitted from the Fukushima accident via multiple synchrotron radiation analyses. Sci. Rep. 10, 11421 (2020).
Nishihara, K., Iwamoto, H. & Suyama, K. Estimation of fuel compositions in Fukushima-Daiichi nuclear power plant. JAEA-Data/Code 2012-018 (2012).
Ono, T. et al. Investigation of the chemical characteristics of individual radioactive microparticles emitted from reactor 1 by the Fukushima Daiichi Nuclear Power Plant accident by using multiple synchrotron radiation X-ray analyses. Bunseki Kagaku 66, 251–261 (2017).
Satou, Y. et al. Analysis of two forms of radioactive particles emitted during the early stages of the Fukushima Dai-ichi nuclear power station accident. Geochem. J. 52, 137–143 (2018).
Miura, H. et al. Discovery of radiocesium-bearing microparticles in river water and their influence on the solid-water distribution coefficient (Kd) of radiocesium in the Kuchibuto River in Fukushima. Geochem. J. 52, 145–154 (2018).
Kubo, A. et al. Radioactive cesium concentrations in coastal suspended matter after the Fukushima nuclear accident. Mar. Pollut. Bull. 131, 341–346 (2018).
Ikenoue, T., Ishii, N., Kusakabe, M. & Takata, H. Contribution of 137Cs-enriched particles to radiocesium concentrations in seafloor sediment: Reconnaissance experiment. PLoS ONE 13, e0204289 (2018).
Zhang, Z. et al. Activity of 90Sr in fallout particles collected in the difficult-to-return zone around the Fukushima Daiichi nuclear power plant. Environ. Sci. Technol. 53, 5868–5876 (2019).
Nakajima, T. et al. Model depiction of the atmospheric flows of radioactive cesium emitted from the Fukushima Daiichi nuclear power station accident. Prog. Earth Planet. Sci. 4, 2 (2017).
Tsuruta, H., Oura, Y., Ebihara, M., Ohara, T. & Nakajima, T. First retrieval of hourly atmospheric radionuclides just after the Fukushima accident by analyzing filter-tapes of operational air pollution monitoring stations. Sci. Rep. 4, 6717 (2014).
Chino, M. et al. Utilization of 134Cs/137Cs in the environment to identify the reactor units that caused atmospheric releases during the Fukushima Daiichi accident. Sci. Rep. 6, 31376 (2016).
Tsuruta, H. et al. Time-series analysis of atmospheric radiocesium at two SPM monitoring sites near the Fukushima Daiichi Nuclear Power Plant just after the Fukushima accident on March 11, 2011. Geochem. J. 52, 103–121 (2018).
International Research Institute for Nuclear Decommissioning (IRID). http://irid.or.jp/_pdf/20170000_01.pdf (2018).
Tanabe, F. A scenario of large amount of radioactive materials discharge to the air from the Unit 2 reactor in the Fukushima Daiichi NPP accident. J. Nucl. Sci. Technol. 49, 360–365 (2012).
Tokyo Electric Power Company (TEPCO). http://www.tepco.co.jp/decommission/information/accident_unconfirmed/pdf/2017/171225j0103.pdf (2017).
Katata, G. et al. Detailed source term estimation of the atmospheric release for the Fukushima Daiichi Nuclear Power Station accident by coupling simulations of an atmospheric dispersion model with an improved deposition scheme and oceanic dispersion model. Atmos. Chem. Phys. 15, 1029–1070 (2015).
Ikehara, R. et al. Novel method of quantifying radioactive cesium-rich microparticles (CsMPs) in the environment from the Fukushima Daiichi Nuclear Power Plant. Environ. Sci. Technol. 52, 6390–6398 (2018).
Taniguchi, K. et al. Transport and redistribution of radiocesium in Fukushima fallout through rivers. Environ. Sci. Technol. 53, 12339–12347 (2019).
Okumura, T., Yamaguchi, N., Dohi, T., Iijima, K. & Kogure, T. Dissolution behaviour of radiocaesium-bearing microparticles released from the Fukushima nuclear plant. Sci. Rep. 9, 3520 (2019).
Nayar, K. G., Sharqawy, M. H., Banchik, L. D. & Lienhard, J. H. Thermophysical properties of seawater: A review and new correlations that include pressure dependence. Desalination 390, 1–24 (2016).
Sharqawy, M. H., Lienhard, V. J. H. & Zubair, S. M. Thermophysical properties of seawater: A review of existing correlations and data. Desalin. Water Treat. 16, 354–380 (2010).
Tsumune, D. et al. Impacts of direct release and river discharge on oceanic 137Cs derived from the Fukushima Dai-ichi Nuclear Power Plant accident. J. Environ. Radioact. 214–215, 106173 (2020).
Morét-Ferguson, S. et al. The size, mass, and composition of plastic debris in the western North Atlantic Ocean. Mar. Pollut. Bull. 60, 1873–1878 (2010).
Ministry of Education, Culture, Sports, Science and Technology (MEXT). Extension Site of Distribution Map of Radiation Dose, etc., GSI Maps. https://ramap.jmc.or.jp/map/eng/.
Otosaka, S. & Kobayashi, T. Sedimentation and remobilization of radiocesium in the coastal area of Ibaraki, 70 km south of the Fukushima Dai-ichi Nuclear Power Plant. Environ. Monit. Assess. 185, 5419–5433 (2013).
Sakaguchi, A. et al. Isotopic determination of U, Pu and Cs in environmental waters following the Fukushima Daiichi Nuclear Power Plant accident. Geochem. J. 46, 355–360 (2012).
Sakaguchi, A. et al. Size distribution studies of 137Cs in river water in the Abukuma Riverine system following the Fukushima Dai-ichi Nuclear Power Plant accident. J. Environ. Radioact. 139, 379–389 (2015).
Acknowledgements
This work was supported by MEXTKAKENHI (a Grant-in-Aid for Scientific Research on Innovative Areas under the A02-04 research team for the ‘Interdisciplinary Study on Environmental Transfer of Radionuclides from the Fukushima Daiichi NPP Accident’; Grant number: 24110005) and by the Environmental Radioactivity Research Network Center (Grant numbers: Y-20-20, F-20-35, Y-19-21, and F19-30). This study was partly supported by the Central Research Institute of the Electric Power Industry.
Author information
Authors and Affiliations
Contributions
H.M., T.I., Y.K., and Y.T. designed the research and produced the main manuscript. T.I., S.O., A.S., and A.K. collected the of samples. Y.I., S.O., A.S., and A.K. performed gamma-ray spectroscopy analysis of bulk samples. H.M. isolated the particulate samples from the bulk samples and performed the SEM analysis. H.M., Y.K., and S.H. performed the gamma-ray spectroscopy analysis of the particulate samples. K.M. calculated the horizontal transport of particulate samples in the ocean. Y.K., D.T., T.I., J.K., and Y.T. supervised the study. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Miura, H., Ishimaru, T., Ito, Y. et al. First isolation and analysis of caesium-bearing microparticles from marine samples in the Pacific coastal area near Fukushima Prefecture. Sci Rep 11, 5664 (2021). https://doi.org/10.1038/s41598-021-85085-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-85085-w
This article is cited by
-
Radiocesium-bearing microparticles discovered on masks worn during indoor cleaning
Scientific Reports (2023)
-
Radiocesium-bearing microparticles found in dry deposition fallout samples immediately after the Fukushima nuclear accident in the Kanto region, Japan
Scientific Reports (2023)
-
Dissolution behavior of radiocesium-bearing microparticles as a function of solution compositions
Scientific Reports (2023)
-
Atmospheric resuspension of insoluble radioactive cesium-bearing particles found in the difficult-to-return area in Fukushima
Progress in Earth and Planetary Science (2022)
-
Characterization of radiocesium-bearing microparticles with different morphologies in soil around the Fukushima Daiichi nuclear power plant
Journal of Radioanalytical and Nuclear Chemistry (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
