
Abstract
Interspecific introgression is considered a potential threat to endangered taxa. One example where this has had a major impact on conservation policy is the lesser white-fronted goose (LWfG). After a dramatic decline in Sweden, captive breeding birds were released between 1981–1999 with the aim to reinforce the population. However, the detection of greater white-fronted goose (GWfG) mitochondrial DNA in the LWfG breeding stock led to the release program being dismantled, even though the presence of GWfG introgression in the actual wild Swedish LWfG population was never documented. To examine this, we sequenced the complete genomes of 21 LWfG birds from the Swedish, Russian and Norwegian populations, and compared these with genomes from other goose species, including the GWfG. We found no evidence of interspecific introgression into the wild Swedish LWfG population in either nuclear genomic or mitochondrial data. Moreover, Swedish LWfG birds are genetically distinct from the Russian and Norwegian populations and display comparatively low genomic diversity and high levels of inbreeding. Our findings highlight the utility of genomic approaches in providing scientific evidence that can help improve conservation management as well as policies for breeding and reinforcement programmes.
Similar content being viewed by others


Introduction
Interspecific introgression has often been highlighted as a potential threat to endangered taxa due to the risk of outbreeding depression1. The reason for this is that the introduction of genes from a different taxon into a small threatened population can reduce the fitness of hybrids1,2. This has led to introgressed individuals being identified as a cause for concern during translocation or reinforcement actions3. At a genetic level, the mechanisms behind outbreeding depression include breakup of coadapted gene complexes and disruption of local adaptations. For example, the introduction of Middle Eastern Ibex individuals to reinforce the Alpine Ibex population in Czechoslovakia resulted in calves being born in the middle of winter, which led to the subsequent disappearance of the Czech population4. In general, when captive populations have been identified as containing hybrids, the management recommendation has often been to exclude these individuals as potential sources for reinforcement programmes5,6,7.
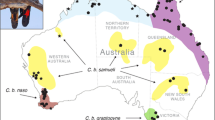
One particular case where concerns about interspecific introgression have had a major impact on conservation policy is the lesser white-fronted goose (Anser erythropus, LWfG) population in Sweden8. Until the early 1900s, the LWfG was a widespread and relatively common breeding bird in arctic and semi-arctic areas of northern Eurasia (e.g. Madsen & Cracknell9). Historically, three different populations, mainly delimited by migration routes and breeding areas, have been identified: the Fennoscandian, the Western Main, and the Eastern Main populations (Fig. 1). During the 20th Century, the Fennoscandian population suffered a dramatic decline to the extent that in the 1980s only 60–90 breeding pairs remained in two small populations, one in Norway and another one in Sweden, with only 20 pairs in the Swedish mountain tundra10,11. This dramatic decline has mainly been explained by increased mortality due to overhunting during migration and in wintering grounds (e.g. Madsen & Cracknell9).

Geographical location of sampling sites for the lesser white-fronted goose. Sampling sites are denoted with white circles. Important sites and main migratory routes for the Western Palearctic LWfG are also depicted (Adapted from Aarvak et al.64). Norwegian migratory routes represent the Fennoscandian population routes; Russian, the Western Main population routes; and Swedish, the westward modified migratory route from the 1989–1999 reinforcement program in Sweden. Map was downloaded from Natural Earth (naturalearthdata.com) and edited in QGIS v2.18.17 (qgis.org) and Inkscape v0.92 (inkscape.org).
As a response to the dwindling population numbers, in 1977 the Swedish Association for Hunting and Wildlife Management initiated an ex-situ breeding programme with the objective to release birds to reinforce the Swedish LWfG population12. The exact origin of the founder birds of this programme is obscure, but it is known that it included wild-caught birds from Swedish Lapland13 as well as captive birds from parks in the Netherlands and England14. From 1981 to 1999, a total of 341 birds were released into an area that was known to hold native LWfG breeding pairs prior to the start of the reinforcement programme11,15.
In the late 1990s, genetic studies suggested that the reinforcement programme may have inadvertently introduced LWfG individuals carrying greater white-fronted goose (Anser albifrons, GWfG) genes into the LWfG Swedish population. Based on short mitochondrial DNA fragments (~ 200 bp), 16% of the adult LWfG captive population was identified as carrying GWfG haplotypes16. Consequently, the original Swedish captive breeding programme was dismantled in 2000 as a precautionary measure. Unfortunately, the juvenile goslings that had been released into the wild were never genetically tested, so the real amount of introgressed individuals released is unknown. An estimate using parentage analysis based on archival data of the breeding project suggested that 5–10% of the released juveniles may have contained GWfG introgressed genes8. Following this, no subsequent genetic studies have been done to investigate the genetic composition and levels of introgression of the present-day LWfG Swedish population. However, inferences of genetic relationships and gene flow based on short mitochondrial DNA sequences can sometimes be problematic. This is due not only to their low resolution in phylogenetic analyses, but also their sensitivity to incomplete lineage sorting. Moreover, previous studies have also indicated a high prevalence of nuclear insertions of mitochondrial DNA (numts) in Anser sp.17, which can further complicate the inferences.
During the last decade, major technological developments in DNA sequencing technology have made it feasible to sequence complete nuclear genomes at reasonable economic costs18. Such whole genome data has revolutionized the possibility to identify introgression among species19,20,21. Together with genomic data, new computational approaches have enabled detection of hybridization even when only a small proportion of the genome has a different ancestry22,23.
The principal aim of this study was to investigate the existence and degree of introgression from GWfG into the wild Swedish LWfG population. To do this, we sequenced 21 high-coverage genomes from LWfG individuals originating from Sweden, Norway and Russia (Table 1, Fig. 1). We then used this dataset combined with genomes from 18 other goose species (Table S1, Supplementary Information 1.3) and several computational approaches to test for introgression. We also examined the amount of genetic differentiation among the Swedish, Norwegian and Russian populations and estimated the levels of genome-wide diversity and inbreeding within each population. Based on the available data from previous studies, and the fact that seven LWfG generations had passed since the release of putatively hybrid geese and when our samples were collected, we predicted a high prevalence of individuals introgressed with GWfG genes in the LWfG Swedish population. Moreover, we expected that such introgression would have resulted in a higher genome-wide diversity and lower inbreeding levels in the Swedish birds compared to the Norwegian and Russian populations.
Results
Sequencing yielded an average of 79 million reads per sample after filtering by quality (Table 1). Of those, an average of 94.87% mapped to the pink-footed goose (Anser brachyrhynchus, PfG) assembly and an average of 83.86% mapped to the more distantly related mallard duck (Anas platyrhynchos). Overall, the newly sequenced genomes had an average coverage of 16.1× (range 10.3–25.5) for the dataset mapped to the PfG, and an average of 13.8× (range 8.7–21.8) for the dataset mapped to the mallard duck. For the goose samples downloaded from Ottenburghs et al.24,25 and remapped, the average coverage was 15.3 × when mapped to PfG and 12.4 × when mapped to the mallard duck (Table S1).
RepeatMasker indicated that up to 7.28% of the PfG assembly was composed from highly repetitive regions (Table S2). After excluding these regions and filtering by read depth (≥ 7), allelic balance (> 0.2 and < 0.8), mapping (> 30) and base quality (> 30), variant calling of all 42 individuals yielded 29,023,687 variable sites (Supplementary Information 1.4). For population genetic analyses of LWfG samples, these sites were linkage-disequilibrium pruned (window size of 50 kb, step size of 5 kb, and a minimum pairwise correlation (r2) of 0.5 for SNPs to be excluded) leaving a final dataset comprising 6,509,393 variants.
The D-statistics suggested that the GWfG has not contributed to the Swedish LWfG population’s gene pool relative to the Norwegian or Russian populations (D = 0 for all comparisons, Fig. 2, Figure S14A). We obtained similar results when extending the test using all the other Anser species in the dataset as donors, i.e. D(SWE, NOR/RUS; X, O), where X is any of the other Anser species candidates to be introgressed into the Swedish population of LWfG, and O is the bar-headed goose (Supplementary Information 2.3.1, Figure S15A). Regardless of the donor assumed, none of the comparisons showed a clear and consistent departure from D = 0, and thus we found no evidence of introgression from any other goose species into the Swedish LWfG relative to Norwegian or Russian populations. To account for any confounding effect of the reference genome used, we repeated all the tests using the dataset mapped to the mallard duck assembly, which yielded very similar results (Figures S13, S14B and S15B).

Estimates of introgression from greater white-fronted goose into Swedish respect to Russian and Norwegian lesser white-fronted goose individuals using the pink-footed goose as outgroup. The tests are in the form D(SWE, RUS/NOR; GWfG, PfG). Error bars depicting 3*SE are displayed. Dots are colored representing the |Z| value of the comparison.
A key question, given the results from the D-statistics, is whether sequencing 12 genomes is enough to detect hybrids in LWfG samples collected in 2010 and onwards. Taking into account that the reinforcement programme may have resulted in a minimum of 5% of all Swedish LWfG birds carrying some sort of GWfG ancestry26, and assuming a generation time of 3–5 years and no selective disadvantage for those birds with hybrid ancestry, the expected proportion of introgressed LWfG individuals at the time of sampling in 2010 is 0.56—0.96 (see Supplementary Information 2.3.2). Using these figures and a hypergeometric distribution27, we estimated that the probability of not detecting GWfG ancestry in any of our 10 birds sampled in 2010, from a finite population of 110, is between P < 0.001 and P < 0.0001. Consequently, it is very unlikely that any meaningful GWfG ancestry had gone undetected given our sampling effort.
To further investigate if any of our sampled LWfG carried GWfG mitochondrial DNA, which could indicate introgression or incomplete lineage sorting, we reconstructed the mitogenomes for the LWfG samples. Accurately reconstructing the mitochondrial genomes was challenging due to the apparent presence of widespread nuclear insertions of mitochondrial DNA (i.e. numts, see Supplementary Information 2.3.3). Nonetheless, regardless of whether we excluded or included sites that indicated presence of numts, the resulting phylogenetic trees grouped all analyzed LWfG and GWfG mitogenomes into two reciprocally monophyletic clades (Figure S17), thus demonstrating that none of our sequenced LWfG carried GWfG mitogenomes.
Principal component analysis (PCA) of the LWfG samples suggested they are structured in two discrete groups, with the majority of Swedish samples in one of them and Norwegian and Russian samples in the other (Fig. 3A,B, Supplementary Information 2.1.3). In fact, the genetic differentiation between these groups is relatively high (Fst = 0.067 SWE-RUS and 0.068 SWE-NOR, Supplementary Information 2.1.4, Table S3). Two Russian and two Swedish samples cluster together and are differentiated from these two main groups. These two pairs were found to be siblings (Figure S3), which is probably affecting their genetic affinities in the PCA (Figure S5) as well as in other analyses based on allele frequencies. Therefore, further analyses were performed removing one individual of each pair of full siblings (see Supplementary Information 2.1.4). Treemix analyses corroborated the PCA results and supported that the Swedish LWfG samples are closely related to each other, but clearly distinct from the group composed of Russian and Norwegian samples (Fig. 3C). The park bird from24,25 shows closer genetic affinity to the sibling Swedish LWfG samples in the PCA and the Treemix analyses.

Genetic structure among the lesser white-fronted goose. (A) Principal component analysis (PCA) of LWfG, GWfG and PfG samples. (B) PCA of the LWfG in this study (excluding first degree relatives). Different colors represent the distinct origins of the samples. (C) TREEMIX best model for LWfG (excluding first degree relatives), GWfG and PfG samples in the dataset and two migration edges (m = 2), including the pairwise residuals. ‘Park Bird’, refers to the LWfG sample from Ottenburghs et al.24,25.
Heterozygosity estimates indicated that all LWfG birds have overall high levels of genomic diversity respect the other goose species (Supplementary Information 2.2.2, Figure S9). When focusing on LWfG alone, we found that the Swedish birds display slightly lower genomic diversity than both Russian and Norwegian LWfG birds (Fig. 4A). These results were also confirmed when analyzing the dataset mapped to the mallard duck (Figure S9).

Genetic diversity and inbreeding in lesser white-fronted goose. (A) Genome wide levels of diversity estimated as number of heterozygote sites per 1kbp in all LWfG samples. (B) Inbreeding values (FROH) for all LWfG birds analyzed in the dataset. (C) Size distribution of runs of homozygosity in each one of the three LWfG sampling groups. Error bars depict the standard deviation within groups. In all cases, pairwise comparisons are done using Tukey's HSD tests (NS: p-value > 0.05, *: p-value < 0.05, and **: p-value < 0.01).
Finally, we estimated an average inbreeding level of 0.052 for all LWfG (5.2% of their genome allocated in runs of heterozygosity), with significantly higher inbreeding levels in the Swedish individuals (FROH = 0.082) than in either the Russian (FROH = 0.01) or the Norwegian (FROH = 0.009) individuals (Fig. 4B). In fact, most Swedish birds were identified as close relatives of different degrees (Supplementary Information 2.1.2). Additionally, we identified that the increased inbreeding in the Swedish birds can be attributed to an excess of both short (< 2 Mb) and long (> 2 Mb) ROHs (Fig. 4C, Figure S12). As in the rest of the analyses, the replicated tests performed on the data mapped to the mallard confirmed the above inbreeding results (Supplementary Information 2.2.2).
Discussion
We found no evidence, in either the nuclear or mitochondrial genomes, suggesting an excess of introgressed alleles from GWfG, or any other goose species, into the wild Swedish LWfG population relative to the Norwegian or Russian ones. Given our sample size and the resulting low probability of non-detection, we therefore consider it highly unlikely that the reinforcement programme between 1981 and 1999 led to the introduction of introgressed genes into the wild Swedish population.
We note that there are several competing hypotheses that can generate a D-statistic value of zero (no introgression) in our nuclear genomic comparisons. First, it is possible that there has been no introgression from the GWfG, or any other goose species, into either the Swedish, Norwegian or Russian LWfG populations. A second possibility is that there has been introgression from GWfG into the LWfG, but that this happened into the ancestral lineage of all extant LWfG populations. This would have resulted in all populations carrying the same amount of GWfG ancestry, and thus D-statistic values of zero in all comparisons. This second scenario would be consistent with the observations by Ottenburghs et al.24, which suggested that introgression during the early stages of speciation has been a common feature among all goose species including the LWfG. A third scenario that could explain our results would be that there have been multiple separate introgressions into all three populations, Sweden, Norway and Russia. Although theoretically possible, we consider this scenario extremely unlikely since the level of introgression into the separate populations must then have been more or less exactly the same in order to result in D-statistic values of zero in all comparisons.
Our mitochondrial analyses also suggested lack of GWfG introgression into LWfG. Although our results grouped GWfG and LWfG into reciprocally monophyletic clades, we note that precise mitochondrial reconstructions were impeded by the presence of seemingly widespread mitochondrial insertions in the nuclear genome (numts). Numts are known to be present in bird species such as ducks and geese28. In fact, one of the first characterizations of a numt in birds was done in snow geese29. Even though the existence of numts has been highlighted in previous mitochondrial-based studies on LWfG17, our results suggest that they could be much more prevalent than previously estimated, possibly spanning the entire mitochondrial sequence. Further analyses, for example making use of long-read DNA sequencing technologies, will be needed to better identify and characterize these numts.
Not finding GWfG introgression into the Swedish LWfG population is contrary to the expectations based on previous studies, which had suggested that GWfG alleles were introgressed in 16% of captive-bred LWfG individuals16. Because these samples included birds from the breeding programme at Öster Malma that were used to reinforce the Swedish wild population between 1981 and 1999, it has been assumed that GWfG alleles were present in the current wild Swedish LWfG population8. However, exactly what proportion of the released LWfG birds actually carried GWfG alleles, and thus acted as vectors to the wild population, was unknown.
We caution that we cannot fully exclude the possibility that some introgression did occur during the release programme between 1981 and 1999, where hybrid birds were at such a selective disadvantage that they left no detectable signature in the population 3–5 generations later. However, we consider it highly unlikely that such a selective disadvantage would have removed also neutral SNPs to the extent that we could not detect any signal in the D-statistic tests, unless first generation hybrids between resident and released birds effectively had zero fitness. Supplementing or translocation projects using stocks that have spent several generations in captivity may indeed show lower success compared to breeding programmes based on wild-caught individuals (e.g. Robert30). In captivity, relaxation from natural selection can affect genetic characteristics as well as learned behaviours31. Consequently, viability, survival and the ability to recruit individuals into existing wild populations decrease with the duration of a reinforcement project32,33. The LWfG released in Sweden in the period 1981–1999, which originated from a stock heavily influenced by captive park birds and collections, probably had low ability to adjust to the wild. Hence, instead of selection against hybrids, we hypothesize that few or no released birds that carried introgressed GWfG genes may have contributed to the Swedish population’s present-day genetic variation, either due to random chance or because they did not adjust to the wild. Finally, it is also possible that the management decision of halting the releases as soon as hybrid birds were detected among the captive-bred LWfG successfully prevented the introduction of any founders with GWfG ancestry into the wild population, which may have eventually occurred had the releases continued.
In addition to no evidence of introgression, we also found that the Swedish population is genetically distinct from both the Russian and Norwegian populations (p < 0.001; FST = 0.069 and 0.068, respectively, Table S3). By comparison, this level of divergence is on par with FST values reported between West African and European human populations34. There could be several different explanations to the distinctiveness of the Swedish LWfG population. For example, it might reflect a different postglacial origin of the Swedish population compared to those in Russia and Norway, a common feature in Anatidae species35. Alternatively, the high divergence could be a consequence of strong genetic drift in the Swedish population during the last 100 years of abrupt population decline. Finally, by incorporating individuals from parks in the Netherlands and England, the release programme between 1981 and 1999 may have resulted in a significant shift in genetic composition of the wild Swedish population. Resolving these questions would be highly interesting but will require a much wider spatial and temporal sampling effort.
In contrast to the high differentiation of the Swedish population, we found that the Norwegian and Russian LWfG birds are genetically indistinguishable (FST = 0, p = 0.471, Table S3). These results suggest that even though the Norwegian (Fennoscandian) and Russian (Western Main) populations have different migratory routes (Fig. 1), they have in fact a very close relationship, probably explained by occasionally shared wintering and breeding grounds36,37 and the less philopatric males pair-bonding and mating with females from other subpopulations, thereby reducing subpopulation differentiation in the nuclear DNA. The lack of differentiation between the Norwegian and Russian samples is consistent with the results from an earlier genetic study on museum specimens38, which identified a temporal increase in genetic diversity in the Norwegian LWfG population from historical times to present day, and suggested this has been caused by an increased male-mediated genetic influx from the Russian populations in the last decades. Interestingly, our results suggest that this increased gene flow does not seem to have affected the Swedish reinforced population.
Our results also showed that the Swedish population had significantly lower genome-wide diversity and higher inbreeding levels compared to the Russian and Norwegian populations. It should be noted here that the LWfG as a species has one of the highest levels of genetic diversity among birds39,40. Nonetheless, the comparatively lower diversity in the Swedish population signifies a reduced potential to adapt to changes in the environment41. Also, the increased amount of inbreeding in the Swedish population, and in particular the excess of long RoHs observed in our analyses, suggest that the Swedish population has been subject to recent mating between close relatives42,43, which may be explained by a generally small population size and/or significant influence from the captive breeding programme.
Lower genetic diversity and higher levels of inbreeding could have suggested that the present-day Swedish population is subject to genetic threats in the form of inbreeding depression and increased genetic load. However, around the time that our Swedish samples were collected, a second reinforcement programme was implemented in Sweden. This second programme has successfully been releasing birds (an average of 46 individuals per year) that originate from the Russian population44. While originally aimed to dilute and reduce the impact of putative GWfG introgression, this second release programme may have resulted in a restoration of genetic diversity and mitigation of inbreeding in the Swedish population, thus mimicking the effect of recent natural gene flow from Russia into Norway38. Since the first evidence of successful reproduction of a released bird with Russian origin was in 2016, and all the released birds are individually ringed, it is certain that none of the Swedish samples analyzed in this study (including the two birds sampled in 2015 and 2016) had any ancestry from Russian birds released during this second reinforcement. Therefore, future studies on samples collected more recently will be needed to monitor the consequences of the current reinforcement programme on the distinctiveness, genetic diversity and inbreeding levels in today's Swedish LWfG population.
Conclusions
Our findings showcase the risk of basing management decisions in translocation and reinforcement projects solely on data from captive breeding stock individuals without carefully monitoring the wild populations. After being released into the wild, captive-bred animals face a set of new challenges, such as severe predation rates45, which critically determine the ability of the newly introduced animals to contribute to the wild recipient population’s gene pool. Conservation legislation and policies, such as the Habitats Directive of the European Union, often mandate the restoration of populations that are extinct or close to extinction, for example through reinforcement46. Low levels of gene flow, even from a divergent population, can provide a substantial demographic and genetic boost to small populations, which may allow them to recover and withstand environmental and genetic stochasticity47,48. Despite this, conservation biologists have generally tended to shy away from routinely crossing populations49. One of the reasons for this, as exemplified by the Swedish LWfG population, is the fear of outbreeding depression and/or genomic contamination through hybridization, which may lead to reduced population viability. In fact, with a growing anthropogenic footprint, there is an increased risk of such human-mediated genomic contamination2,50,51. However, as shown in this study, population genomics offers a highly powerful way to assess the levels of hybridization in threatened populations. A genomic approach should also make it possible to manage the risk of introducing hybrids into the wild through genetic testing of captive individuals before releasing them. In addition, genomics can also be used to carefully monitor the wild population during and after such releases. We think this should be the norm, not the exception, whenever these kinds of projects are planned.
Methods
Samples and dataset
DNA samples used in this study were collected from blood extracts of live birds during conservation efforts. Sampling permits were issued by the Swedish Board of Agriculture (Jordbruksverket), Nordens Ark and the Swedish Association for Hunting and Wildlife Management (Jägareförbundet), and were approved by the ethical boards Uppsala Djurförsöksetiska Nämnd and Göteborgs Djurförsöksetiska Nämnd with permit numbers C171/10, 257/2011, and 140/2014. All sampling was carried out in accordance with the relevant guidelines and regulations.
We collected blood samples from 21 LWfG birds from three different populations: 12 at known stop over sites of the Swedish population (Figure S1); 5 from Russian breeding grounds at the Nentsien Autonom Okrug; and 4 from Norway from local nesting birds (Table1, Supplementary Information 1.1). The blood samples from the Norwegian birds were obtained from the existing collections at BirdLife Norway (collection IDs VA7, VA8, VA12, and VA14, Table 1). Genomic DNA was extracted using a KingFisher Cell and Tissue DNA Kit (Thermo Fisher Scientific, MA, USA) and genomic libraries for whole genome sequencing built using the TruSeq PCR-Free protocol (Illumina, CA, USA). Genomic libraries were then sequenced on five Illumina HiSeqX lanes using 2 × 150 bp paired-end read settings.
Raw reads were mapped against the pink-footed goose genome52 using the BWA mem algorithm53 with default parameters (Table 1, Supplementary Information 1.3). In order to detect possible biases in some analyses, we also mapped the raw reads to the mallard duck (Anas platyrhynchos, CAU_duck_1.054) genome, which has a better quality assembly but is much more distantly related. The publicly available raw data from 22 other goose samples from 18 different species24,25 and the raw reads from the PfG sample that was used to generate the de novo assembly52 were mapped to both references using the same settings (Table S1). Thus, the final datasets contained 42 genomes from 18 different goose species (Table 1, Table S1).
Variant discovery
We identified variants in each sample using bcftools mpileup and bcftools call55 and filtered those calls by keeping only biallelic variants on positions covered at least seven times (i.e. minimum read depth 7), they are present in more than 90% of the samples, and with genotype quality > 30 (Supplementary Information 1.4). Heterozygote variants were only kept if they had an allelic balance > 0.2 and < 0.8. We then used RepeatMasker56 and RepeatModeler57 to identify repetitive regions in both reference genomes, and excluded all variants allocated on those regions as well.
Computational analyses
We used the normalized ratio of reads mapping to chromosome Z and the ones mapping to an autosome of similar size (chromosome 4) to determine the sex of each newly sequenced LWfG bird58 (Supplementary Information 2.1.1). Kinship within the LWfG samples was investigated using the tool –relatedness2 from vcftools59 (Supplementary Information 2.1.2).
We used principal component analysis (PCA) to explore the broad genetic affinities among the LWfG, GWfG and PfG samples in the dataset, and also within the LWfG samples alone (Supplementary Information 2.1.3). Genomic differentiation (FST) between the three LWfG populations was estimated using the tool -weir-fst-pop in vcftools, which estimates differentiation values between groups of samples using a weighted FST60, and assessed significance using a permutation test (Supplementary Information 2.1.4). Genetic affinities and possible admixture events among GWfG, PfG and LWfG were further explored using the maximum likelihood approach of TreeMix61 (Supplementary Information 2.1.5).
We estimated per sample genome-wide heterozygosity directly counting heterozygote and homozygote genotype calls (‘hard-calls’) from the filtered VCF files (Supplementary Information 2.2.1). Long runs of homozygosity (> 100 kb), stretches of the genome with none or very limited number of heterozygote sites, were identified in each sample using the –homozyg option in Plink62 (see Supplementary Information 2.2.2 for details on settings and thresholds), and used to estimate per individual inbreeding coefficients (FROH).
In order to identify introgression from other goose species into the Swedish LWfG birds we estimated D-statistics, also known as ABBA-BABA tests19 (Supplementary Information 2.3.1), in popstats63. We first investigated GWfG introgression in Swedish LWfG samples respect to the Norwegian or Russian ones by performing tests of the form D(SWE, RUS/NOR; GWfG, PfG), where SWE are all possible Swedish LWfG birds, RUS and NOR are all possible Russian and Norwegian birds respectively, the GWfG sample is used as donor, and the pink-footed goose as outgroup. We then checked for possible reference biases by performing the same tests using the bar-headed goose (Anser indicus, BhG) as outgroup (i.e. D(SWE, RUS/NOR, GWfG, BhG)), and repeated all tests using the dataset mapped to the mallard duck (Supplementary Information 2.3.1). Additionally, we explored evidence of introgression from all the other Anser species in the dataset into the Swedish LWfG population using tests of the form D(SWE, RUS/NOR; X, BhG), where X is any of the possible Anser species tested as donors, and the bar-headed goose as outgroup. We used the same settings as above for all tests and replicated them using both the data mapped to the PfG assembly and to the mallard duck assembly (Supplementary Information 2.3.1).
Finally, we reconstructed the mitochondrial genomes of our LWfG samples from the shotgun sequencing data using the mitogenome of a GWfG as reference (NC_004539.1). We then built a maximum-likelihood tree of the LWfG mitogenomes together with the previously published mitogenomes of two bar-headed geese and four GWfG (Supplementary Information 2.3.3).
Data availability
All the raw data generated for this manuscript are available at the European Nucleotide Archive (ENA), accession number PRJEB40857.
References
Allendorf, F. W., Leary, R. F., Spruell, P. & Wenburg, J. K. The problems with hybrids: setting conservation guidelines. Trends Ecol. Evol. 16, 613–622 (2001).
Todesco, M. et al. Hybridization and extinction. Evol. Appl. 9, 892–908 (2016).
IUCN. Guidelines for reintroductions and other conservation translocations. Gland. Switz. Camb. UK IUCN SSC Re-Introd Spec Group (2013).
Greig, J. C. Principles of genetic conservation to wildlife management in Southern Africa. S. Afr. j. wildl. res. 9, 57–78 (1979).
Olech, W. & Perzanowski, K. A genetic background for reintroduction program of the European bison (Bison bonasus) in the Carpathians. Biol. Conserv. 108, 221–228 (2002).
Thévenon, S., Bonnet, A., Claro, F. & Maillard, J.-C. Genetic diversity analysis of captive populations: the Vietnamese sika deer (Cervus nippon pseudaxis) in zoological parks. Zoo. Biol. 22, 465–475 (2003).
Landa, A. et al. The endangered Arctic fox in Norway—the failure and success of captive breeding and reintroduction. Polar Res. 36, 9 (2017).
Jones, T., Martin, K., Barov, B. & Nagy, S. International single species action plan for the conservation of the Western Palearctic population of the Lesser White-fronted Goose Anser erythropus. AEWA technical series. (2008).
Madsen, J. & Cracknell, G. Goose populations of the Western Palearctic: a review of status and distribution (National Environmental Research Institute, Denmark, 1999).
Norderhaug, A. & Norderhaug, M. Status of the lesser white-fronted goose, Anser erythropus. Swed. Wildl. Res. 13, 171–185 (1984).
Andersson, Å. & Holmqvist, N. The Swedish population of Lesser White-fronted Goose Anser erythropus–supplemented or re-introduced. Ornis Svec. 20, 202–206 (2010).
von Essen, L. A note on the Lesser White-fronted goose. Ardea 79, 305–306 (1991).
Tegelström, H. & von Essen, L. DNA fingerprinting of captive breeding pairs of lesser white-fronted geese (Anser erythropus) with unknown pedigrees. Biochem. Genet. 34, 287–296 (1996).
Ruokonen, M., Andersson, A. C. & Tegelström, H. Using historical captive stocks in conservation. The case of the lesser white-fronted goose. Conserv. Genet. 8, 197–207 (2007).
Mooij, J. H., Hansson, P., Kampe-Persson, H. & Nilsson, L. Analysis of historical observations of Fennoscandian Lesser White-fronted Geese Anser erythropus in Sweden and the West Palearctic. Vogelwelt 129, 269–280 (2008).
Ruokonen, M., Kvist, L., Tegelström, H. & Lumme, J. Goose hybrids, captive breeding and restocking of the Fennoscandian populations of the Lesser White-fronted goose (Anser erythropus). Conserv. Genet. 1, 277–283 (2000).
Ruokonen, M., Kvist, L. & Lumme, J. Close relatedness between mitochondrial DNA from seven Anser goose species. J. Evol. Biol. 13, 532–540 (2000).
Leffler, E. M. et al. Revisiting an old riddle: what determines genetic diversity levels within species?. PLoS Biol. 10, e1001388 (2012).
Green, R. E. et al. A Draft Sequence of the Neandertal Genome. Science 328, 710–722 (2010).
Durand, E. Y., Patterson, N., Reich, D. & Slatkin, M. Testing for ancient admixture between closely related populations. Mol. Biol. Evol. 28, 2239–2252 (2011).
Kulathinal, R. J., Stevison, L. S. & Noor, M. A. F. The genomics of speciation in Drosophila: diversity, divergence, and introgression estimated using low-coverage genome sequencing. PLoS Genet. 5, e1000550 (2009).
Patterson, N. et al. Ancient admixture in human history. Genetics 192, 1065–1093 (2012).
Reich, D., Thangaraj, K., Patterson, N., Price, A. L. & Singh, L. Reconstructing Indian population history. Nature 461, 489–494 (2009).
Ottenburghs, J. et al. A history of hybrids? Genomic patterns of introgression in the True Geese. BMC Evol. Biol. 17, 201 (2017).
Ottenburghs, J. et al. A tree of geese: A phylogenomic perspective on the evolutionary history of True Geese. Mol. Phylogenet. Evol. 101, 303–313 (2016).
Naturvårdsverket. Åtgärdsprogram för fjällgås 2011–2015. (Naturvårdsverket, 2011).
Sjögren, P. & Wyöni, P.-I. Conservation genetics and detection of rare alleles in finite populations. Conserv. Biol. 8, 267–270 (1994).
Sorenson, M. D. & Quinn, T. W. Numts: a challenge for avian systematics and population biology. Auk 115, 214–221 (1998).
Quinn, T. W. The genetic legacy of Mother Goose–phylogeographic patterns of lesser snow goose Chen caerulescens caerulescens maternal lineages. Mol. Ecol. 1, 105–117 (1992).
Robert, A. Captive breeding genetics and reintroduction success. Biol. Conserv. 142, 2915–2922 (2009).
Champagnon, J., Elmberg, J., Guillemain, M., Gauthier-Clerc, M. & Lebreton, J.-D. Conspecifics can be aliens too: a review of effects of restocking practices in vertebrates. J. Nat. Conserv. 20, 231–241 (2012).
Bouzat, J. L. et al. Beyond the beneficial effects of translocations as an effective tool for the genetic restoration of isolated populations. Conserv. Genet. 10, 191–201 (2009).
Robert, A. et al. Defining reintroduction success using IUCN criteria for threatened species: a demographic assessment. Anim. Conserv. 18, 397–406 (2015).
Bhatia, G., Patterson, N., Sankararaman, S. & Price, A. L. Estimating and interpreting FST: the impact of rare variants. Genome Res. 23, 1514–1521 (2013).
Ploeger, P.L. Geographical differentiation in arctic Anatidae as a result of isolation during the last glacial. (Brill Archive, 1968).
Marchant, J. H. & Musgrove, A. J. Review of European flyways of the Lesser White-fronted Goose Anser erythropus. BTO Res. Rep. 595, 2 (2011).
Lorentsen, S.-H., Øien, I. J. & Aarvak, T. Migration of Fennoscandian lesser white-fronted geese Anser erythropus mapped by satellite telemetry. Biol. Conserv. 84, 47–52 (1998).
Ruokonen, M., Aarvak, T., Chesser, R. K., Lundqvist, A.-C. & Merilä, J. Temporal increase in mtDNA diversity in a declining population. Mol. Ecol. 19, 2408–2417 (2010).
Eo, S. H., Doyle, J. M. & DeWoody, J. A. Genetic diversity in birds is associated with body mass and habitat type. J. Zool. 283, 220–226 (2011).
Evans, S. R. & Sheldon, B. C. Interspecific patterns of genetic diversity in birds: correlations with extinction risk. Conserv. Biol. 22, 1016–1025 (2008).
Mable, B. K. Conservation of adaptive potential and functional diversity: integrating old and new approaches. Conserv. Genet. 20, 89–100 (2019).
Rehder, C. W. et al. American College of Medical Genetics and Genomics: standards and guidelines for documenting suspected consanguinity as an incidental finding of genomic testing. Genet. Med. 15, 150–152 (2013).
Pemberton, T. J. et al. Genomic patterns of homozygosity in worldwide human populations. Am. J. Hum. Genet. 91, 275–292 (2012).
Kampe-Persson, H. Swedish Lesser White-fronted Geese Anser erythropus in the Baltic States. Ornis Svecica 25 (2015).
Moseby, K. E. et al. Predation determines the outcome of 10 reintroduction attempts in arid South Australia. Biol. Conserv. 144, 2863–2872 (2011).
López-Bao, J. V., Fleurke, F., Chapron, G. & Trouwborst, A. Legal obligations regarding populations on the verge of extinction in Europe: conservation, restoration, recolonization reintroduction. Biol. Conserv. 227, 319–325 (2018).
Fitzpatrick, S. W. et al. Gene flow from an adaptively divergent source causes rescue through genetic and demographic factors in two wild populations of Trinidadian guppies. Evol. Appl. 9, 879–891 (2016).
Tallmon, D. A., Luikart, G. & Waples, R. S. The alluring simplicity and complex reality of genetic rescue. Trends Ecol. Evol. 19, 489–496 (2004).
Waller, D. M. Genetic rescue: a safe or risky bet?. Mol. Ecol. 24, 2595–2597 (2015).
Baveja, P., Tang, Q., Lee, J. G. H. & Rheindt, F. E. Impact of genomic leakage on the conservation of the endangered Milky Stork. Biol. Conserv. 229, 59–66 (2019).
Veale, A. J. & Russello, M. A. Sockeye salmon repatriation leads to population re-establishment and rapid introgression with native kokanee. Evol. Appl. 9, 1301–1311 (2016).
Pujolar, J. M., Dalén, L., Olsen, R. A., Hansen, M. M. & Madsen, J. First de novo whole genome sequencing and assembly of the pink-footed goose. Genomics 110, 75–79 (2018).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Huang, Y. et al. The duck genome and transcriptome provide insight into an avian influenza virus reservoir species. Nat. Genet. 45, 776–783 (2013).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011).
Smit, A. F. A., Hubley, R. & Green, P. 1996--2010. RepeatMasker Open-3.0. (2017).
Smit, A. & Hubley, R. RepeatModeler Open-1.0. (2015).
Pečnerová, P. et al. Genome-based sexing provides clues about behavior and social structure in the Woolly Mammoth. Curr. Biol. 27, 3505-3510.e3 (2017).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Weir, B. S. & Cockerham, C. C. Estimating f-statistics for the analysis of population structure. Evolution 38, 1358–1370 (1984).
Pickrell, J. K. & Pritchard, J. K. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 8, 1 (2012).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Skoglund, P. et al. Genetic evidence for two founding populations of the Americas. Nature 525, 104–108 (2015).
Aarvak, T., Øien, I. J. & Shimmings, P. A critical review of lesser white-fronted goose release projects. NOF Rep. 6, 218 (2016).
Acknowledgements
The authors would like to thank Gerard Muskens, Helmut Kruckenberg, Åke Andersson and Hakon Kampe-Persson for their work in collecting the Swedish and Russian samples. We thank Edana Lord, Pontus Skoglund, Thomas Källman and Per-Ivan Wyoni for their help in data analysis, and Christine Kowallik and Kees Koffijberg for collating the reports of ringed LWfG birds and designing the map for Figure S1. We also thank Jouni Aspi, Marja E. Heikkinen, Ingar Jostein Øien and Tomas Aarvak for access to the Norwegian LWfG samples. Ingar Jostein Øien (ingar@birdlife.no) and Tomas Aarvak (tomas@birdlife.no) can be contacted for any additional details or questions about these samples. We acknowledge support from Science for Life Laboratory, the National Genomics Infrastructure, and UPPMAX for providing assistance in massive parallel sequencing and computational infrastructure.
Funding
Open Access funding provided by Stockholm University. This project received funding from the Swedish Environmental Protection Agency and the Swedish Association for Hunting and Wildlife Management. D.D.dM. was supported through a Carl Trygger’s scholarship (grant nr. CTS17:109). L.D. acknowledges funding from FORMAS (2015-676).
Author information
Authors and Affiliations
Contributions
D.D.dM., N.G. and L.D. conceived and designed the study. N.L. and F.W. collected samples. N.G. and J.v.S. conducted laboratory analyses. Computational analyses were done by D.D.dM., with assistance from Jv.S. and P.S.G. D.D.dM., L.D., N.L., N.G. and F.W. wrote the manuscript, with input from MS. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Díez-del-Molino, D., von Seth, J., Gyllenstrand, N. et al. Population genomics reveals lack of greater white-fronted introgression into the Swedish lesser white-fronted goose. Sci Rep 10, 18347 (2020). https://doi.org/10.1038/s41598-020-75315-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-75315-y
This article is cited by
-
Insights into genetic diversity and phenotypic variations in domestic geese through comprehensive population and pan-genome analysis
Journal of Animal Science and Biotechnology (2023)
-
GenErode: a bioinformatics pipeline to investigate genome erosion in endangered and extinct species
BMC Bioinformatics (2022)
-
New developments in the field of genomic technologies and their relevance to conservation management
Conservation Genetics (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
