
Abstract
Sophora tonkinensis belongs to genus Sophora of the Fabaceae family. It is mainly distributed in the ridge and peak regions of limestone areas in western China and has high medicinal value and important ecological functions. Wild populations of S. tonkinensis are in danger and need urgent conservation. Furthermore, wild S. tonkinensis resources are very limited relative to the needs of the market, and many adulterants are present on the market. Therefore, a method for authenticating S. tonkinensis and its adulterants at the molecular level is needed. Chloroplast genomes are valuable sources of genetic markers for phylogenetic analyses, genetic diversity evaluation, and plant molecular identification. In this study, we report the complete chloroplast genome of S. tonkinensis. The circular complete chloroplast genome was 154,644 bp in length, containing an 85,810 bp long single-copy (LSC) region, an 18,321 bp short single-copy (SSC) region and two inverted repeat (IR) regions of 50,513 bp. The S. tonkinensis chloroplast genome comprised 129 genes, including 83 protein-coding genes, 38 transfer RNA (tRNA) genes, and 8 ribosomal RNA (rRNA) genes. The structure, gene order and guanine and cytosine (GC) content of the S. tonkinensis chloroplast genome were similar to those of the Sophora alopecuroides and Sophora flavescens chloroplast genomes. A total of 1,760 simple sequence repeats (SSRs) were identified in the chloroplast genome of S. tonkinensis, and most of them (93.1%) were mononucleotides. Moreover, the identified SSRs were mainly distributed in the LSC region, accounting for 60% of the total number of SSRs, while 316 (18%) and 383 (22%) were located in the SSC and IR regions, respectively. Only one complete copy of the rpl2 gene was present at the LSC/IRB boundary, while another copy was absent from the IRA region because of the incomplete structure caused by IR region expansion and contraction. The phylogenetic analysis placed S. tonkinensis in Papilionoideae, sister to S. flavescens, and the genera Sophora and Ammopiptanthus were closely related. The complete genome sequencing and chloroplast genome comparative analysis of S. tonkinensis and its closely related species presented in this paper will help formulate effective conservation and management strategies as well as molecular identification approaches for this important medicinal plant.
Similar content being viewed by others


Introduction
Sophora tonkinensis is an important medicinal plant species in the genus Sophora, which belongs to Papilionoideae, a subfamily of the Fabaceae. This species is distributed mainly in Baise city, Jinchengjiang city, and Donglan County of Guangxi province as well as Guizhou and Yunnan Provinces of China1. S. tonkinensis grows mostly in ridge and peak regions of limestone areas in the wild and is used as an efficient species for the ecological restoration of karst rocky desertification areas (Fig. 1A,B)2. S. tonkinensis is effective in curing acute pharyngolaryngeal infection, eczema, colpitis, sore throat, gastrointestinal haemorrhage, and acute dysentery diseases3. The active ingredients in S. tonkinensis Gagnep mainly consist of alkaloids, saponins, flavonoids, and polysaccharides. Studies have shown that S. tonkinensis has various pharmacological effects, such as antitumour, anti-inflammatory, anti-arrhythmic, anti-diarrhoea, analgesic, and immune regulatory effects, as well as anti-hepatic fibrosis and liver-protective activities4.

(A) The wild habitat of S. tonkinensis. (B) Habitat destruction of S. tonkinensis. (C) S. tonkinensis is used as an efficient species for the ecological restoration of karst rocky desertification areas.
Due to the unique habitat and extremely low reproductive capacity of S. tonkinensis, its wild resources are very limited relative to the needs of the market5. The species is under threat due to commercial overexploitation and serious habitat destruction (Fig. 1C), and its wild populations have been seriously shrinking. However, little is known regarding its genetic background. The plant chloroplast genome, with a length of 110–160 kb, is a valuable source of genetic markers for phylogenetic analyses, genetic diversity evaluation, and plant molecular identification due to its conserved structure and comparatively high substitution rate6,7. Therefore, a good understanding of chloroplast genomic information will make it easy to study genetic variation in and design reasonable conservation strategies for wild populations of S. tonkinensis.
Furthermore, there are many adulterants of S. tonkinensis on the market, and it is difficult to distinguish them according to outward appearance8, indicating an urgent need for a molecular approach with which to differentiate S. tonkinensis species from other adulterating species. DNA barcode sequence analysis, a molecular identification technology, can provide a rapid, accurate, and automatable method of species identification using a standardized piece of DNA sequence9,10,11. Chloroplast non-coding regions have been successfully applied in DNA barcoding research. Yao et al. found that the psbA-trnH intergenic spacer region could be used as a barcode to distinguish various Dendrobium species and to differentiate them from adulterating species12. Chen et al. tested the discrimination ability of ITS2 in more than 6,600 plant samples belonging to 4,800 species from 753 distinct genera and found that the rate of successful identification with ITS2 was 92.7% at the species level13. Chloroplast genomic information for S. tonkinensis will provide candidate DNA barcodes for the authentication of S. tonkinensis and the identification of its adulterants.
In the present study, we assembled and analysed the chloroplast genome sequence of S. tonkinensis based on Illumina paired-end (PE) sequencing data. The sequence was also compared with other known chloroplast genome sequences using bioinformatics analysis, and the evolutionary position of S. tonkinensis among the Papilionoideae was confirmed.
Results
Genome sequencing and assembly
In this study, PE DNA sequencing was carried out using the Illumina MiSeq sequencing platform. In total, 17,594,210*2 PE reads and 5,313,451,420 bases were obtained, and a nucleotide quality score greater than 20 (Q20) was achieved at a rate of 96.92%. After quality filtering, 16,892,769*2 PE reads, 663,584 single reads, and 5,058,544,355 bases were obtained. According to the total length of the assembled sequence, number of scaffolds and scaffold N50, the assembly results for multiple K-mers were evaluated comprehensively, and then the optimal-K-mer data were selected as the final assembly results. We obtained 1 scaffold with a length of 154,644 bp. These data demonstrated a high-quality assembly. The complete chloroplast genome sequence of S. tonkinensis was deposited in the Sequence Read Archive (SRA) (accession number: SRR8434290).
General features of the S. tonkinensis chloroplast genome
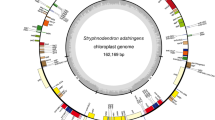
Overall, the S. tonkinensis chloroplast genome was 154,644 bp in length and presented a complete circular structure, including a pair of inverted repeats (IRs) (50,513 bp) that divided the genome into two single-copy regions (long single-copy (LSC) region of 85,810 bp; short single-copy (SSC) region of 18,321 bp) (Fig. 2). Coding regions (60,756 bp) accounted for 39.3% of the genome, and intergenic regions (93,888 bp) comprised the remaining 60.7%. The percentages of guanine and cytosine bases (GC %) in the gene regions (37.8%) were higher than those in the intergenic regions (35.4%). The average gene length and gene density were 732 bp and 0.484, respectively. The frequencies of the four bases adenine (A), thymine (T), cytosine (C), and guanine (G) in the S. tonkinensis chloroplast genome were 49,139, 49,198, 27,915, and 28,392, accounting for 31.7%, 31.8%, 18.1%, and 18.4% of the genome, respectively (Table 1). The chloroplast genome of S. tonkinensis contained a total of 129 genes, including 83 protein-coding genes, 38 transfer RNA (tRNA) genes, and 8 ribosomal RNA (rRNA) genes (Table 2). Of the 83 protein-coding genes, nine (rps16, rpoC1, atpF, petB, petD, rpl16, rpl2, ndhA and ndhB) contained one intron, while clpP and ycf3 possessed two introns (Table S1). The S. tonkinensis chloroplast genome contained 64 types of codons encoding 21 types of amino acids (Fig. 3). The number of codons differed from 247 to 2,320, with a fraction ranging from 0.08 to 1. The amino acids Met and Trp had only one codon, while the remaining amino acids possessed 2–6 codons.

Chloroplast genome map of S. tonkinensis. Genes shown in the inner side of the circle are transcribed clockwise and those located on the outside of the circle are transcribed counter-clockwise. Genes belonging to different functional groups are colour-coded. Dashed area in the inner circle indicates the GC content of the chloroplast genome.

Relative synonymous codon usage (RSCU) of 20 amino acid and stop codons in all protein-coding genes of the chloroplast genome of S. tonkinensis.
Comparison to the chloroplast genomes of other Sophora species
The size of the S. tonkinensis chloroplast genome was found to be similar to those of the Sophora alopecuroides and Sophora flavescens chloroplast genomes14,15 (Table 3). However, the S. tonkinensis chloroplast genome had the longest LSC region (85,809 bp), whereas the S. alopecuroides chloroplast genome had the shortest LSC region (84,221 bp). As shown in Table 3, S. tonkinensis had the lowest GC content (36.4%), while S. alopecuroides and S. flavescens had the same higher GC content (36.6%). A total of 129 genes and 8 rRNA genes (four rRNA species) were identified in every species. S. tonkinensis and S. alopecuroides contained 83 protein-coding genes, whereas S. flavescens contained 84. The S. tonkinensis and S. alopecuroides chloroplast genomes possessed 38 tRNA genes, whereas the S. flavescens chloroplast genome possessed 37.
Simple sequence repeat (SSR) and tandem repeat analyses
SSRs are molecular markers with high variation within the same species that are used in population genetic and polymorphism studies. The types, presence, and distribution of SSRs in the chloroplast genome of S. tonkinensis were studied. A total of 1,760 SSRs were identified, including 1,639 (93.1%) mononucleotide, 30 (1.7%) dinucleotide, 79 (4.5%) trinucleotide, 6 (0.3%) tetranucleotide, and 6 (0.3%) pentanucleotide repeats, and the mononucleotide A and T repeat units accounted for the largest portion, with a percentage of 81.9%. Moreover, the SSRs were mainly distributed in the LSC region, accounting for 60% of the total SSRs, while 316 (18%) and 383 (22%) were located in the SSC and IR regions, respectively (Fig. 4A). Of these, 21 dinucleotide, 55 trinucleotide, 6 tetranucleotide, and 4 pentanucleotide repeats were identified in the LSC region; 3 dinucleotide repeats, 5 trinucleotide repeats, and 1 pentanucleotide repeats were found in the SSC region; and 6 dinucleotide repeats, 19 trinucleotide repeats, and 1 pentanucleotide repeat were observed in the IR region (Fig. 4B–D). The size and location of the tetra- and pentapolymers are shown in Table S2. Of these repeats, 10 and 2 were localized in intergenic spacers and coding regions, respectively, and none were found in introns.

The distribution, type and presence of simple sequence repeats (SSRs) in the chloroplast genome of S. tonkinensis. (A) Presence of SSRs in the LSC, SSC, and IR regions. (B) Presence of polymers in the LSC regions. (C) Presence of polymers in the SSC regions. (D) Presence of polymers in the IR regions.
Tandem repeat sequences play a crucial role in genome rearrangement and phylogenetic analysis16. In the current study, a total of 23 tandem repeats were identified in the S. tonkinensis chloroplast genome (Table S3), which was smaller than the numbers observed in S. alopecuroides (49), Ammopiptanthus mongolicus (39), and Maackia floribunda (64) of Papilionoideae . Most of the tandem repeats were distributed in intergenic spacers and introns (19 (82.7%) in the intergenic spacers and 1 in the intron of clpP), and just 2 and 1 were located in the protein-coding regions of ycf2 and ndhF, respectively.
Comparative analysis of the S. tonkinensis chloroplast genome
Three published sequences representing Sophora (S. alopecuroides), Ammopiptanthus (A. mongolicus), and Maackia (M. floribunda) of Papilionoideae were selected for comparison with the sequence of S. tonkinensis to estimate the sequence divergence of different regions of these plastomes. The overall sequence identities of the four Papilionoideae chloroplast genomes were plotted using mVISTA with the annotation of S. tonkinensis as the reference, and we observed approximately identical gene orders and organizations among them (Fig. 5). The coding regions were found to be more highly conserved than the non-coding regions, and the two IR regions were less divergent than the LSC and SSC regions. The most divergent coding regions of the four chloroplast genomes were ycf1, ndhF, accD, rpoC2, and rpoB, and the four rRNA genes (rrn4.5, rrn5, rrn16, and rrn23) were the most conserved.

Comparison of the chloroplast genome sequences of S. tonkinensis, S. alopecuroides, A. mongolicus, and M. floribunda generated with mVISTA. Grey arrows indicate the position and direction of each gene. Red and blue areas indicate the intergenic and genic regions, respectively. The vertical scale indicates the percentage of identity, ranging from 50 to 100%.
IRs are the most conserved regions in the chloroplast genome, and contraction and expansion at their boundaries are common evolutionary events, representing one of the main factors affecting chloroplast genome size. Using Nicotiana tabacum as the reference species, we compared the IR/LSC and IR/SSC borders of the chloroplast genomes of S. tonkinensis, S. alopecuroides, A. mongolicus, and M. floribunda of Papilionoideae (Fig. 6). The results showed that S. tonkinensis had size differences in the LSC, SSC and IR regions compared with those in other closely related chloroplast genomes of Papilionoideae species. In all of these species, the rps19 gene was located in the LSC region. The rpl2 gene of S. tonkinensis spanned the LSC and IRB regions, while the rpl2 genes of the other species were all observed in the IRB region, with a 4–5 bp distance from the LSC/IRB border. The ycf1 pseudogene spanned the IRB/SSC boundary in all chloroplast genes, while the yfc1 pseudogene and nadH gene overlapped in A. mongolicus. The nadH gene was present in the SSC region of all genomes, with a 7–74 bp distance from the IRB/SSC junction. Expansion and contraction of the ycf1 gene were observed in the boundary regions of the SSC/IRA. Size variation in ycf1 from 5,318 to 5,708 bp was identified in all chloroplast genomes. The trnH gene was found in the LSC region of all genomes but was located 2 to 138 bp from the IRA/LSC boundary. In S. tonkinensis, the rpl2 gene was absent in the IRA region because of the incomplete gene structure caused by the expansion and contraction of IR regions.

Comparison of the border regions among LSC, IR and SSC in the chloroplast genomes of S. tonkinensis, S. alopecuroides, A. mongolicus, and M. floribunda. JLB: junction line between LSC and IRb; JSB: junction line between IRb and SSC; JSA: junction line between SSC and IRa; JLA: junction line between IRa and LSC.
Synonymous (KS) and non-synonymous (KA) substitution rate analysis
A total of 70 genes in the chloroplast genome of S. tonkinensis were used to calculate the KA/KS ratio relative to the chloroplast genome of S. alopecuroides and S. flavescens (Fig. 7). The KA/KS ratios of most of the genes in S. tonkinensis Vs. those in S. flavescens and S. alopecuroides were consistent with negative (or purifying) selection (KA/KS < 1), while six genes (matK, psbE, psbF, psbM, psaI, and rpl36) displayed positive selection (KA/KS > 1). Notably, the KA/KS ratios of psbE, psbF, psbM, psaI, and rpl36 in the S. tonkinensis Vs. S. flavescens and S. alopecuroides comparisons were as high as 50, which indicated great evolutionary divergence in these genes. The rps2 and rpl32 genes were differentially selected: rps12 did not differ in the S. tonkinensis Vs. S. flavescens comparison, but it was positively selected in the S. tonkinensis Vs. S. alopecuroides comparison (KA/KS = 9.25). rpl32 exhibited no difference in the S. tonkinensis Vs. S. alopecuroides comparison but was negatively selected in the S. tonkinensis Vs. S. flavescens (KA/KS = 0.32) comparison.

KA/KS values of 70 protein-coding genes in the S. tonkinensis Vs. S. alopecuroides and S. tonkinensis Vs. S. flavescens comparisons. Orange coloured bars indicate S. tonkinensis Vs S. flavescens, and blue coloured bars indicate S. tonkinensis Vs. S. alopecuroides.
Single nucleotide polymorphism (SNP) analysis
SNP loci are very useful resources for phylogenetic analysis and species identification17. To determine the differences between S. tonkinensis and the two other Sophora species S. alopecuroides and S. flavescens at the chloroplast genome level, SNP analysis was carried out with the chloroplast genome of S. tonkinensis as the reference sequence. The results revealed 805 SNPs were found in the intergenic region, and 485 SNPs, including 236 non-synonymous SNPs and 249 synonymous SNPs were identified in 64 protein-coding genes. Of these genes, ycf1 contained the most SNP sites (Fig. 8).

Variation analysis between S. tonkinensis and S. alopecuroides or S. flavescens. Green coloured bars indicate non-synonymous SNPs and purple coloured bars indicate synonymous SNPs.
Phylogenetic analysis
In the present study, we aligned 20 complete chloroplast genomes of Papilionoideae to reveal the phylogenetic position of S. tonkinensis (Fig. 9). The phylogenetic positions of these 20 chloroplast genomes were successfully resolved with full bootstrap support across almost all nodes. We found that S. tonkinensis was grouped into Sophora with S. flavescens and S. alopecuroides and S. tonkinensis exhibited the closest relationship with S. flavescens. A close relationship among the genera Sophora, Salweenia and Ammopiptanthus was also uncovered.

Phylogenetic tree of S. tonkinensis and 19 species of subfamily Papilionoideae using Maximum likelihood based on the complete chloroplast genomes. The bootstrap value based on 1,000 replicates is shown on each node.
Discussion
Since the first sequenced plant chloroplast genome was isolated from tobacco18, thousands of chloroplast genomes from various species have been sequenced. As of 2019, more than 3,300 chloroplast genome sequences had been recorded in the National Center for Biotechnology Information (NCBI) database. In recent years, DNA barcoding has become a powerful tool for species identification. In plants, commonly used DNA barcodes include the chloroplast genes rbcL, matK and psba-trnh and nuclear genes ITS and ITS219. Of these, ITS2 has been suggested as a universal DNA barcode for medicinal plants due to its strong identification ability12. However, DNA barcodes do not have enough variation information for species identification of some medicinal plants, such as Epimedium and Fritillariae20,21. Compared with a standardized piece of DNA sequence, the whole chloroplast genome contains more mutation sites and is more efficient in identification. The whole chloroplast genome is used as a superbarcode and has been successfully applied in species identification of some medicinal plants and their closely related species. Cui et al. found that three medicinal and edible Amomum taxa (A. villosum, A. villosum var. xanthioides and A. longiligulare) could be accurately identified using their whole chloroplast genomes22. Chen et al. discovered that the complete chloroplast genome can be used as a superbarcode to identify six Ligularia species23. The chloroplast genome could distinguish C. indicum from its closely related species and might become a potential superbarcode for the identification of these species24. Zhu et al. found that the complete plastome sequence dataset had the highest discriminatory power for D. officinale and its closely related species, indicating that complete plastome sequences can be used to accurately authenticate Dendrobium species25. The whole chloroplast genome of S. tonkinensis and its hypervariable region, including the most divergent regions (ycf1, ndhF, accD, and rpoC2), which are also the genes containing the most SNP sites, and the six positively selected genes (matK, psbE, psbF, psbM, psaI, and rpl36) could be selected as potential DNA barcodes for identification of species in future studies.
Genetic variation plays an important role in the ability of plants to maintain their evolutionary potential to adapt to the ever-changing environment, therefore the maintenance of genetic variation is the main goal of the conservation strategies for most endangered species26. SSRs, also known as microsatellites, have high polymorphism rate at the species level27,28,29,30. Therefore, they have been widely used as effective molecular markers in population genetic and evolution studies31,32. Yang et al. used eight SSR primer to assess the genetic diversity and structure of 22 natural populations of the endangered medicinal plant Phellodendron amurense in China, and proposed proper conservation measures for this species33. An ex situ conservation measure for conserving genetically distant populations to maximize the genetic diversity of Eucommia ulmoides is recommended based on the genetic analysis diversity within and among the semi-wild and cultivated populations of E. ulmoides using two cpSSR loci34. In the S. tonkinensis chloroplast genome, five types of SSRs (mono-, di-, tri-, tetra-, and penta-nucleotide repeats) and a total of 150 SSR loci with a length of at least 10 bp were identified (Table S4). The mononucleotide repeats were the most abundant SSR. Most of the mononucleotide and dinucleotides are composed of multiple copies of A/T and AT/TA repeats, respectively, this result is similar to that of previous study on S. alopecuroides35. These SSRs of the S. tonkinensis chloroplast genome could be useful biomarkers for genetic diversity studies of wild populations of S. tonkinensis, which will help to formulate effective conservation and management strategies for this important medicinal plant.
Conclusions
In conclusion, the chloroplast genome of S. tonkinensis was sequenced on the Illumina HiSeq 2000 platform in this study. SSRs and tandem repeats were identified and 1,760 SSRs were found, most of which were mononucleotides, in the chloroplast genome of S. tonkinensis. SSR analysis can provide valuable information for developing highly variable DNA markers for population genetic surveys and other ecological and evolutionary studies of S. tonkinensis. Further, we performed phylogenetic analysis of 20 chloroplast genomes and collinearity analysis of three closely related species of S. tonkinensis. The contraction and expansion of the IR regions of the three closely related species were also compared. The results of the above analyses provide valuable reference information that will help formulate effective conservation and management strategies as well as molecular identification approaches for this important medicinal plant.
Materials and methods
Sample preparation and DNA extraction
Sophora tonkinensis leaves were obtained from 2-year-old plants in the Guangxi Botanical Garden of Medicinal Plants (Nanning, China). Genomic DNA was extracted using the DNeasy Plant Mini Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions.
Genome assembly and annotation
DNA was randomly fragmented by a Covaris M220 apparatus. After adding the poly “A” tail, the DNA fragments with desired lengths (400–500 bp) were ligated to adapters and purified using the TruSeq™ DNA Sample Prep Kit for Illumina MiSeq sequencing. Before assembly, raw reads were filtered, and the reads with adapters, reads containing too many uncalled bases (“N” characters, ≥ 10%), the reads showing a quality score below 20 (Q < 20), and the duplicated sequences were removed. The optimized sequence was first assembled by using SOAP de Novo v2.04 software (https://soap.genomics.org.cn/)36. Second, GapCloser v1.12 software was used to fill the gaps in the assembly results and for base correction. Annotation of the chloroplast genome was conducted using Dual Organellar GenoMe Annotator (DOGMA) software (https://dogma.ccbb.utexas.edu/)37, and artificial correction was carried out to predict the genes, rRNAs, and tRNAs in the genome. A circular chloroplast genome map was drawn using the OGDRAW program (https://chlorobox.mpimp-golm.mpg.de/OGDraw.html)38.
Codon usage analysis
RSCU (Relative Synonymous Codon Usage) was computed from the protein-coding gene sequences of the S. tonkinensis cp genome. The online program CodonW 1.4.2 (https://codonw.sourceforge.net/) was employed for RSCU and codon frequency analysis39.
Analysis of simple sequence repeats (SSRs) and tandem repeats
The SSRs in the complete chloroplast genome of S. tonkinensis were detected using MIcroSAtellite identification tool (MISA) software (https://pgrc.ipk-gatersleben.de/misa/)40. The repeat sequences with repeating units of 5, 5, 3, 3, and 3 bp were considered as SSRs for mononucleotide, dinucleotides, trinucleotides, tetranucleotides, and pentanucleotide, respectively. Tandem Repeats Finder (TRF) v4.04 was used to identify tandem repeats41.
Comparative genomic analysis of the S. tonkinensis chloroplast genome
The complete chloroplast genome of S. tonkinensis was compared with that of three other Papilionoideae species, namely, S. alopecuroides [MH_748034], A. mongolicus [NC_034742], and M. floribunda [KX_388160], in the NCBI plastid database using the mVISTA program with a shuffle-LAGAN model42. S. tonkinensis was set as the reference.
Synonymous (KS) and non-synonymous (KA) substitution rate analysis
The chloroplast genome sequence of S. tonkinensis was compared with those of S. alopecuroides [MH_748034] and S. flavescens [MK_114100] in the NCBI plastid database. The same functional protein-coding exons were extracted and aligned separately to analyse the KA and KS substitution rates using Geneious v7.1.9 software. The aligned sequences were translated into protein sequences and then analysed. The KA and KS substitution rates for each protein-coding exon were calculated in DnaSP43. The ratios KA/KS > 1, KA/KS = 1 and KA/KS < 1 indicate positive selection, neutral selection and negative selection, respectively44.
Phylogenetic analysis
The phylogenetic analysis was based on the complete chloroplast genome sequence of S. tonkinensis and 19 species of Papilionoideae downloaded from the NCBI database (https://www.ncbi.nlm.nih.gov/), including two species of Sophora species, S. alopecuroides and S. flavescens, and 17 chloroplast genomes from the other basic lineage of Papilionoideae. The maximum likehood method was used to infer the phylogenetic relationship with 1,000 bootstrap replicates in MEGA 5.045.
References
Zhou, H. W. Research progress and tendency of Sophora Tonkinensis Gapnep. J. Guangxi Agric. 29, 43–47 (2014).
Peng, F. H. et al. Research progress of high-yield cultivation techniques of Sophora Tonkinensis Gapnep. in Guizhou rock desertification area. Tillage Cultiv. 39, 24–26. https://doi.org/10.13605/j.cnki.52-1065/s.2019.03.007 (2019).
Zheng, L. N., Sun, H., Xie, Y. Z. & Sun, R. Research progress on chemical compositions of Sophorae Tonkinensis Radix et Rhizoma related to its efficacy and toxicity. Food Drug 13, 205–209 (2011).
Wu, J. N. An Illustrated Chinese Materia Medica. 600–601 (Oxford University Press, Oxford, 2005).
Shen, L., Luo, Y., Zhang, P. G. & Huang, R. S. The progress of resource status and quality standard in Sophora tonkinensis. Da Zhong Ke Ji 5, 145–146 (2011).
Dong, W. P., Liu, J., Yu, J., Wang, L. & Zhou, S. L. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE 7, e35071. https://doi.org/10.1371/journal.pone.0035071 (2012).
Dong, W. et al. A chloroplast genomic strategy for designing taxon specific DNA mini-barcodes: A case study on ginsengs. BMC Genet. 15, 138. https://doi.org/10.1186/s12863-014-0138-z (2014).
Hua, Y. S. Identification of Shandougen and its seven common conterfeits. West. J. Tradit. Chin. Med. 28, 21–24 (2015).
Hebert, P. D. N., Cywinska, A., Ball, S. L. & DeWaard, J. R. Biological identifications through DNA barcodes. Proc. R. Soc. B-Biol. Sci. 270, 313–321. https://doi.org/10.1098/rspb.2002.2218 (2003).
Kress, W. J., Wurdack, K. J., Zimmer, E. A., Weigt, L. A. & Janzen, D. H. Use of DNA barcodes to identify flowering plants. Proc. Natl. Acad. Sci. USA 102, 8369–8374. https://doi.org/10.1073/pnas.0503123102 (2005).
Schindel, D. E. & Miller, S. E. DNA barcoding a useful tool for taxonomists. Nature 435, 17. https://doi.org/10.1038/435017b (2005).
Yao, H. et al. Identification of dendrobium species by a candidate DNA barcode sequence: The chloroplast psbA-trnH intergenic region. Planta Med. 75, 667–669. https://doi.org/10.1055/s-0029-1185385 (2009).
Chen, S. L. et al. Validation of the ITS2 region as a novel DNA barcode for identifying medicinal plant species. PLoS ONE 5, e8613. https://doi.org/10.1371/journal.pone.0008613 (2010).
Zhang, W. L., Li, L. & Guo, H. L. Characterization of the complete chloroplast genome of shrubby sophora (Sophora flavescens Ait.). Mitochondrial DNA Part B 3, 1282–1283. https://doi.org/10.1080/23802359.2018.1532839 (2018).
Duan, N. et al. The complete chloroplast genome of Sophora alopecuroides (Fabaceae). Mitochondrial DNA Part B 4, 1336–1337. https://doi.org/10.1080/23802359.2019.1596760 (2019).
Cavalier-Smith, T. Chloroplast evolution: Secondary symbiogenesis and multiple losses. Curr. Biol. 12, R62-64. https://doi.org/10.1016/s0960-9822(01)00675-3 (2002).
Tonti-Filippini, J., Nevill, P. G., Dixon, K. & Small, I. What can we do with 1000 plastid genomes?. Plant J. 90, 808–818. https://doi.org/10.1111/tpj.13491 (2017).
Shinozaki, K. et al. The complete nucleotide sequence of the tobacco chloroplast genome: Its gene organization and expression. EMBO J. 5, 2043–2049 (1986).
Hollingsworth, P. M., Li, D. Z., van der Bank, M. & Twyford, A. D. Telling plant species apart with DNA: From barcodes to genomes. Philos. Trans. R. Soc. Lond. B. Biol. Sci. https://doi.org/10.1098/rstb.2015.0338 (2016).
Guo, M., Xu, Y., Ren, L., He, S. & Pang, A. X. A systematic study on DNA barcoding of medicinally important genus Epimedium L. (Berberidaceae). Genes https://doi.org/10.3390/genes9120637 (2018).
Luo, K. et al. Molecular identification of Fritillariae Cirrhosae Bulbus and its adulterants. World Sci. Technol. Modern. Tradit. Chin. Med. Materia Medica 14, 1153–1158 (2012).
Cui, Y. et al. Comparison and phylogenetic analysis of chloroplast genomes of three medicinal and edible Amomum species. Int. J. Mol. Sci. https://doi.org/10.3390/ijms20164040 (2019).
Chen, X. et al. Identification of Ligularia herbs using the complete chloroplast genome as a super-barcode. Front. Pharmacol. 9, 695. https://doi.org/10.3389/fphar.2018.00695 (2018).
Xia, Y. et al. The complete chloroplast genome sequence of Chrysanthemum indicum. Mitochondrial DNA A DNA Mapp. Seq. Anal. 27, 4668–4669. https://doi.org/10.3109/19401736.2015.1106494 (2016).
Zhu, S. et al. Accurate authentication of Dendrobium officinale and its closely related species by comparative analysis of complete plastomes. Acta Pharm. Sin. B 8, 969–980. https://doi.org/10.1016/j.apsb.2018.05.009 (2018).
Livingston, K. Conservation genetics. Case histories from nature. Science 272, 364–364 (1996).
Powell, W., Morgante, M., McDevitt, R., Vendramin, G. G. & Rafalski, J. A. Polymorphic simple sequence repeat regions in chloroplast genomes: Applications to the population genetics of pines. Proc. Natl. Acad. Sci. USA 92, 7759–7763. https://doi.org/10.1073/pnas.92.17.7759 (1995).
Provan, J., Corbett, G., McNicol, J. W. & Powell, W. Chloroplast DNA variability in wild and cultivated rice (Oryza spp.) revealed by polymorphic chloroplast simple sequence repeats. Genome 40, 104–110. https://doi.org/10.1139/g97-014 (1997).
Pauwels, M. et al. Nuclear and chloroplast DNA phylogeography reveals vicariance among European populations of the model species for the study of metal tolerance, Arabidopsis halleri (Brassicaceae). New Phytol. 193, 916–928. https://doi.org/10.1111/j.1469-8137.2011.04003.x (2012).
Xue, J., Wang, S. & Zhou, S. L. Polymorphic chloroplast microsatellite loci in Nelumbo (Nelumbonaceae). Am. J. Bot. 99, e240-244. https://doi.org/10.3732/ajb.1100547 (2012).
Dong, W. et al. Comparative analysis of the complete chloroplast genome sequences in psammophytic Haloxylon species (Amaranthaceae). PeerJ 4, e2699. https://doi.org/10.7717/peerj.2699 (2016).
Yang, Y. et al. Comparative analysis of the complete chloroplast genomes of five Quercus species. Front Plant. Sci. 7, 959. https://doi.org/10.3389/fpls.2016.00959 (2016).
Yang, H. S. et al. Genetic diversity and population structure of the endangered medicinal plant Phellodendron amurense in China revealed by SSR markers. Biochem. Syst. Ecol. 66, 286–292. https://doi.org/10.1016/j.bse.2016.04.018 (2016).
Yao, X. H., Deng, J. Y. & Huang, H. W. Genetic diversity in Eucommia ulmoides (Eucommiaceae), an endangered traditional Chinese medicinal plant. Conserv. Genet. 13, 1499–1507. https://doi.org/10.1007/s10592-012-0393-3 (2012).
Zha, X., Wang, X., Li, J., Gao, F. & Zhou, Y. Complete chloroplast genome of Sophora alopecuroides (Papilionoideae): Molecular structures, comparative genome analysis and phylogenetic analysis. J. Genet. 99, 13 (2020).
Luo, R. et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18. https://doi.org/10.1186/2047-217X-1-18 (2012).
Wyman, S. K., Jansen, R. K. & Boore, J. L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. https://doi.org/10.1093/bioinformatics/bth352 (2004).
Lohse, M., Drechsel, O. & Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 52, 267–274. https://doi.org/10.1007/s00294-007-0161-y (2007).
Sharp, P. M., Tuohy, T. M. & Mosurski, K. R. Codon usage in yeast: Cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 14, 5125–5143. https://doi.org/10.1093/nar/14.13.5125 (1986).
Beier, S., Thiel, T., Munch, T., Scholz, U. & Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 33, 2583–2585. https://doi.org/10.1093/bioinformatics/btx198 (2017).
Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. https://doi.org/10.1093/nar/27.2.573 (1999).
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M. & Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 32, W273-279. https://doi.org/10.1093/nar/gkh458 (2004).
Librado, P. & Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. https://doi.org/10.1093/bioinformatics/btp187 (2009).
Yang, Z. & Nielsen, R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol. Biol. Evol. 19, 908–917. https://doi.org/10.1093/oxfordjournals.molbev.a004148 (2002).
Tamura, K. et al. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. https://doi.org/10.1093/molbev/msr121 (2011).
Acknowledgements
This work was supported by Guangxi Natural Science Foundation (2018GXNSFBA294016, 2018GXNSFBA281050), Guangxi Innovation-Driven Development Project (GuiKe AA18242040), “Guangxi Bagui Scholars” and Research Innovation Team Project (GuiYaoChuang2019005) and the National Public Welfare Special Project of China “Quality Guarantee system of Chinese herbal Medicines” (201507002).
Author information
Authors and Affiliations
Contributions
J.M. conceived this study. F.W. performed the experiments and drafted the manuscript. K.W. and Y.Z. contributed to materials management and collection of samples. D.T. contributed to data interpretation and preparation of the manuscript. M.H.K. helped to revise the manuscript. F.Q., L.L., Y.L. and A.K. provided suggestions for the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wei, F., Tang, D., Wei, K. et al. The complete chloroplast genome sequence of the medicinal plant Sophora tonkinensis. Sci Rep 10, 12473 (2020). https://doi.org/10.1038/s41598-020-69549-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-69549-z
This article is cited by
-
Applications of some advanced sequencing, analytical, and computational approaches in medicinal plant research: a review
Molecular Biology Reports (2024)
-
Characterization of the complete chloroplast genome of purple flowering stalk (Brassica campestris var. purpuraria) and comparisons with related species
Genetic Resources and Crop Evolution (2024)
-
Complete chloroplast genome and phylogenetic analysis of Anemone shikokiana
Molecular Biology Reports (2023)
-
Comparative plastomes and phylogenetic analysis of seven Korean endemic Saussurea (Asteraceae)
BMC Plant Biology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
