
Abstract
Heat shock transcription factor (Hsf) plays a transcriptional regulatory role in plants during heat stress and other abiotic stresses. 31 non-redundant ZmHsf genes from maize were identified and clustered in the reference genome sequenced by Single Molecule Real Time (SMRT). The amino acid length, chromosome location, and presence of functional domains and motifs of all ZmHsfs sequences were analyzed and determined. Phylogenetics and collinearity analyses reveal gene duplication events in Hsf family and collinearity blocks shared by maize, rice and sorghum. The results of RNA-Seq analysis of anthesis and post-anthesis periods in maize show different expression patterns of ZmHsf family members. Specially, ZmHsf26 of A2 subclass and ZmHsf23 of A6 subclass were distinctly up-regulated after heat shock (HS) at post-anthesis stage. Nanopore transcriptome sequencing of maize seedlings showed that alternative splicing (AS) events occur in ZmHsf04 and ZmHsf17 which belong to subclass A2 after heat shock. Through sequence alignment, semi-quantitative and quantitative RT-PCR, we found that intron retention events occur in response to heat shock, and newly splice isoforms, ZmHsf04-II and ZmHsf17-II, were transcribed. Both new isoforms contain several premature termination codons in their introns which may lead to early termination of translation. The ZmHsf04 expression was highly increased than that of ZmHsf17, and the up-regulation of ZmHsf04-I transcription level were significantly higher than that of ZmHsf04-II after HS.
Similar content being viewed by others

Introduction
To address the agriculture and food needs, research has been focused on understanding abiotic stress responses in plants1. In particular, attention is being paid to heat stress in connection to global warming. During growth and development, especially the reproductive stage, crop is sensitive to extreme heat waves that consequently influence grain yield and quality2,3. Heat stress often reduces photosynthesis rates, primarily by changing the structure of thylakoid membranes4. The floral organs are sensitive to high temperature at anthesis, with poor pollination affecting seed development5. To survive under environmental stresses, plants have evolved intricate signal pathways and gene expression regulation. As a critical step in gene expression, transcriptional regulation has been extensively studied6. Transcription factors (TFs) can recognize and bind specific cis-elements so as to activate or repress target genes expression at specific times and locations7.
58 types of TFs, have been identified in plants currently, some of them are involved in heat shock (HS)8. The heat shock transcription factors (Hsfs) are important group of eukaryotic stress responsive TFs, which have been identified in several plant species after being firstly discovered in yeast9,10,11. The structure of Hsf protein is highly conserved, which indicating an important, conserved function of the family genes and providing a convenient opportunity for research11,12. Typical Hsf structure includes an N-terminal DNA binding domain (DBD), which has landmark central ‘wing’ helix-turn-helix (HTH) motif; and an oligomerization domain (OD), which has a bipartite heptad pattern of hydrophobic amino acid residues (HR-A/B)13,14,15. The DBD, which including three α helical bundles and four β stranded antiparallel sheets, can bind to heat shock elements (HSEs) with high selectivity11,16. The HSEs of most downstream target genes of eukaryotic cell contain repetitive palindromic motifs (5′-nGAAnnTTCn-3′) by which HSEs interact with Hsf trimers17. Hsf trimers are the active forms in plants. The HR-A and HR-B regions of the OD are responsible for forming specific homo-oligomeric or hetero-oligomeric combinations18. The number of amino acid between HR-A and HR-B regions is the basis to sort Hsf family into three classes: A, B and C. Members of the class A and C contain 21 and 7 inserted residues, respectively, and no inserted residues in class B11. Unlike the highly conserved N-terminal domain, C-terminal activation domains (CTAD) of different Hsf classes contain diversified motifs, in the class A, the aromatic and highly hydrophobic amino acid residues (AHA) motif is located in the CTAD to assist with transcriptional activation19. Some Hsf members contain nuclear localization signals (NLS) and nuclear export signals (NES) that dictating cellular localization.
Hsf can activate many target genes in response to several environment stresses, including high temperature, heavy metals, oxidants and drought20,21. Hsfs of subclass A1, constitutively-expressed genes, have been proved to be the major regulators and can specific bind affinity to Hsfs of subclass A222. Typically, the Hsfs of subclass A2 are more dramatically induced by HS than other members23,24. AtHsfA2 sustains the expression of Hsp genes to extend the duration of acquired thermotolerance in Arabidopsis25. The up-regulation of HsfA4a was proved to enhance Cd tolerance of wheat and rice26. The transcriptional activation activity of HsfA6 has been connected to HS response in rice and wheat27,28. HsfB, such as AtHsfB1 and AtHsfB2b, were considered to be transcriptional repressors that negatively regulate heat responsive genes29. But in both tomato and chickpea, HsfB1 and HsfB2 are co-activators which can interact with HsfA genes to produce positive HS response30,31. Lacking of AHA motifs, Hsfs both Class B and C are thought to have no transcriptional activation activity31. However, yeast one-hybrid assays of OsHsfC1a and OsHsfC1b, suggest that transcriptional activation of Hsfs may not require the AHA motif in rice32.
Using genomic techniques, Hsf family genes from various non-model plants have been characterized, such as maize, Chinese cabbage, pepper, grape and Triticum aestivum24,33,34,35,36. The Hsfs belong to typical multigene family. Previous studies have identified orthologous and paralogous Hsf genes in different species23, and gene functions have been assigned using homology comparison and gene collinearity analysis37. After several rounds of genome and segmental duplication, maize genome is diverse38. If based on second-generation sequencing technology, the maize reference genome may be fragmented and many complex repeat regions will be missed39. The latest released maize reference genome was assembled with PacBio Single Molecule Real Time (SMRT) sequencing and features a 52-fold increase in contig length compared to previous assemblies39. Our previous studies have cloned and characterized several ZmHsfs from maize and found variations in the expression profile, transcript activation and gene function of maize Hsf family40,41,42. In this work, we identified and classified putative ZmHsfs by blasting all Hsf genes of Arabidopsis and rice against the newly maize reference genome. Phylogenetics and collinearity analyses were carried out using data from Arabidopsis, rice, sorghum and maize. Further, transcriptome analyses of maize leaves at anthesis and post-anthesis stages were performed and possible alternative splicing events were identified by full-length transcriptome sequencing.
Results
Identification and sequence analysis of ZmHsfs
A total of 58 ZmHsf proteins were found by HMM and Blastp searches in the latest released maize genome. Among these, 31 non-redundant ZmHsf proteins containing DBD and HR-A/B regions were identified by the SMART program. 25 identified ZmHsfs were previously numbered according to their chromosomal locations33. Our newly identified Hsfs, ZmHsf26, 27, 28, 29, 30 and 31, were also named according to their chromosomal location too. The WoLFPSORT website analysis of their amino acid sequences predicts that most of the ZmHsfs are localized in the nucleus, ZmHsf23, ZmHsf10 and ZmHsf11 are located to the cytoplasm, chloroplast and ER, respectively. The length of ZmHsfs proteins varies from 250 to 622 amino acids, the molecular weight is between 27309.07~68553.79 Da and the pI value varies from 4.7 to 9.53 (Table 1).
Multiple sequence alignment illustrates the conservation of the DBD domain and HR-A/B regions of ZmHsfs (Fig. 1). The secondary structure of all ZmHsf proteins contains 3 α-helical structures and 4 β-fold structures in the DBD domain. Based on the length of flexible linkers between the HR-A and HR-B regions, 16, 10 and 5 members of ZmHsf were assigned to class A, B and C, respectively. There are 21 inserted residues in class A and 7 in class C between the HR-A and HR-B regions and no insertion in class B.

Multiple sequence alignment of DBD and HR-A/B domains of maize Hsfs. Arranged the ZmHsf proteins in the order according to the homology comparison with rice. The secondary structure elements of DBD are shown above the alignment.
Conserved protein motifs and gene structure of ZmHsfs
The results of phylogenetic tree, motif identification and gene structure analysis of 31 ZmHsf proteins were merged using TBtools to assess conserved motifs and gene structure. All ZmHsfs contain motifs 1, 2, 3 and 4 except ZmHsf31, which is lack of motif 4. All members of class A include motif 5 and 6 except ZmHsf15, which does not have motif 6. All members of class C contain motif 6 (Fig. 2). Motifs 1, 2 and 3 function in the DBD domain, motif 4 functions in the HR-B domain, and motif 6 functions in the NLS domain. The Hsf domains were found in all ZmHsf proteins. Most class A members have longer introns and a larger number of exons than members of class B and C (Fig. 2).

The conserved protein motifs and gene structures of maize Hsfs. ZmHsf proteins are displayed in order based on the phylogenetic analysis. Twenty-five motifs and thirteen domains identified in ZmHsf proteins marked by different colors.
Analysis of phylogenetic tree and collinearity of AtHsfs, OsHsfs, SbHsfs and ZmHsfs
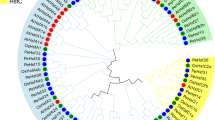
A phylogenetic tree was constructed from a multiple sequence alignment of full-length proteins from 31 maize ZmHsfs, 21 Arabidopsis AtHsfs, 25 rice OsHsfs and 25 sorghum SbHsfs (Fig. 3). The class and subclass of the ZmHsfs are highlighted with different background colors and different line colors, respectively (Fig. 3). There are 16, 10 and 5 ZmHsf proteins in class A, B and C, respectively. Same to the two Gramineae species, class A has more members than class B or C in maize, and the subclass A2 has the most members in class A. Compared with the dicot Arabidopsis, the three Gramineae genomes lack members of subclass A7, A8 and B3, and contain fewer members of subclass A1 and more subclass A2 and class C (Fig. 3).

Phylogenetic tree of Hsf family proteins among Arabidopsis, rice, sorghum and maize. The Hsf family proteins are classified three classes (A–C) which marked by three underground colors. The font colors of Hsf ID from Arabidopsis, rice, sorghum and maize were blue, green, red and black respectively.
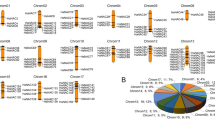
Through homologous blast of maize amino acid sequences performed with MCScan toolkit, 7783 collinear genes, 1152 tandem repeat genes and 415 collinear blocks were identified (Table S3). Chromosome 1 contains the most ZmHsfs of class A and B. Chromosomes 2, 4 and 10 only contain class B genes. The majority of ZmHsfs were located at the end of chromosomes (Fig. 4a). Pairs of paralogous ZmHsf gene arose from whole genome duplication or segmental duplication in subclass A1, A2, B1, B2, C1 and C2, respectively (Fig. 4a). The most members might arise from transposition and have no tandem repeat genes. The results of homologous blast show the collinearity among maize, rice and sorghum (Fig. 4b). There are more collinear genes and blocks between maize and sorghum than between maize and rice (Table S4 and S5). Most ZmHsfs distribute in the regions in which there are more than 10 consecutive collinear genes (highlighted by blue lines), excepting ZmHsf03, ZmHsf11 and ZmHsf19 of subclass B2 and ZmHsf24 of subclass A9 (Fig. 4b). Maize chromosome 1 shares the most collinear blocks with chromosome 1 of sorghum bicolor and chromosome 3 of rice (Fig. 4b).

The collinearity analysis of maize itself and among maize, rice and sorghum. (A) The duplicated ZmHsf genes in maize based on the collinearity analysis of all the chromosomes from maize. The pairs of duplicated genes are connected with lines. (B) The collinearity analysis of the chromosomes from maize, rice and sorghum. The collinearity blocks include 10 successive homologous genes are connected with blue lines. The red flags represent the 31 ZmHsfs located on different chromosomes of maize.
Expression analysis of ZmHsfs under heat stress
Transcriptome sequencing analysis of the ZmHsf family was performed by measuring plants TPM levels both anthesis and post-anthesis stage under HS (Fig. 5). Most of ZmHsf members responded to HS, and expression patterns are similar in the same subclass. Total eight members of subclass A2 (ZmHsf26, ZmHsf04, ZmHsf01, ZmHsf05) and subclass B2 (ZmHsf25, ZmHsf03, ZmHsf11, ZmHsf19) were induced by both HS1 and HS2 treatments. In addition, the expressions of such genes were all improved after HS treatment: ZmHsf23 of subclass A6, ZmHsf02 of subclass A9, ZmHsf08 of subclass B1 and ZmHsf29 of subclass C2. The transcription levels of ZmHsf17 of subclass A2 and ZmHsf13 of subclass C2, were up-regulated in the anthesis stage but down-regulated in the post-anthesis stage after HS treatments. To further confirm transcription changes, qRT-PCR was done for 24 ZmHsfs responsive to HS, and the results of qRT-PCR were consistent with that of transcriptome sequencing analysis (Fig. S1).

The transcription profiles of ZmHsf family genes in maize leaves at anthesis and post-anthesis stages after HS treatment. Different color correspond to the TPM levels from RNA-seq data, and number 0 to 12 represent the range of TPM levels (from the lowest to highest). Groups CK1/HS1 and CK2/HS2 represent anthesis and post-anthesis stages respectively.
Alternative splicing (AS) analysis of ZmHsfs
The third generation full length transcriptome sequencing was used for AS analysis in the leaves of maize seedlings after HS treatment. The results showed that AS events occurred only in ZmHsf04 and ZmHsf17 in the form of intron retention, causing ZmHsf04-II and ZmHsf17-II with the retention lengths of intron 229 bp and 141 bp, respectively. New splicing sites were identified in the first intron both ZmHsf04 and ZmHsf17 genes. The sequences of retained regions contain several new ‘TAG’ and ‘TAA’ stop codons (Fig. 6A,B). The intron retention probably generated small peptides which retain only a truncated DBD domain, because the first intron was between the third α-helical and β-fold structures of the DBD domain. Compared the C-terminal amino acid sequences of ZmHsf04-II and ZmHsf17-II hypothetical small peptides, an additional Leucine-rich hydrophobic motif was found in ZmHsf04-II but not in ZmHsf17-II (Fig. S2)

The structures of two transcripts from ZmHsf04 and ZmHsf17 are analyzed based on the alternative splice events after HS treatment. (A,B) sequence alignment of two transcripts from ZmHsf04 and ZmHsf17 respectively, the premature termination codons within the partial introns, (C,D) semi quantitative RT-PCR after HS treatment at three development stages, (E,F) quantitative RT-PCR after HS treatment at three development stages. The expression levels of CK0, CK1 and CK2 are set as ‘1’ respectively. CK0 and HS0: seedling stage, CK1 and HS1: anthesis stage, CK2 and HS2: post-anthesis stage.
The AS events in leaves were confirmed at seedling, anthesis and post-anthesis stages by semi-quantity RT-PCR and quantity RT-PCR experiments. Specific primers were designed in both ends of ZmHsf04 and ZmHsf17 genes and used with semi-quantitative PCR assays to assess the response of different isoforms to HS treatment. Two pairs of primers which can amplify two different isoforms respectively were designed and used for quantitative RT-PCR. Both isoforms of ZmHsf04 and ZmHsf17 were significantly induced by HS treatment (Fig. 6C,D), and the up-regulation were most significant at the seedling stages (Fig. 6E,F). The ZmHsf04 expression have a far higher increase than ZmHsf17 after HS treatment. The expression increasing in ZmHsf04-I were higher than that in ZmHsf04-II (Fig. 6C,E). Differently, the ZmHsf17-I expression decreased after HS treatment at post-anthesis stage, and the ZmHsf17-II expressions were up-regulated slightly after HS treatment at both anthesis and post-anthesis stages (Fig. 6D,F).
Discussion
Since three Hsfs were identified from HS treated tomato cells, the Hsf family genes has been studied as one of key regulators of plant thermotolerance43. Sessile plants possess more Hsf family members than other eukaryotes. With the development of sequencing technology, most Hsfs have been identified, such as, 21 AtHsfs in Arabidopsis11,44, 26 SlyHsfs in tomato23, 25 OsHsfs in rice45, 25 SbHsfs in sorghum46 and 82 TaHsfs in wheat24. All genes identified by HMM research or BLASTP possess canonical DBD domain and OD domains. Recently the maize B73 inbred line was genome-sequenced and assembled using single-molecule PacBio sequencing technology, making it more precise for gene annotation and discovery of new genes30.
Base on homology comparison with rice, 31 ZmHsfs were identified from the maize ‘B73RefGen_V4′ and assigned into three class A, B and C, which contain 7, 3 and 2 subclasses, respectively. Similar to rice and sorghum bicolor, class A of maize has more members than the other classes, and subclass A2, with 5 members, is the most abundant subclass. In wheat, 18 members of TaHsfA2 were recently identified24. Differently, Arabidopsis has only one HsfA2 gene, but four HsfA1 genes44. As the dominate Hsf, HsfA2 is strongly induced by HS and has been improved to be a key regulator of abiotic stress response by recent research24,45,46. Speaking from structure (Fig. 2), all members of the subclass A2 possess the AHA motif (motif 7), which has a transcriptional activation function. Meanwhile the AHA motifs are different in subclass A1, A4 and A6. Subcellular localization predictions indicate that ZmHsf23 and ZmHsf10 of subclass A6 are not localized to the nucleus. Previously, AtHsfA6a has been simultaneously found in both cytoplasm and nucleus and appeared to be translocated into the nucleus after salt stress47.
Compared to the model dicot Arabidopsis, class B and C were relatively more abundant in monocots24,33,45,46,48, this was verified in maize in this study (Fig. 3). Curiously, in the maize B73 genome, two Hsfs are aimed to subclasses A1, A4d, A9, B1, B2b and C1a, respectively, but there is only one member of these subclasses in both rice and sorghum bicolor (Fig. 3). These perhaps because the maize genome underwent a whole genome duplication event about 5 to 12 mya and has continuously expanded over the last 3 million years via long terminal repeat retrotransposons38. Similarly, in the wheat genome, duplication events may be one important means of expanding Hsf family37. Gene loss and retention after duplication events are presumably the results of enhanced resistance to environmental stress49, redundant duplicates of ZmHsfs in subclasses A1 and A2 may be associated with the thermotolerance-regulating. Our previous research found that obvious differences existed in the regulating function and expression patterns of two highly homologous HsfA1 genes, ZmHsf1250 and ZmHsf0640.
The flowering, pollination and grain filling of maize are all highly susceptible to high temperature51. Transcriptome analysis of maize leaves under HS in both anthesis and post-anthesis stages showed that the expression levels of some ZmHsfs changed significantly. The three most distinctly up-regulated subclasses were A2, A6 and B2, similarly in wheat24. These can be proved by the close phylogenetic relationship of subclass A2 and A6 in our phylogenetic tree (Fig. 3). As one of key regulators for heat stress response, the up-regulation of ZmHsfA2 was expected. Subclass A2 were also induced by HS in rice and sorghum45,46. The ZmHsf05 of subclass A2 was induced by HS treatment in maize, and rescued the reduced thermotolerance of the athsfa2 mutant in Arabidopsis. In ZmHsf05-overexpressing lines of Arabidopsis, the basal and acquired thermotolerance of plants were all enhanced41. Up-regulations of the four ZmHsfs of subclass B2 were observed in maize after heat treatment, similar to the expression of both SlyHsf03 and SlyHsf10 of subclass B2 in tomato, which increased dramatically after heat treatment for 1–2 h23. The ZmHsf12 and ZmHsf06 of subclass A1 were down-regulated in both anthesis and post-anthesis stages, differing from previous studies in maize seedlings33. The expression levels of TaHsfA1a was down-regulated after heat or drought stress in wheat seedlings too24. In Arabidopsis, HsfA1s, act as transcriptional activator, can be induced at the early HS response stage25,52. Those suggest that Hsf can demonstrate different expression responses during different developmental stages. In our experiment, ZmHsf17 of subclass A2 and ZmHsf13 of subclass C2 were observed to have different responses to HS at anthesis and post-anthesis stages, both transcriptional levels of two genes increased in anthesis stage and decreased in post-anthesis stage under HS treatment. Different express patterns suggest diversity and complexity of regulating roles.
Almost all Hsfs have the conserved DBD domain which containing two independent exons separated by one intron53. The conserved intron locates in the end of the central helix-turn-helix motif (H2-T-H3) which is necessary for specific recognition of the palindromic HSEs in plant53. AS of pre-messenger RNA is the important post-transcriptional regulation mechanism that often widely happen during various developmental stages of plants54. Previous studies verified that intron retention is the predominant form of AS in plants and exon skipping is the most frequent AS event in mammals55. More than 6000 Arabidopsis genes and 1000 grape genes have displayed AS patterns under salt and high-temperature stress, respectively56,57. In Arabidopsis, three spliced variants of AtHsfA2 were identified after different HS treatment. Except for full spliced AtHsfA2-I, AtHsfA2-II and AtHsfA2-III have different C-terminal amino acid sequences due to different parts of the first intron were retained58,59. AtHsfA2-II without Leucine-rich hydrophobic motif (LRM) in C-terminal was degraded by nonsense-mediated mRNA decay (NMD)58. However, AtHsfA2-III with the LRM in C-terminal could be translated into a small truncated AtHsfA2 protein at HS recovered stage. This “small AtHsfA2” protein was shown to bind to the TATA box-proximal clusters of HSE in the HsfA2 promoter to activate the transcription of HsfA259.
In our experiment, only ZmHsf04 and ZmHsf17 were found to have AS events after HS, in the form of intron retention. ZmHsf04-II and ZmHsf17-II retained different partial introns which containing several premature termination codons. The two truncated isoforms of ZmHsf04 and ZmHsf17 might be involved in different transcriptional regulation process. Based on the amino acid sequences, we speculated that ZmHsf17-II without the LRM may be degraded by NMD, like the AtHsfA2-II of Arabidopsis58. However, ZmHsf04-II with the LRM in C-terminal could be translated and activate the transcription of ZmHsf04-I, like the AtHsfA2-III of Arabidopsis59. In previous reports, overexpression of ZmHsf04-I in Arabidopsis up-regulated the expression of heat and other related genes, the thermotolerance and salt tolerance of transgene plants were all enhanced42. At the same time, Quantitative RT-PCR showed that the expression increase of ZmHsf04 were far higher than ZmHsf17 after HS in three development periods, the expression of ZmHsf17 were also observed in normal plants, these verify above speculation further. The full spliced HsfA2 protein contain typical complete functional domains and motifs and have been considered as the major transcriptional regulatory forms57. In rice, the full spliced OsHsfA2d-I could participated in the unfolded protein response by regulating expression of OsBiP1, but OsHsfA2d-II could not, because of its incomplete DBD domain and LRM motif60. The regulating functions of different isoforms both ZmHsf04 and ZmHsf17 need more deeply research.
Conclusion
There is remarkable functional diversity in the members of plant Hsf family after the long-term adaptation to high temperature11. We identified and classified all possible ZmHsfs and found the orthologous and paralogous genes through amino acid sequence alignment, the results were valuable for the functional study of ZmHsfs. We analyzed the expression pattern of all the ZmHsfs responsive to heat stress at anthesis and post-anthesis stages through RNA-seq and quantitative RT-PCR, and found some up-regulated Hsf genes from subclass A2, A6 and B2. Intron retention events which often occurring within DBD domain were found in ZmHsf04 and ZmHsf17 of subclass A2 through full length transcriptome sequencing. The two truncated isoforms both ZmHsf04 and ZmHsf17 have different expression patterns after HS treatment in plants of seedlings, anthesis and post-anthesis stages. We infer that different spliced variants of Hsf genes in maize may involve in different transcriptional regulation process.
Materials and Methods
Plant culture and heat stress
Maize inbred variety ‘H21(♀ Huangzao 4 × H84 ♂)’ were selected for this study. Maize inbred variety ‘H21’ is one of the main inbred lines of China and it is widely used as a parent in breeding hybrid. Inbred H21 has typical characteristics like early maturing, compact plant type and strong combining ability, meanwhile it has good drought and disease resistance61,62. The plants were grown in large pots containing nutritive soil in a greenhouse and used for RNA sequencing experiments. When the seedlings grew to two-leaves-old, some plants were subjected to HS at 42 °C for 30 min (HS0). When pollens were released, some plants were subjected to same HS as seedlings (HS1). After 3 weeks, the other plants were subjected to HS either (HS2), and the plants growing under the normal conditions of different stages were performed as CK0, CK1 and CK2. The second leaves or the flag leaves were sampled quickly and frozed in liquid nitrogen and stored until RNA extraction.
Identification and bioinformatic analysis of Hsfs in maize
The maize reference genome (Maize B73 RefGen_V4) was downloaded from the MaizeGDB database on the website ftp://ftp.ensemblgenomes.org/pub/release-41/plants/fasta/zea_mays/dna/. 47 non-redundant Hsfs amino acid sequences were downloaded from PlantTFDB and blasted against the amino acid sequences retrieved from the maize reference genome. Additionally, the Hsf-type DBD domain (Pfam: PF00447) was used as a query in BLASTP (P = 0.001) search for probable Hsf protein sequences in the maize genome reference data base. 58 maize Hsf proteins were collected after homologous comparison and HMM research. Results from the two searches described above were blasted, respectively, against the UniprotKB/Swiss-prot data base on the NCBI blastp suite and the NCBI batch CDD (https://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi). Redundant sequences were discarded. Using SMART, sequences without a DBD domain or HR-A/B domain were eliminated. Motif elicitation of Hsf proteins was completed using the MEME suite (http://meme-suite.org/tools/meme). The pI and MW of identified Hsf proteins were calculated using the Expasy website (https://web.expasy.org/compute_pi/). WoLFPSORT was used to predict subcellular localization motifs in amino acid sequences of the identified Hsf proteins (https://wolfpsort.hgc.jp/).
Multiple sequence alignment and phylogenetic tree
The amino acid sequences of Hsf proteins from Arabidopsis, Oryza sativa, Sorghum bicolor and Zea mays were examined together. MEGA 7.0 was used for multiple sequence alignment and the phylogenetic tree was constructed using the Neighbor joining method with a bootstrap value of 1000. The phylogenetic tree was modified using Figtree software. The physical location and gene duplication events were assigned based on the maize genome annotation results from the MCScan toolkit in TBtools63. MCScan toolkit was also used to analyze the collinearity of rice, sorghum and maize Hsf proteins and plot collinear genes and blocks on the chromosomes63.
RNA extraction and RNA sequencing analysis
Total RNA of leaves from different treatments were extracted and purified using the Total RNA Extractor kit and RNase-free DNase I (Sangon, China). The RNA quality and integrity were estimated with an Agilent 2100 Bioanalyzer (Agilent, USA), and the RNA quantity was measured with a NanoDrop ND-1000 spectrophotometer (Thermo scientific, USA). The cDNA libraries were constructed using 2 μg of fragmented RNA from different samples with slight modifications of previously published method24. Refer to the instruction manual of VAHTS mRNA-seq V3 library prep Kit for Illumina (Vazyme, China) for the specific construction process. The four cDNA libraries were sequenced using Hiseq XTen sequencers (Illumina, USA). A data quality assessment of the raw reads was attained with FastQC and Trimmomatic. The clean reads were mapped to the maize reference genome sequence (Maize B73 RefGen_V4), and the transcription-level expression was analyzed with HISAT, StringTie and Ballgown according to a previously reported standard process64. Gene expression levels were calculated in TPM (Transcripts per million) using StringTie and Ballgown, and the heat map of gene expression levels was plotted using TBtools.
Quantitative RT-PCR for validation of RNA-Seq
The first strand cDNA was synthesized with 1 μg RNA by reverse transcription PCR. A SYBR Premix ExTaqTM kit (Takara, Japan) was used for quantitative RT-PCR assays in the ABI 7500 (Applied Biosystem, USA) according to the manufacturer instructions. The reaction procedure included pre-denaturation at 95 °C for 10 min, followed by 40 cycles of denaturation at 95 °C for 5 s and annealing/extension at 60 °C for 40 s. Three biological replicates were performed for each group. After the reaction, the data were analyzed using the 2−ΔΔCt method65,66 and plotted with Microsoft Excel 2010. The expression level of CK1 was set as 1. For statistical analysis, each dataset was repeated at least three times. The Actin2 gene was used as an endogenous control. All of the primers used in quantitative RT-PCR are listed in Table S1.
Nanopore sequencing and alternative splicing analysis
The second leaves of two-leaf-old maize seedlings were sampled after HS for nanopore sequencing experiments. RNA extraction, cDNA library construction and long-read sequencing were performed according to the standard protocols of Oxford Nanopore Technologies (ONT)67,68. Ribosomal RNA and low-quality raw reads less than 500 bp in length were removed. Minmap2 software was used to map all full-length reads to the reference genome and remove the redundant transcript reads were removed. The alternative splicing events in both CK0 and HS0 treatments were identified using Astalavista software69. The structures of different isoforms were analyzed by sequence alignment in DNAman software. The RNA samples of maize leaves from seedling, anthesis and post-anthesis stages were used to analyze the transcriptional levels of different isoforms. The transcription abundances of the cDNA from different isoforms were tested by semi-quantity and quantity RT-PCR methods, respectively. According to the operation manual, the 2 × Taq Plus Master Mix II (Dye Plus) kits were used for semi-quantity RT-PCR experiments. The primers used in semi-quantity and quantity RT-PCR were listed in Table S2.
References
Moustafa, K. Food and sustainability challenges under climate changes. Sci. Eng. Ethics. 22, 1831–1836 (2016).
Gourdji, S. M., Sibley, A. M. & Lobell, D. B. Global crop exposure to critical high temperatures in the reproductive period: historical trends and future projections. Environ. Res. Letters 8, 024041, https://doi.org/10.1088/1748-9326/8/2/024041 (2013).
Puteh, A., Thuzar, M., Mondal, M. M. A., Abdullah, N. P. B. & Halim, M. R. A. Soybean [Glycine max (L.) Merrill] seed yield response to high temperature stress during reproductive growth stages. Aust. J. Crop Sci. 7, 1472–1479 (2013).
Murthy, S. D. S. & Prasuna, A. B. Altered thylakoid membrane photo functions under high temperature stress in wheat primary leaves. J. Phytol. Res. 19, 11–13 (2006).
Vara Prasad, P. V., Pisipati, S. R., Ristic, Z., Bukovnik, U. & Fritz, A. K. Impact of nighttime temperature on physiology and growth of spring wheat. Crop Sci. 48, 2372–2380 (2008).
Roelofs, D., Morgan, J. & Stürzenbaum, S. The significance of genome-wide transcriptional regulation in the evolution of stress tolerance. Evol. Ecol. 24, 527–539 (2010).
Lambert, S. A. et al. The human transcription factors. Cell 172, 650–665 (2018).
Jin, J. et al. PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 45, D1040–D1045 (2017).
Sorger, P. K. & Pelham, H. R. Yeast heat shock factor is an essential DNA-binding protein that exhibits temperature-dependent phosphorylation. Cell 54, 855–864 (1988).
Nover, L. et al. Arabidopsis and the heat stress transcription factor world: how many heat stress transcription factors do we need? Cell Stress Chaperon. 6, 177–189 (2001).
Scharf, K. D., Berberich, T., Ebersberger, I. & Nover, L. The plant heat stress transcription factor (Hsf) family: structure, function and evolution. Biochim. Biophys. Acta 1819, 104–119 (2012).
Liu, X. D., Liu, P. C., Santoro, N. & Thiele, D. J. Conservation of a stress response: human heat shock transcription factors functionally substitute for yeast HSF. Embo. J. 16, 6466–6477 (1997).
Damberger, F. F., Pelton, J. G., Harrison, C. J., Nelson, H. C. & Wemmer, D. E. Solution structure of the DNA-binding domain of the heat shock transcription factor determined by multidimensional heteronuclear magnetic resonance spectroscopy. Protein Sci. 3, 1806–1821 (1994).
Schultheiss, J. et al. Solution structure of the DNA-binding domain of the tomato heat-stress transcription factor HSF24. Eur. J. Biochem. 236, 911–921 (1996).
Peteranderl, R. et al. Biochemical and biophysical characterization of the trimerization domain from the heat shock transcription factor. Biochemistry 38, 3559–3569 (1999).
Sakurai, H. & Enoki, Y. Novel aspects of heat shock factors: DNA recognition, chromatin modulation and gene expression. FEBS J. 277, 4140–4149 (2010).
Guo, L. et al. Isolation of heat Shock factor HsfA1a-binding sites in vivo revealed variations of heat shock elements in Arabidopsis thaliana. Plant Cell Physiol. 49, 1306–1315 (2008).
Chan-Schaminet, K. Y., Baniwal, S. K., Bublak, D., Nover, L. & Scharf, K. D. Specific Interaction between tomato HsfA1 and HsfA2 creates hetero-oligomeric superactivator complexes for synergistic activation of heat stress gene expression. J. Biol. Chem. 284, 20848–20857 (2009).
Czarnecka-Verner, E., Yuan, C. X., Scharf, K. D., Englich, G. & Gurley, W. B. Plants contain a novel multi-member class of heat shock factors without transcriptional activator potential. Plant Mol. Biol. 43, 459–471 (2000).
Swindell, W. R., Huebner, M. & Weber, A. P. Transcriptional profiling of Arabidopsis heat shock proteins and transcription factors reveals extensive overlap between heat and non-heat stress response pathways. BMC Genomics 8, 125, https://doi.org/10.1186/1471-2164-8-125 (2007).
Zhang, H. Z. et al. Identification and expression analysis of the heat shock transcription factor (HSF) gene family in Populus trichocarpa. Plant Omics 6, 415–424 (2013).
Scarf, K. D. et al. The tomato hsf system: HsfA2 needs interaction with HsfA1 for efficient nuclear import and may be localized in cytoplasmic heat stress granules. Mol. Cell. Biol. 18, 2240–2251 (1998).
Yang, X., Zhu, W., Zhang, H., Liu, N. & Tian, S. Heat shock factors in tomatoes: genome-wide identification, phylogenetic analysis and expression profiling under development and heat stress. Peer J 4, e1961, https://doi.org/10.7717/peerj.1961 (2016).
Duan, S., Liu, B., Zhang, Y., Li, G. & Guo, X. Genome-wide identification and abiotic stress-responsive pattern of heat shock transcription factor family in Triticum aestivum L. BMC Genomics 20, 257, https://doi.org/10.1186/s12864-019-5617-1 (2019).
Charng, Y. Y. et al. A heat-inducible transcription factor, HsfA2, is required for extension of acquired thermotolerance in Arabidopsis. Plant Physiol. 143, 251–262 (2007).
Shim, D. et al. Orthologs of the class A4 heat shock transcription factor HsfA4a confer cadmium tolerance in wheat and rice. Plant Cell 21, 4031–4043 (2009).
Singh, G., Sarkar, N. K. & Grover, A. Mapping of domains of heat stress transcription factor OsHsfA6a responsible for its transactivation activity. Plant Sci. 274, 80–90 (2018).
Xue, G. P., Drenth, J. & Mcintyre, C. L. TaHsfA6f is a transcriptional activator that regulates a suite of heat stress protection genes in wheat (Triticum aestivum L.) including previously unknown Hsf targets. J. Exp. Bot. 66, 1025–1039 (2015).
Ikeda, M., Mitsuda, N. & Ohme-Takagi, M. Arabidopsis HsfB1 and HsfB2b act as repressors of the expression of heat-inducible Hsfs but positively regulate the acquired thermotolerance. Plant Physiol. 157, 1243–1254 (2011).
Ma, H. et al. CarHSFB2, a Class B heat shock transcription factor, is involved in different developmental processes and various stress responses in Chickpea (Cicer Arietinum L.). Plant Mol. Biol. Rep. 34, 1–14 (2015).
Bharti, K. et al. Tomato heat stress transcription factor HsfB1 represents a novel type of general transcription coactivator with a histone-like motif interacting with the plant CREB binding protein ortholog HAC1. Plant Cell 16, 1521–1535 (2004).
Lavania, D., Dhingra, A. & Grover, A. Analysis of transactivation potential of rice (Oryza sativa L.) heat shock factors. Planta 247, 1–10 (2018).
Lin, Y. X. et al. Genome-wide identification, classification and analysis of heat shock transcription factor family in maize. BMC Genomics 12, 76, https://doi.org/10.1186/1471-2164-12-76 (2011).
Ma, J. et al. Genome-wide analysis of HSF family transcription factors and their responses to abiotic stresses in two Chinese cabbage varieties. Acta Physiol. Plant. 36, 513–523 (2014).
Guo, M. et al. Genome-wide analysis, expression profile of heat shock factor gene family (CaHsfs) and characterisation of CaHsfA2 in pepper (Capsicum annuum L.). BMC Plant Biol. 15, 151, https://doi.org/10.1186/s12870-015-0512-7 (2015).
Liu, G. et al. Genome-wide identification and classification of HSF family in grape, and their transcriptional analysis under heat acclimation and heat stress. Hortic. Plant J. 4, 133–143 (2018).
Zhou, M. et al. Genome-wide identification, phylogenetic and expression analysis of the heat shock transcription factor family in bread wheat. BMC Genomics 20, 505, https://doi.org/10.1186/s12864-019-5876-x (2019).
Schnable, P. S. et al. The B73 maize genome: complexity, diversity, and dynamics. Science 326, 1112–1115 (2009).
Jiao, Y. et al. Improved maize reference genome with single-molecule technologies. Nature 546, 524–527 (2017).
Li, H. et al. Expression of maize heat shock transcription factor gene ZmHsf06 enhances the thermotolerance and drought-stress tolerance of transgenic Arabidopsis. Funct. Plant Biol. 42, p1080, https://doi.org/10.1071/FP15080 (2015).
Li, G. L. et al. ZmHsf05, a new heat shock transcription factor from Zea mays L. improves thermotolerance in Arabidopsis thaliana and rescues thermotolerance defects of the athsfa2 mutant. Plant sci. 283, 375–384 (2019).
Jiang, Y., Zheng, Q., Chen, L., Liang, Y. & Wu, J. Ectopic overexpression of maize heat shock transcription factor gene ZmHsf04 confers increased thermo and salt-stress tolerance in transgenic Arabidopsis. Acta Physiol. Plant. 40, 9, https://doi.org/10.1007/s11738-017-2587-2 (2018).
Scharf, K. D., Rose, S., Zott, W., Schoffl, F. & Nover, L. Three tomato genes code for heat stress transcription factors with a region of remarkable homology to the DNA binding domain of the yeast HSF. EMBO J. 9, 4495–4501 (1990).
Sultan, S., Ali, M., Nawaz, S., Ali, M. A. & Shahzad, A. Genome wide analysis of heat shock factors (HSF) gene family of Arabidopsis Thaliana. J. Biol. Agric. Healthc. 6, 69–77 (2016).
Chauhan, H., Khurana, N., Agarwal, P. & Khurana, P. Heat shock factors in rice (Oryza sativa L.): genome-wide expression analysis during reproductive development and abiotic stress. Mol. Genet. Genomics 286, 171–187 (2011).
Nagaraju, M. et al. Genome-wide scanning and characterization of Sorghum bicolor L. heat shock transcription factors. Curr. Genomics 16, 279–291 (2015).
Hwang, S. M. et al. Functional characterization of Arabidopsis HsfA6a as a heat-shock transcription factor under high salinity and dehydration conditions. Plant Cell Environ. 37, 1202–1222 (2014).
Xiang, J. et al. Heat shock factor OsHsfB2b negatively regulates drought and salt tolerance in rice. Plant Cell Rep. 32, 1795–1806 (2013).
Maere, S. et al. Modeling gene and genome duplications in eukaryotes. Proc. Natl. Acad. Sci. USA 102, 5454–5459 (2005).
Li, G. et al. Characteristics and regulating role in thermotolerance of the heat shock transcription factor ZmHsf12 from Zea mays L. J. Plant Biol. 62, 329–341 (2019).
Neiff, N., Trachsel, S., Valentinuz, O. R., Balbi, C. N. & Andrade, F. H. High temperatures around flowering in maize: effects on photosynthesis and grain yield in three genotypes. Crop Sci. 56, 2702–2712 (2016).
Liu, H. C., Liao, H. T. & Charng, Y. Y. The role of class A1 heat shock factors (HSFA1s) in response to heat and other stresses in Arabidopsis. Plant Cell Environ. 34, 738–751 (2011).
Nover, L. et al. Arabidopsis and the Hsf world: How many heat stress transcription factors do we need? Cell Stress Chapteron. 6, 177–189 (2001).
Laloum, T., Martín, G. & Duque, P. Alternative splicing control of abiotic stress responses. Trends in Plant Sci. 23, 140–150 (2018).
Barbazuk, W. B., Fu, Y. & Mcginnis, K. M. Genome-wide analyses of alternative splicing in plants: opportunities and challenges. Genome Res. 18, 1381–1392 (2008).
Feng, J., Li, J. & Gao, Z. SKIP confers osmotic tolerance during salt stress by controlling alternative gene splicing in Arabidopsis. Mol. Plant. 8, 1038–1052 (2015).
Jiang, J. et al. Integrating omics and alternative splicing reveals insights into grape response to high temperature. Plant Physiol. 173, 1502–1518 (2017).
Sugio, A., Dreos, R., Aparicio, F. & Maule, A. J. The cytosolic protein response as a subcomponent of the wider heat shock response in Arabidopsis. Plant Cell 21, 642–654 (2009).
Liu, J. et al. An auto regulatory loop controlling Arabidopsis HsfA2 expression: role of heat shock-induced alternative splicing. Plant Physiol. 162, 512–521 (2013).
Cheng, Q. et al. An alternatively spliced heat shock transcription factor, OsHSFA2dI, functions in the heat stress-induced unfolded protein response in rice. Plant Biol. 17, 419–429 (2015).
Liu, E., Shi, X., Zhu, J. & Li, C. Studies on breeding new inbred lines of maize better than “Huangzao 4”. J. Laiyang Agr. Coll. 7, 87–91 (1990).
Su, Z. et al. Evaluation of drought tolerance of commonly used maize inbred lines in China. J. Maize Sci. 5, 19–24 (2009).
Chen, C., Xia, R., Chen, H. & He, Y. TBtools, a toolkit for biologists integrating various HTS-data handling tools with a user-friendly interface. BioRxiv 289660; https://doi.org/10.1101/289660 (2018).
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T. & Salzberg, S. L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667 (2016).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔct method. Methods 25, 402–408 (2001).
Schmittgen, T. D. & Livak, K. J. Analyzing real-time PCR data by the comparative CT method. Nat Protoc 3, 1101–1108 (2008).
Volden, R. et al. Improving nanopore read accuracy with the R2C2 method enables the sequencing of highly multiplexed full-length single-cell cDNA. Proc. Natl. Acad. Sci. USA 115, 9726–9731 (2018).
Fleming, M. B. et al. Exploring the fate of mRNA in aging Seeds: Protection, Destruction, or Slow Decay? J. Exp. Bot. 69, 4309–4321 (2018).
Foissac, S. & Sammeth, M. Analysis of alternative splicing events in custom gene datasets by Astalavista. Methods Mol. Biol. 1269, 379–392 (2015).
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2018YFD0300504), Natural Science Foundation of Hebei Province (C2017301065), the Technological Innovation Basal Research Fund of Hebei Academy of Agriculture and Forestry Sciences (2018110101) and the Technological Innovation Project of Modern Agriculture of Hebei Province (494-0402-JBN-C7GQ).
Author information
Authors and Affiliations
Contributions
X.G. and D.H. conceived the idea and supervised the study. C.F. and S.D. performed the qRT-PCR and semi qRT-PCR. H.Z. and G.L. analyzed the data and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, H., Li, G., Fu, C. et al. Genome-wide identification, transcriptome analysis and alternative splicing events of Hsf family genes in maize. Sci Rep 10, 8073 (2020). https://doi.org/10.1038/s41598-020-65068-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-65068-z
This article is cited by
-
The complex transcriptional regulation of heat stress response in maize
Stress Biology (2024)
-
Genome-wide identification and comprehensive analysis heat shock transcription factor (Hsf) members in asparagus (Asparagus officinalis) at the seeding stage under abiotic stresses
Scientific Reports (2023)
-
Heat shock transcription factor (Hsf) gene family in common bean (Phaseolus vulgaris): genome-wide identification, phylogeny, evolutionary expansion and expression analyses at the sprout stage under abiotic stress
BMC Plant Biology (2022)
-
Mapping responsive genomic elements to heat stress in a maize diversity panel
Genome Biology (2022)
-
Identification and Transcriptional Analysis of Zinc Finger-Homeodomain (ZF-HD) Family Genes in Cucumber
Biochemical Genetics (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
