
Abstract
Heat shock transcription factors (HSFs) are central elements in the regulatory network that controls plant heat stress response. They are involved in multiple transcriptional regulatory pathways and play important roles in heat stress signaling and responses to a variety of other stresses. We identified 41 members of the HSF gene family in moso bamboo, which were distributed non-uniformly across its 19 chromosomes. Phylogenetic analysis showed that the moso bamboo HSF genes could be divided into three major subfamilies; HSFs from the same subfamily shared relatively conserved gene structures and sequences and encoded similar amino acids. All HSF genes contained HSF signature domains. Subcellular localization prediction indicated that about 80% of the HSF proteins were located in the nucleus, consistent with the results of GO enrichment analysis. A large number of stress response–associated cis-regulatory elements were identified in the HSF upstream promoter sequences. Synteny analysis indicated that the HSFs in the moso bamboo genome had greater collinearity with those of rice and maize than with those of Arabidopsis and pepper. Numerous segmental duplicates were found in the moso bamboo HSF gene family. Transcriptome data indicated that the expression of a number of PeHsfs differed in response to exogenous gibberellin (GA) and naphthalene acetic acid (NAA). A number of HSF genes were highly expressed in the panicles and in young shoots, suggesting that they may have functions in reproductive growth and the early development of rapidly-growing shoots. This study provides fundamental information on members of the bamboo HSF gene family and lays a foundation for further study of their biological functions in the regulation of plant responses to adversity.
Similar content being viewed by others


Introduction
Plant growth is influenced by a variety of adverse environmental factors such as high temperature, salt stress, drought, and other abiotic stresses, as well as by biotic stresses such as diseases and pests. High temperature and drought are particularly important abiotic stresses that can promote chlorophyll degradation, damage chloroplast membrane mechanisms, and reduce photosynthetic efficiency1, thereby profoundly restricting plant growth, development, and function. Because of their sessile nature, plants cannot actively escape from stress and must rely on physiological and biochemical mechanisms to survive external stresses2. Therefore, they have evolved a series of complex and effective strategies to maintain normal physiology, metabolism, and growth under adverse conditions3. Transcription factor (TF) gene expression plays an important role in plant stress resistance4. For example, ABRE Binding Factor (ABF) and MYC TFs are directly involved in abscisic acid (ABA) and jasmonate (JA) signaling pathways5, and heat shock transcription factors (HSFs) are involved in the regulation of reactive oxygen species (ROS), calcium signaling, and other signal transduction pathways6. HSFs, a class of transcription factors widely found in eukaryotes, also have important roles in the receipt and transmission of signals, the recognition of heat shock elements (HSEs) and regulation of downstream genes, the stimulation of plant stress responses, and the promotion of heat tolerance7.
HSF transcription factors contain five basic functional domains: an N-terminal DNA-binding domain (DBD), an oligomerization domain (OD or HR-A/B), a nuclear localization signal (NLS), a nuclear export signal (NES), and in some cases, a C-terminal transcriptional activation domain (CTAD)8. The highly conserved DNA-binding domain (also known as the HSF domain) consists of three α-helix bundles and four strands of reverse-parallel β-folded layers that form a tight sphere. This structure ensures the specific binding of HSFs to HSEs in target gene promoters, thereby regulating the transcription and expression of heat shock genes9. The OD consists of two hydrophobic 7-peptide repeat regions, HR-A and HR-B, and HSFs are divided into three subfamilies (HsfA–C) based on differences in the number of amino acids inserted between HR-A and HR-B. HsfAs and HsfCs have 21 and 7 amino acid residues between HR-A and HR-B, respectively, whereas HsfBs have none10,11. The nuclear localization signal (NLS) is composed of basic amino acids and directs the transport of HSFs into the nucleus12, whereas the leucine-rich nuclear export signal (NES) facilitates their export from the nucleus to the cytoplasm13. The CTAD is the least conserved region and contains AHA motifs that consist of aromatic, large hydrophobic, and acidic amino acid residues. The AHA sequence is specific to the class A subfamily, and it is generally required for transcriptional activation. HsfBs and HsfCs do not contain AHA motifs and thus lack a transcriptional activation function14.
The first plant HSF gene was cloned from tomato in 1990. As more completed plant genome sequences became available, HSF family members were identified in Arabidopsis (21 HSF genes)10, tomato (26)15, cabbage (52)16, pepper (25)17, poplar (31)18, maize (25)19, and rice (25)20. Detailed functional studies of individual HSF subfamilies have been performed in Arabidopsis, tomato, and other model plants, and experiments have confirmed that HSFs have important roles in plant responses to heat and other abiotic stresses. Arabidopsis HsfAs were shown to be transcriptional activators, and AtHsfA1a regulates the synthesis of heat shock proteins (Hsps) during the heat-response phase to minimize plant growth damage at high temperatures21. The overexpression of Arabidopsis AtHsfA222 and tomato SlHsfA323 significantly increased both basal and acquired heat tolerance. The expression of AtHsfA6a increased nearly 150-fold under salt stress compared with control conditions24. By contrast, HsfB1 and HsfB2b from subfamily B were shown to be transcriptional repressors, and rice OsHsfB2b was found to significantly reduce plant salt tolerance after NaCl treatment20,25. Under drought stress, plants with upregulated expression of AtHsfA1b showed significant increases in yield and harvest index. Plants that overexpressed HsfA1b also had enhanced resistance to pathogenic bacteria, indicating that this gene is involved in the regulation of both abiotic and biotic stress resistance26.
Moso bamboo (Phyllostachys edulis) is a non-timber forestry species from the subfamily Bambusoideae of the family Poaceae and is native to China27. It has a wide distribution area and a high economic value, and it plays an important role in soil and water conservation, soil carbon sequestration and oxygen dynamics, and climate regulation28. However, water availability in moso bamboo forests is entirely dependent on natural precipitation, and its two most important water-demanding growth stages, ‘asparagus gestation’ in the autumn and ‘asparagus emergence’ in the spring, are most sensitive to heat and drought stress28,29. High temperatures and drought not only damage moso bamboo growth and development, thereby reducing the yield of bamboo shoots, but also reduce the carbon stocks of moso bamboo30. The intensification of global climate change is predicted to significantly alter the level and quality of ecological services to forests31, which will aggravate the effects of water stress on moso bamboo and significantly limit its yield.
To date, there have been no studies of the HSF transcription factor family in moso bamboo. Here, we used a bioinformatics approach to identify and characterize HSF genes and their encoded proteins in the moso bamboo genome32,33; we analyzed their evolutionary relationships, conserved domains, gene structures, promoter elements, duplication patterns, tissue-specific expression, and responses to external hormone treatments. Our results lay the groundwork for further study of heat stress response mechanisms in bamboo.
Materials and methods
Identification and sequence analysis of HSF proteins from moso bamboo
Genomic data from moso bamboo were downloaded from the P. edulis genome database (http://parrot.genomics.cn/gigadb/pub/10.5524/100001_101000/100,498/assembly_fasta/Bamboo.HIC.genome.fasta.gz). A hidden Markov model of the HSF DBD (PF00447) was obtained from the Pfam database (http://pfam.xfam.org/) and used as the seed model for an HMMER3 search (http://hmmer.janelia.org/) of the downloaded bamboo protein sequence data (E ≤ 10−20)34, and redundant genes were removed to produce a set of preliminary HSF candidate sequences. To verify that these candidates were HSFs, we used the normal mode of SMART (http://smart.embl-heidelberg.de/)35 sequence analysis with default search parameters and a Batch search (E < 0.001) of the Pfam (http://pfam.xfam.org)36 database to filter out sequences that lacked complete HSF_DNA-bind (PF00447.17) domains. The confirmed HSF genes were renamed according to their positions on the moso bamboo chromosomes.
Subcellular localization predictions were generated using CELLO with default parameters (http://cello.life.nctu.edu.tw/)37, and the ExPASy ProtParam tool (https://web.expasy.org/protparam/)38 was used with default parameters to predict protein physicochemical parameters such as molecular weight (MW) and isoelectric point (pI).
Sequence alignment and phylogenetic tree construction
Whole genome information for Arabidopsis and rice was downloaded from the TAIR10 database (http://www.arabidopsis.org/index.jsp) and the Rice Genome Annotation Project database (http://rice.plantbiology.msu.edu). Maize and pepper genomic data were downloaded from the Ensembl database (http://asia.ensembl.org/index.html). Twenty-one Arabidopsis HSF proteins and 25 rice HSF proteins were identified from HMMER3 searches of the corresponding local protein databases34. The Arabidopsis and rice HSF sequences were combined with those from moso bamboo, a multiple protein sequence alignment was produced with ClustalX 2.0 (http://www.clustal.org/clustal2/). The comparison parameter is the multiple comparison mode (other parameters are default), and the resulting alignment was used to construct a maximum likelihood (ML) phylogenetic tree in MEGA 7.0 with 1000 bootstrap replicates39. Intraspecific classification of the moso bamboo HSF sequences was based on this interspecific phylogenetic tree.
The amino acid sequences of conserved domains were compared and edited using Jalview software (V2.10.5) (http://www.jalview.org/)40, and the Jalview output was submitted to JPred (http://www.compbio.dundee.ac.uk/jabaws) for protein secondary structure prediction using default parameters41.
Gene structures, motif identification, and conserved domains
The intron–exon distributions of the moso bamboo HSF genes were obtained using GFF annotation files from the moso bamboo genome. Conserved amino acid sequences of HSF proteins were analyzed using the online MEME tool (http://meme-suite.org/)42. MEME analysis parameters included a minimum width ≥ 6, a maximum width of 50, and a motif number of 10; all other parameters were set to default values. Conserved domains in the HSFs were predicted using the NCBI Conserved Domain Database (https://www.ncbi.nlm.nih.gov/cdd/) (E-value < 0.001, other parameters set to defaults), and DOG 2.0 (http://dog.biocuckoo.org) was used for protein structure visualization with default parameters43.
Chromosomal locations, genomic duplications, and Ka/Ks ratios
Chromosome lengths and gene locations were obtained from the moso genome annotation file, and MG2C v.2 (http://mg2c.iask.in/mg2c_v2.0/) was used to visualize the gene locations on chromosomes44. The moso bamboo protein sequences were aligned to one another or to the protein sequences from Arabidopsis, rice, maize, or pepper using TBtools software45. MCScanX46 was used with default parameters to identify gene duplication events and syntenic relationships among the HSF proteins, and the results were visualized using Circos and Dual Synteny Plot in TBtools45.
For Ka/Ks analysis, thirteen homologous gene pairs were identified by BLASTn using two criteria: (1) > 75% sequence similarity and (2) an alignable region > 75% of the length of the longer sequence47. KaKs_Calculator2.0 was used to calculate the synonymous substitution rate (Ks), nonsynonymous substitution rate (Ka), and Ka/Ks ratio between homologous gene pairs48. Evolutionary divergence times within the bamboo HSF gene family were calculated using the bamboo-specific divergence time formula T = Ks/2λ (where λ = 6.5 × 10−9)32.
Identification of cis-acting elements
PlantCARE (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/) was used to identify cis-acting elements in the 1,500-bp promoter region upstream of each gene’s transcription start site, and the results were visualized using TBtools49.
Expression analysis of the HSF genes
Replicated transcriptomic data from different plant organs (roots, rhizomes, panicles, and leaves), bamboo shoots at different germination stages (20, 50, and 100 cm), and seedling root tissues treated with 5 μM gibberellic acid (GA) or 5 μM naphthalene acetic acid (NAA) were obtained from the NCBI Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra) and the European Nucleotide Archive (https://www.ebi.ac.uk/ena/browser/home). In total, 31 transcriptomic datasets were downloaded (accession numbers SRP119924, SRP012682, SRP109631, ERR105067, ERR105068, ERR105069, ERR105070, ERR105071, ERR105072, ERR105073, ERR105074, ERR105075, and ERR105076.) Transcriptome data in the form of transcripts per million reads (TPM) were log10-transformed and imported into TBtools, where Amazing Heatmap was used to generate expression heatmaps.
Three-dimensional (3D) structural modeling of HSF family proteins
The PDB database (http://www.rcsb.org/) was used to retrieve protein models homologous to those of the HSF proteins. Swiss Model (https://www.swissmodel.expasy.org/) was then used with default parameters to predict the protein tertiary structures by homology modeling, and the quality of the resulting models was assessed using SAVES v.5.0 (http://servicesn.mbi.ucla.edu/SAVES/).
Protein interaction network predictions and GO enrichment analyses
The HSF protein sequences were uploaded to the STRING database (https://string-db.org/) for node comparison, and relationships among important proteins were predicted based on rice protein interactions. Cytoscape (V3.7.1) was used to visualize the resulting network50.
GOATOOLS (http://github.com/tanghaibao/GOatools)51 was used to assign GO annotations to HSFs, and Fisher's exact test was used to identify biological functions enriched in the PeHsfs relative to the full GO database. A Bonferroni52 multiple testing correction was used to minimize false positives, and functions were considered to be significantly enriched when their Bonferroni-corrected P-values (Padjust) were < 0.05.
Results
Identification of HSF genes in moso bamboo
Forty-four putative HSF candidate genes were obtained from an HMMER3 search of the bamboo protein database using the plant HSF-type DBD model (Pfam PF00447) with an E-value threshold of ≤ 10−20. We removed redundant genes and verified the presence of conserved domains and motifs to arrive at a final set of 41 HSF family members. The genes were renamed PeHsf01–PeHsf41 based on their positions on the chromosomal scaffolds (Table 1).
The predicted physicochemical properties of the amino acid sequences showed that the 41 HSF genes encoded proteins containing 159 (PeHsf03) to 512 (PeHsf30) amino acids and their molecular weights ranged from 17.49 (PeHsf03) to 56.33 kDa (PeHsf32). Approximately 90% of the HSF proteins had molecular weights of 30–50 kDa. Their predicted isoelectric points (pI) ranged from 4.72 (PeHsf04) to 9.31 (PeHsf39). Instability index calculations predicted that 39 (95%) of the HSF proteins were stable in vitro, with the exceptions of PeHsf05 and PeHsf21, which had instability indices of 32.96 and 37.27, respectively. Aliphatic amino acid indices showed that the thermal stability of the proteins ranged from 57.01 to 83.25, indicating that differences in their thermal stability were relatively minor. The grand average of hydropathicity (GRAVY) scores of all HSF proteins were negative, indicating that they were hydrophilic proteins. CELLO subcellular localization predictions suggested that about 80% of the HSF proteins were located in the nucleus.
Phylogenetic analysis and sequence alignment of the PeHsf proteins
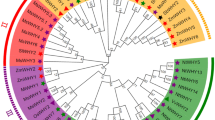
To clarify the evolutionary relationships among the HSF proteins, amino acid sequences of 41 moso bamboo HSFs, 25 rice HSFs, and 21 Arabidopsis HSFs were used to construct an ML phylogenetic tree53 (Fig. 1 and Supplementary Table S1). Based on well-established Arabidopsis and rice HSF family classifications54, the HSF proteins were divided into three major subfamilies, HsfA (blue), HsfB (green), and HsfC (yellow). The HsfA subfamily was the largest, with 48 members across the three species, whereas the HsfC subfamily was the smallest, with 12 members. There was only one Arabidopsis HsfC family member (AtHsfC1), suggesting that the C subfamily had expanded in monocots. The interspecific phylogenetic tree indicated that the PeHsfs included members of all three subfamilies: 20 HsfAs, 14 HsfBs, and 7 HsfCs.

Phylogenetic analysis of full-length HSF protein sequences from Phyllostachys edulis (Pe, bamboo), Arabidopsis thaliana (At, Arabidopsis), and Oryza sativa (Os, rice). ClustalX was used to build a multiple sequence alignment, and MEGA7.0 (https://www.megasoftware.net/) was used to construct a maximum likelihood (ML) phylogenetic tree with 1000 bootstrap replicates. Blue, red, and green circles indicate bamboo, Arabidopsis, and rice sequences, respectively. Evolview (https://evolgenius.info//evolview-v2/#login) online URL for beautification.
A multiple sequence alignment of the 41 PeHsf proteins was generated to investigate the presence and locations of conserved protein domains. All HSF family members contained a highly conserved DBD at the amino terminus that consisted of approximately 100 amino acids (Fig. 2). The protein secondary structure was predicted to contain three α-helix bundles (α1–α3) and four reverse-parallel β-folds (β1–β4). The DBD domain can specifically recognize and precisely localize heat stress elements, and similar phenomena are found in other plants.

Multiple sequence alignment of the DNA-binding domains (DBDs) of HSF proteins from moso bamboo. Secondary structure elements of the DBDs (α1-β1-β2-α2-α3-β3-β4) based on JNet structure predictions are shown above the alignment (see Methods). α-helices are indicated by red bars, and β-folds are indicated by green arrows. The amino acid sequences of conserved domains were compared and edited using Jalview software (V2.10.5) (http://www.jalview.org/)2, and the Jalview output was submitted to JPred (http://www.compbio.dundee.ac.uk/jabaws) for protein secondary structure prediction using default parameters(Supplementary Table S2) .
Gene structures, motif identification, and conserved domain analysis
The number of introns in each moso bamboo HSF gene ranged from one to three (Fig. 3). Approximately 75% of the genes contained a single intron, whereas six genes contained two introns and four contained three introns. With the exception of PeHsf31, all HsfCs contained only one intron. Structures of HSF genes from the same subfamily were generally similar.

Intron–exon structure of the HSF genes in moso bamboo. Green boxes represent exons (CDS), black lines represent introns, and light blue boxes represent 5′ and 3′ untranslated regions. Visualized by TBtools (v1.0697): Visualize Gene Structure (https://github.com/CJ-Chen/TBtools/releases).
We identified up to 10 highly conserved motifs in each HSF protein using MEME (Fig. 4). All HSFs contained motifs 1–3, which constituted the most highly conserved portions of the DBD. The relative positions of the motifs were similar for most sequences, with the exception of the HsfB PeHsf03, which lacked motif 2. In addition, both class B and C subfamily members did not contain motif 6, which is associated with transcriptional activation.

Distribution of conserved motifs of HSF proteins from moso bamboo. The scale bar at the bottom indicates the protein lengths, and sequence logos for each conserved motif are shown at right. TBtools (v1.0697) (https://github.com/CJ-Chen/TBtools/releases): Visualize MEME/MAST Motif pattern for visualization. The XML file obtained by the MEME online tool is obtained by visualizing the Batch MEME Motifs Viz plugin in TBtools (v1.0697).
The structures of the PeHsf proteins were further characterized using the NCBI Conserved Domain Database (Fig. 5). All PeHsf proteins contained highly conserved DBDs at the N terminus between amino acids 7 and 155, the presence of which served as confirmation of their identity as HSF proteins. Some proteins contained additional conserved domains. For example, PeHsf10, PeHsf27, and PeHsf41 contained a bZIP-like transcription factor domain, and PeHsf17 and PeHsf36 contained a PRK10246 domain. Therefore, multiple functions may have evolved in some members of the moso bamboo HSF family.

Conserved domain predictions for 41 PeHsf proteins. The gray bars represent the length of each protein sequence, and conserved domains are represented by colored boxes. DOG 2.0 (http://dog.biocuckoo.org) was used for protein structure visualization with default parameters.
Chromosomal distribution and synteny analysis of the PeHsf genes
The PeHsf genes were non-uniformly distributed across the 19 chromosome scaffolds of moso bamboo (Fig. 6). The largest number were found on scaffold 21 (5), followed by scaffolds 14, 15, and 16 (4), scaffolds 17, 18, and 20 (3), and scaffolds 3, 13, and 22 (2). All other chromosomes contained a single PeHsf gene. Small gene clusters were found on scaffolds 3 and 18, based on the definition of gene clusters55.

The distribution of PeHsf genes on the moso bamboo chromosome scaffolds. MG2C v.2 (http://mg2c.iask.in/mg2c_v2.0/) was used to visualize the gene locations on chromosomes.
Gene duplication events are prevalent in all species; they can give rise to new functional genes and drive species evolution56. We therefore used MCScanX genome synteny analysis to explore duplications within the moso bamboo HSF gene family (Fig. 7). Twenty-seven gene pairs appeared to have arisen from segmental duplications, and only PeHsf07 and PeHsf31 had no duplicates in the genome. Previous studies have shown that two or more highly similar genes located in close proximity to one another are likely to be tandem duplicates57. As shown in Fig. 6, two such pairs, PeHsf01–PeHsf02 and PeHsf28–PeHsf29, were present on scaffolds 3 and 18, respectively.

Chromosomal distribution and interchromosomal relationships of PeHsf genes. Gray lines indicate syntenic blocks within the moso bamboo genome, and red lines represent duplicate PeHsf gene pairs. Visualization using Circos in TBtools (v1.0697) (https://github.com/CJ-Chen/TBtools/releases).
To further investigate gene duplications in the HSF gene family, we performed genome-to-genome synteny analysis between moso bamboo and four representative plants (Supplementary Table S3). These included two dicots, Arabidopsis and Capsicum annuum (pepper) (Fig. 8a), and two monocots, O. sativa (rice) and Zea mays (maize) (Fig. 8b). Four, 3, 58, and 52 moso bamboo HSF genes were syntenic with those of Arabidopsis, pepper, rice, and maize, respectively. Furthermore, the rice HSF genes all had corresponding orthologs in moso bamboo, and most of them had more than two orthologs, suggesting that moso bamboo has undergone additional whole-genome duplication events during its evolution.

Synteny analysis of the moso bamboo genome with two monocot (a) and two dicot (b) plant genomes. The gray lines represent aligned blocks between the paired genomes, and the red lines indicate syntenic HSF gene pairs. Visualization was performed using Dual Synteny Plot in TBtools (v1.0697).
To investigate evolutionary constraints and selection pressures on the PeHsf genes, we calculated Ka, Ks, and Ka/Ks for 13 homologous PeHsf gene pairs (Table 2). The synonymous substitution rate (Ks) represents the background base substitution rate, and Ks values can therefore be used to predict the timing of whole-genome duplication events. The Ks values of the PeHsf gene pairs ranged from 0.1023 to 1.0833, indicating that a large-scale PeHsf gene duplication event occurred as early as 83.33 million years ago (MYA) and as recently as 7.86 MYA. The Ka/Ks values of the gene pairs were all less than 1.0, and these genes may have undergone strong purifying selection during evolution.
Promoter cis-element analysis of PeHsf genes
cis-acting elements are non-coding DNA sequences in gene promoters that regulate the transcription of their associated genes. We identified ten cis-acting elements in the 1500 base pairs upstream of the PeHsf genes using PlantCARE software (Fig. 9). Most were stress-response elements, and all promoters except that of PeHsf09 contained at least one MYB drought stress response element. The next most common element was the ABRE element, which is involved in abscisic acid responses. Other elements present in the PeHsf promoters included the MBS element (involved in drought, high salt, and low temperature responses), the TC-rich repeat (involved in defense and stress responses), the SP1 light response element, the P-box and TGA gibberellin response elements, the TCA salicylic acid response element, and the CGTCA motif (involved in methyl jasmonate response). These results suggest that the expression of moso bamboo HSF genes is regulated by cis-elements associated with plant developmental processes and abiotic stress responses.

Diagram of cis-acting elements identified in the 1,500-bp upstream promoter regions of the PeHsf genes. Rectangular boxes of different colors represent individual cis-acting elements, and some elements overlap with one another. PlantCARE (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/) was used to identify cis-acting elements in the 1500-bp promoter region upstream of each gene’s transcription start site, and the results were visualized using TBtools (v1.0697).
Transcription profiles of the PeHsf genes
We used published transcriptome data to investigate the expression patterns of the PeHsf genes (1) in seedling roots treated with gibberellic acid (GA) or naphthalene acetic acid (NAA), (2) in roots, rhizomes, panicles, and leaves, and (3) in young shoots of different heights (20, 50, and 100 cm). In (Fig. 10 and Supplementary Table S4), blue indicates low transcript abundance and red indicates high transcript abundance. Under GA treatment (Fig. 10a), the expression of PeHsf17, PeHsf24, PeHsf26, PeHsf06, PeHsf22, PeHsf33, PeHsf20, PeHsf15, PeHsf08, and PeHsf36 was downregulated compared with the control, indicating that GA repressed the expression of these genes. By contrast, the expression of PeHsf27, PeHsf01, and PeHsf31 was elevated under GA treatment but reduced under NAA treatment (Fig. 10b), indicating that the root expression of individual PeHsf genes differed between the hormone treatments. Similarly, PeHsf expression differed markedly among plant organs (Fig. 10c). In general, more than half of the genes were highly expressed in panicles, and four genes (PeHsf27, PeHsf11, PeHsf10, and PeHsf41) had expression levels greater than 100 TPM in this organ. Seven genes (PeHsf33, PeHsf13, PeHsf03, PeHsf35, PeHsf09, PeHsf16, and PeHsf29) were highly expressed only in roots, suggesting that they may participate in the development and/or function of bamboo roots. PeHsf36 and PeHsf30 were highly expressed only in leaves, and a large proportion of PeHsf genes were either not expressed or expressed only at low levels in the rhizome. Expression analysis of bamboo shoots at different germination stages (Fig. 10d) showed that many PeHsfs were expressed highly in 20-cm shoots, at intermediate levels in 50-cm shoots, and at the lowest level in 100-cm shoots. In general, these PeHsfs were specifically expressed during rapid shoot growth, suggesting that they may have important functions during this time.

Heat maps of PeHsf gene expression (log10-transformed TPM values) in multiple tissues and in response to hormone treatments. (a) GA treatment. (b) NAA treatment. (c) Roots, rhizomes, panicles, and leaves. (d) Young bamboo shoots of different heights. CK, control group. Each tissue or treatment was replicated three times and its gene expression levels averaged. The relative expression levels are depicted according to the color scale, where a change from blue to red indicates transcript abundance from low to high. Amazing Heatmap in TBtools (v1.0697) is used to generate expression heatmaps.
GO enrichment analysis of the PeHsf proteins
Plants have evolved complex mechanisms to perceive and respond to biotic and abiotic stresses, and HSFs are important components of these defense systems2. To further investigate the biological functions of the moso bamboo HSFs, we performed gene ontology (GO) annotation and enrichment analysis of the 41 PeHsf proteins (Fig. 11 and Supplementary Table S5). Eight molecular function, one cellular component, and eleven biological process GO terms were enriched in the PeHsfs relative to the complete GO database. Thirty-six proteins were annotated with the enriched GO term heat response, which had an enrichment factor of 0.09, confirming that heat response is a primary function of the PeHsf proteins. GO enrichment results suggested that PeHsf transcription factors were also involved in macromolecule biosynthesis, RNA biosynthesis, nitrogen compound metabolism, cellular biosynthetic processes, primary metabolic processes, and the regulation of RNA metabolic processes. The largest number of genes (37) were associated with the term “regulation of cellular macromolecule biosynthetic process”.

GO enrichment analysis of the PeHsf proteins relative to the GO database. The horizontal axis indicates the enrichment factor, and the size of the circle indicates the number of genes annotated with a given GO term. GOATOOLS (http://github.com/tanghaibao/GOatools) was used to assign GO annotations to HSFs, and Fisher's exact test was used to identify biological functions enriched in the PeHsfs relative to the full GO database. Visualization were performed using the Majorbio online platform (https://cloud.majorbio.com).
Construction of a PeHsf protein interaction network
We used the STRING database to predict potential interactions among the PeHsf proteins (Fig. 12 and Supplementary Table S6). There were 19 nodes in the PeHsf protein interaction network, each of which interacted with multiple other nodes. Some proteins exhibited direct interactions, e.g. PeHsf14 and PeHsf16, and others showed more complex multi-gene interactions, e.g. PeHsf01, PeHsf16, and PeHsf23. PeHsf01, PeHsf24, PeHsf25, PeHsf27, and PeHsf40 were predicted to be central nodes, radiating numerous connections to other nodes.

Interaction network of the HSF proteins in moso bamboo. Nodes represent proteins, central nodes are indicated in red, and black lines indicate interactions between nodes. Cytoscape (V3.7.1) (https://cytoscape.org/) was used to visualize the resulting network.
Homology modeling of PeHsf tertiary structures
PeHsf15 from subfamily HsfA, PeHsf28 from subfamily HsfB, and PeHsf26 from subfamily HsfC were selected for tertiary structural homology modeling. The three-dimensional structures of the three subfamilies shared a number of similarities (Fig. 13). The highly conserved DNA binding domain (DBD), a distinctive feature of HSFs, was visible as a spherical region formed from three α-helix bundles and four strands of reverse-parallel β-folded layers. Different sequences had different convoluted structures. In PeHsf15, ARG91 was linked to the substrate via hydrogen bonds (Fig. 13a). By contrast, PeHsf28 (Fig. 13b) and PeHsf26 (Fig. 13c), representing the HsfB and HsfC families, showed no hydrogen bonding with the substrate owing to their lack of AHA motifs.

Predicted three-dimensional structures of the moso bamboo HSF protein sequences. (a) PeHsf15 was selected to represent the structure of the HsfA subfamily. (b) PeHsf28 was selected to represent the structure of the HsfB subfamily. (c) PeHsf26 was selected to represent the structure of the HsfC subfamily. Models were constructed using SWISS-MODEL. ARG, arginine. GOL, substrate. Predicted model structures were visualized and manipulated with Discovery Studio, version 2016 (BIOVIA) (http://www.discoverystudio.net/).
Discussion
HSFs are a specific class of transcription factors that play an important role in plant resistance to various stress injuries58,59,60. The HSF gene family has been studied in Arabidopsis10, tomato15, cabbage16, pepper17, poplar18, maize19, and rice20, and specific molecular functions of the HSFs have been validated in the model plants Arabidopsis and rice. However, HSFs have not previously been investigated in the economically important and widely distributed graminaceous species moso bamboo. At present, the genome draft of moso bamboo is almost complete, enabling the comprehensive characterization of important gene families33,34. Here, we identified 41 PeHsf genes from moso bamboo and classified them into three subfamilies (A–C) based on phylogenetic analysis (Fig. 1). Gene structure analysis revealed that some members within the same subfamily had structural differences. For example, PeHsf31 in the HsfC subfamily contained three introns, whereas the other HsfCs had only one. PeHsf31 may therefore have experienced the splicing or insertion of gene fragments during its evolution61,62. Nonetheless, similar conserved sequences and gene structures among HSF subfamily members suggests that genes within a subfamily may generally have similar biological functions.
The highly conserved plant HSF DBD is located at the N terminus, where it enables the precise localization and recognition of the heat stress element (HSE) in target gene promoters63,64. Here, multiple sequence alignment and secondary structure prediction showed that a DBD consisting of three α-helices and four β-folds was present in all PeHsf proteins (Fig. 2). Interestingly, other conserved domains were also present in some PeHsf proteins, and further experimental verification is needed to determine whether this is a manifestation of gene family functional diversification. Tertiary structure analysis demonstrated that the portion of the transcription factors that interacts with nucleic acids was conserved among subfamilies. HsfB and HsfC members did not contain AHA motifs and therefore probably lack transcriptional activation function, consistent with the results of previous studies14.
Gene duplication plays an important role in evolution by facilitating the generation of new genes and gene functions. There are three main evolutionary modes of gene duplication10: segmental duplication, tandem duplication, and translocation events. Segmental and tandem duplication most commonly underlie the expansion of plant gene families57,65. The number of HSF genes was significantly higher in moso bamboo (41) than in the model plants Arabidopsis (21) and rice (25), suggesting that gene duplication had occurred, consistent with previous reports of whole-genome duplication in moso bamboo32,66. We therefore performed intra- and inter-genomic collinearity analyses of the HSFs. Evolutionary analysis has shown that whole genome duplication (WGD) and segmental duplication play an important role in the expansion of the HSF gene family. Within the moso bamboo genome, there were 29 duplicated gene pairs among the HSF genes, including 27 segmental duplicate pairs and two tandem duplicate pairs. Segmental duplications therefore dominated the expansion of the HSF gene family in moso bamboo. By comparison, a previous study reported nine segmental duplicate pairs in 25 rice HSF genes54. Synteny analysis of the moso bamboo genome with four other sequenced plant genomes showed that there was significant collinearity of HSF family members between bamboo and the monocots maize and rice. Only a few HSF members were collinear between bamboo and the dicots Arabidopsis and pepper. This result is consistent with the evolutionary relationship between dicot and monocot plants.
There are four main fates of duplicated genes67. First, both genes may retain their original function, leading to functional redundancy. Alternatively, duplicates with the same ancestral function may gradually develop different functions in a process referred to as subfunctionalization. In a less common scenario, one of the duplicates may acquire a wholly new function (neofunctionalization). Finally, the intermediate evolutionary stage between subfunctionalization and neofunctionalization preserves genes that are essential for plant growth. We found that many PeHsf homologous gene pairs were expressed at similar levels (Fig. 10), suggesting that duplication of the HSF genes in moso bamboo has primarily resulted in functional redundancy.
Ka and Ks values were calculated for thirteen homologous, segmentally duplicated gene pairs. Ks values indicated that large-scale PeHsf gene duplication events had occurred between ~ 7.86 and 83.33 MYA. Interestingly, about 62% of the homologous gene pairs appeared to have undergone duplication far earlier than 12 MYA, consistent with the occurrence of a separate whole-genome duplication (WGD) event prior to the WGD previously reported to have occurred 7–12 MYA32. In addition, all the homologous gene pairs had Ka/Ks values less than 1, indicating that the PeHsf genes were subject to purifying selection during their evolutionary history.
Flowering is the most important life history trait in angiosperm plants, and bamboo is no exception. Unlike many other bamboos, moso bamboo produces a single flowering spike approximately every fifty years, blooms sporadically, and dies after flowering. Several studies have demonstrated that HSFs modulate the expression of stress-related proteins such as heat shock proteins (HSPs) that play important roles in plant stress response68. HSPs such as HSP70 and HSP90 are involved in flowering time regulation and vernalization pathways that promote flowering by inhibiting FLC expression69. Based on HSF expression profiling in different bamboo tissues (Fig. 10c), we found that many PeHsfs showed relatively high expression levels in the panicle. For example, orthologs of rice OsHsfB2b (PeHsf27, PeHsf10, and PeHsf38) were highly expressed in panicles, and a previous study has shown that OsHsfB2b acts as a negative regulator of plant drought response20. These PeHsf genes may therefore be involved not only in panicle development but also in the response of moso bamboo to drought stress. Some PeHsf genes showed little to no expression in any tissue tested, suggesting that they may act in other tissues or at other developmental stages. Interestingly, some PeHsfs were highly expressed in leaves but expressed at low levels in early 20-cm bamboo shoots, suggesting that they are mainly involved in cell differentiation and elongation, rather than in the process of lignification.
Multiple cis-acting elements located in gene promoters play a crucial role in signaling, and synergistic interactions among them can regulate complex biological processes. The drought-responsive MYB element was present in the promoter regions of almost all PeHsf genes, as were ABRE, MBS, P-box, TC-rich repeat, and SP1 elements, indicating that PeHsf expression is likely to be affected by abiotic factors such as high temperature, drought, high salinity, and light. Examination of expression profiles of seedling roots under NAA and GA treatments revealed that most PeHsf genes were expressed at different levels under treated and control conditions.
Some genes, (e.g., PeHsf40, PeHsf31, and PeHsf13) were upregulated by GA treatment, whereas others (e.g., PeHsf33, PeHsf06, PeHsf03, PeHsf35, and PeHsf26) were downregulated. Interestingly, the promoter analysis (Fig. 9) indicated that these genes all contained cis-elements involved in ABA response. In rice, multiple HSF genes are induced by exogenous gibberellin 3 (GA3) and abscisic acid (ABA)70, and multiple ABRE and P-box elements responsive to ABA and GA, respectively, were present in the PeHsf promoters. Promoter analysis and transcriptomic results therefore suggest that HSF genes are involved not only in the regulation of heat response, but also in responses to drought, salt, and various exogenous hormones (NAA, GA, and ABA), making them important transcription factors for plant resistance to multiple stresses.
Conclusions
We systematically identified and analyzed 41 moso bamboo HSF genes and divided them into three subfamilies, each of which had similar gene structures and sequences. Evolutionary analysis indicated that segmental duplications associated with whole-genome duplication events were responsible for much of the expansion of the moso bamboo HSF gene family. Transcriptomic analyses confirmed that expression of some PeHsfs responded to exogenous GA and NAA application, and the high expression of other PeHsfs in 20-cm shoots suggested that they may function in the rapid growth and early development of bamboo shoots. The PeHsf gene family appears to be functionally diverse, with roles in bamboo growth and development, stress response, and the functions of specific tissues and organs. These results provide a foundation for the subsequent exploration of bamboo HSF gene functions.
References
Bakshi, A. et al. Ectopic expression of Arabidopsis Target of Rapamycin (AtTOR) improves water-use efficiency and yield potential in rice. Sci. Rep. 7, 42835 (2017).
Guo, M. et al. The plant heat stress transcription factors (HSFs): Structure, regulation, and function in response to abiotic stresses. Front. Plant Sci. 7, 114 (2016).
Zhou, R., Bing, L., Liu, H. & Sun, D. Progress in the participation of Ca2+–calmodulin in heat shock signal transduction. Progr. Nat. Sci. 10, 1201–1208 (2009).
Nakashima, K., Takasaki, H., Mizoi, J., Shinozaki, K. & Yamaguchi-Shinozaki, K. NAC transcription factors in plant abiotic stress responses. Biochim. Biophys. Acta 1819, 97–103 (2012).
Yoon, Y. et al. The role of stress-responsive transcription factors in modulating abiotic stress tolerance in plants. Agronomy 10, 788 (2020).
Xue, G. P., Sadat, S., Drenth, J. & McIntyre, C. L. The heat shock factor family from Triticum aestivum in response to heat and other major abiotic stresses and their role in regulation of heat shock protein genes. J. Exp. Bot. 65, 539–557 (2014).
Ohama, N., Sato, H., Shinozaki, K. & Yamaguchi-Shinozaki, K. Transcriptional regulatory network of plant heat stress response. Trends Plant Sci. 22, 53–65 (2017).
Scharf, K. D., Berberich, T., Ebersberger, I. & Nover, L. The plant heat stress transcription factor (Hsf) family: Structure, function and evolution. Biochim. Biophys. Acta. 1819, 104–119 (2012).
Sangster, T. A. & Queitsch, C. The HSP90 chaperone complex, an emerging force in plant development and phenotypic plasticity. Curr. Opin. Plant Biol. 8, 86–92 (2005).
Nover, L. et al. Arabidopsis and the heat stress transcription factor world: How many heat stress transcription factors do we need?. Cell Stress Chaperones 6, 177–189 (2001).
Baniwal, S. K. et al. Heat stress response in plants: A complex game with chaperones and more than twenty heat stress transcription factors. J. Biosci. 29, 471–487 (2004).
Lyck, R. et al. Intracellular distribution and identification of the nuclear localization signals of two plant heat-stress transcription factors. Planta 202, 117–125 (1997).
Heerklotz, D., Döring, P., Bonzelius, F., Winkelhaus, S. & Nover, L. The balance of nuclear import and export determines the intracellular distribution and function of tomato heat stress transcription factor HsfA2. Mol. Cell. Biol. 21, 1759–1768 (2001).
Kotak, S., Port, M., Ganguli, A., Bicker, F. & von Koskull-Döring, P. Characterization of C-terminal domains of Arabidopsis heat stress transcription factors (Hsfs) and identification of a new signature combination of plant class A Hsfs with AHA and NES motifs essential for activator function and intracellular localization. Plant J. 39, 98–112 (2004).
Yang, X., Zhu, W., Zhang, H., Liu, N. & Tian, S. Heat shock factors in tomatoes: Genome-wide identification, phylogenetic analysis and expression profiling under development and heat stress. PeerJ 4, e1961 (2016).
Ma, J. et al. Genome-wide analysis of HSF family transcription factors and their responses to abiotic stresses in two Chinese cabbage varieties. Acta Physiol. Plant 36, 513–523 (2014).
Guo, M. et al. Genome-wide analysis, expression profile of heat shock factor gene family (CaHsfs) and characterisation of CaHsfA2 in pepper (Capsicum annuum L.). BMC Plant Biol. 15, 151 (2015).
Zhang, H. et al. Identification and expression analysis of the heat shock transcription factor (HSF) gene family in Populus trichocarpa. Plant Omics 6, 415–424 (2013).
Lin, X. Y. et al. Genome-wide identification, classification and analysis of heat shock transcription factor family in maize. BMC Genom. 12, 76 (2011).
Xiang, J. et al. Heat shock factor OsHsfB2b negatively regulates drought and salt tolerance in rice. Plant Cell Rep. 32, 1795–1806 (2013).
Busch, W., Wunderlich, M. & Schöffl, F. Identification of novel heat shock factor-dependent genes and biochemical pathways in Arabidopsis thaliana. Plant J. 41, 1–14 (2005).
Ogawa, D., Yamaguchi, K. & Nishiuchi, T. High-level overexpression of the Arabidopsis HsfA2 gene confers not only increased thermotolerance but also salt/osmotic stress tolerance and enhanced callus growth. J. Exp. Bot. 58, 3373–3383 (2007).
Liu, H. C. & Charng, Y. Y. Common and distinct functions of Arabidopsis class A1 and A2 heat shock factors in diverse abiotic stress responses and development. Plant Physiol. 163, 276–290 (2013).
Miller, G. & Mittler, R. Could heat shock transcription factors function as hydrogen peroxide sensors in plants?. Ann. Bot. 98, 279–288 (2006).
Ikeda, M., Mitsuda, N. & Ohme-Takagi, M. Arabidopsis HsfB1 and HsfB2b act as repressors of the expression of heat-inducible Hsfs but positively regulate the acquired thermotolerance. Plant Physiol. 157, 1243–1254 (2011).
Ulrike, B. et al. Arabidopsis HEAT SHOCK TRANSCRIPTION FACTORA1b overexpression enhances water productivity, resistance to drought, and infection. J. Exp. Bot. 64, 3467–3481 (2013).
Biswas, P., Chakraborty, S., Dutta, S., Pal, A. & Das, M. Bamboo flowering from the perspective of comparative genomics and transcriptomics. Front. Plant Sci. 7, 1900 (2016).
Karhu, K., Mattila, T., Bergstrm, I. & Regina, K. Biochar addition to agricultural soil increased CH4 uptake and water holding capacity: Results from a short-term pilot field study. Agric. Ecosyst. Environ. 140, 309–313 (2011).
Yeasmin, L., Ali, M. N., Gantait, S. & Chakraborty, S. Bamboo: An overview on its genetic diversity and characterization. 3 Biotech 5, 1–11 (2015).
Yuan, W. et al. Severe summer heatwave and drought strongly reduced carbon uptake in Southern China. Sci. Rep. 6, 18813 (2016).
IUFRO Adaptation of forests and people to climate change: A global assessment report. In IUFRO World Series Vol. 22 (eds Seppälä, R. et al.) 224 (IUFRO, 2009).
Peng, Z. et al. The draft genome of the fast-growing non-timber forest species moso bamboo (Phyllostachys heterocycla). Nat. Genet. 45, 456–461 (2013).
Zhao, H. et al. Chromosome-level reference genome and alternative splicing atlas of moso bamboo (Phyllostachys edulis). Gigascience 7, giy115 (2018).
Finn, R. D., Clements, J. & Eddy, S. R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37 (2011).
Letunic, I., Doerks, T. & Bork, P. SMART 7: Recent updates to the protein domain annotation resource. Nucleic Acids Res. 40, D302–D305 (2011).
Finn, R. D. et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 44(D1), D279–D285 (2016).
Huang, B. & Xu, C. Identification and characterization of proteins associated with plant tolerance to heat stress. J. Integr. Plant Biol. 50, 1230–1237 (2008).
Wilkins, M. R. et al. Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 112, 531–552 (1999).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 7, 1870–1874 (2016).
Waterhouse, A. M., Procter, J. B., Martin, D. & Barton, G. Jalview: Visualization and analysis of molecular sequences, alignments, and structures. BMC Bioinform. 6, P28 (2005).
Troshin, P. V. et al. JABAWS 2.2 Distributed web services for bioinformatics: Protein disorder, conservation and RNA secondary structure. Bioinformatics 11, 1939–1940 (2018).
Bailey, T. L. et al. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208 (2009).
Ren, J. et al. DOG 1.0: Illustrator of protein domain structures. Cell Res. 19, 271–273 (2009).
Chao, J., Kong, Y., Qian, W., Sun, Y. & Liu, G. MapGene2Chrom, a tool to draw gene physical map based on Perl and SVG languages. Yi Chuan 37, 91–97 (2015).
Chen, C. et al. TBtools - an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202 (2020).
Wang, Y. et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49 (2012).
Gu, Z., Cavalcanti, A., Chen, F. C., Bouman, P. & Li, W. H. Extent of gene duplication in the genomes of Drosophila, nematode, and yeast. Mol. Biol. Evol. 19, 256–262 (2002).
Wang, D. P., Wan, H. L., Zhang, S. & Yu, J. GAMMA-MYN: A new algorithm for estimating Ka and Ks with consideration of variable substitution rates. Biol. Direct 4, 20 (2009).
Lescot, M. et al. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 30, 325–327 (2002).
Otasek, D., Morris, J. H., Bouças, J., Pico, A. R. & Demchak, B. Cytoscape Automation: Empowering workflow-based network analysis. Genome Biol. 20, 185 (2019).
Klopfenstein, D. V. et al. GOATOOLS: A Python library for Gene Ontology analyses. Sci. Rep. 8, 10872 (2018).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300 (1995).
Duan, S., Liu, B., Zhang, Y., Li, G. & Guo, X. Genome-wide identification and abiotic stress-responsive pattern of heat shock transcription factor family in Triticum aestivum L. BMC Genom. 20, 257 (2019).
Guo, J. et al. Genome-wide analysis of heat shock transcription factor families in rice and Arabidopsis. J. Genet. Genomics 35, 105–118 (2008).
Bai, J. et al. Diversity in nucleotide binding site–leucine-rich repeat genes in cereals. Genome Res. 12, 1871–1884 (2002).
Lynch, M. & Conery, J. S. The evolutionary fate and consequences of duplicate genes. Science 290, 1151–1155 (2000).
Zhu, Y. et al. Soybean (Glycine max) expansin gene superfamily origins: Segmental and tandem duplication events followed by divergent selection among subfamilies. BMC Plant Biol. 14, 93 (2014).
Kotak, S., Vierling, E., Bäumlein, H. & von Koskull-Döring, P. A novel transcriptional cascade regulating expression of heat stress proteins during seed development of Arabidopsis. Plant Cell 19, 182–195 (2007).
Wang, J., Sun, N., Deng, T., Zhang, L. & Zuo, K. Genome-wide cloning, identification, classification and functional analysis of cotton heat shock transcription factors in cotton (Gossypium hirsutum). BMC Genom. 15, 961 (2014).
Wang, P. et al. Genome-wide dissection of the heat shock transcription factor family genes in Arachis. Front. Plant Sci. 8, 106 (2017).
Staiger, D. & Brown, J. W. S. Alternative splicing at the intersection of biological timing, development, and stress responses. Plant Cell 25, 3640–3656 (2013).
Li, J. et al. Gene replacements and insertions in rice by intron targeting using CRISPR-Cas9. Nat. Plants 2, 16139 (2016).
Cicero, M. P. et al. The wing in yeast heat shock transcription factor (HSF) DNA-binding domain is required for full activity. Nucleic Acids Res. 29, 1715–1723 (2001).
Schultheiss, J. et al. Solution structure of the DNA-binding domain of the tomato heat-stress transcription factor HSF24. Eur. J. Biochem. 236, 911–921 (1996).
Cannon, S. B., Mitra, A., Baumgarten, A., Young, N. D. & May, G. The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 4, 10 (2004).
Opanowicz, M., Vain, P., Draper, J., Parker, D. & Doonan, J. H. Brachypodium distachyon: Making hay with a wild grass. Trends Plant Sci. 13, 172–177 (2008).
Charon, C., Bruggeman, Q., Thareau, V. & Henry, Y. Gene duplication within the Green Lineage: The case of TEL genes. J. Exp. Bot. 63, 5061–5077 (2012).
Guo, H., Li, Z., Zhou, M. & Cheng, H. cDNA-AFLP analysis reveals heat shock proteins play important roles in mediating cold, heat, and drought tolerance in Ammopiptanthus mongolicus. Funct. Integr. Genom. 14, 127–133 (2014).
Kumar, S. V. & Wigge, P. A. H2A.Z-containing nucleosomes mediate the thermosensory response in Arabidopsis. Cell 140, 136–147 (2010).
Watanabe, H., Takahashi, K. & Saigusa, M. Morphological and anatomical effects of abscisic acid (ABA) and fluridone (FLU) on the growth of rice mesocotyls. Plant Growth Regul. 34, 273–275 (2001).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC, Project grant: 31770721). We are grateful to the members of the lab for their assistance and helpful discussions. The authors would like to thank TopEdit (www.topeditsci.com) for linguistic assistance during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
B.H. performed data collection and processing, participated in study design and interpretation, performed experiments, and wrote the manuscript. Z.H. participated in some of the experiments. R.M. and J.C. assisted in the interpretation of the results. Z.Z. and K.Y. are responsible for the completeness of the data and accuracy of the data analysis. The manuscript was reviewed and edited by all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huang, B., Huang, Z., Ma, R. et al. Genome-wide identification and analysis of the heat shock transcription factor family in moso bamboo (Phyllostachys edulis). Sci Rep 11, 16492 (2021). https://doi.org/10.1038/s41598-021-95899-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-95899-3
This article is cited by
-
Genome-wide analysis of the KNOX gene family in Moso bamboo: insights into their role in promoting the rapid shoot growth
BMC Plant Biology (2024)
-
Genome-wide identification and comprehensive analysis heat shock transcription factor (Hsf) members in asparagus (Asparagus officinalis) at the seeding stage under abiotic stresses
Scientific Reports (2023)
-
Biotechnological interventions in bamboo plants
Plant Cell, Tissue and Organ Culture (PCTOC) (2023)
-
Heat shock transcription factor (Hsf) gene family in common bean (Phaseolus vulgaris): genome-wide identification, phylogeny, evolutionary expansion and expression analyses at the sprout stage under abiotic stress
BMC Plant Biology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
