
Abstract
The integrative aspect on carbon fixation and lipid production is firstly implemented in cyanobacterium Synechocystis sp. PCC 6803 using metabolic engineering approach. Genes related to Calvin–Benson–Bassham (CBB) cycle including rbcLXS and glpD and free fatty acid recycling including aas encoding acyl-ACP synthetase were practically manipulated in single, double and triple overexpressions via single homologous recombination. The significantly increased growth rate and intracellular pigment contents were evident in glpD-overexpressing (OG) strain among all strains studied under normal growth condition. The triple aas_glpD_rbcLXS-overexpressing (OAGR) strain notably gave the highest contents of both intracellular lipids and extracellular free fatty acids (FFAs) of about 35.9 and 9.6% w/DCW, respectively, when compared to other strains at day 5 of cultivation. However, the highest intracellular lipid titer and production rate were observed in OA strain at day 5 (228.7 mg/L and 45.7 mg/L/day, respectively) and OG strain at day 10 (358.3 mg/L and 35.8 mg/L/day, respectively) due to their higher growth. For fatty acid (FA) compositions, the main saturated fatty acid of palmitic acid (C16:0) was dominantly found in both intracellular lipid and secreted FFAs fractions. Notably, intracellular FA proportion of myristic acid (C14:0) was induced in all engineered strains whereas the increase of stearic acid (C18:0) composition was found in extracellular FFAs fraction. Altogether, these overexpressing strains efficiently produced higher lipid production via homeostasis balance on both its lipid synthesis and FFAs secretion.
Similar content being viewed by others

Introduction
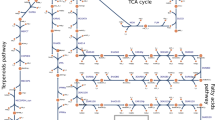
Fatty acids and lipids, which are present at significant levels in cyanobacteria, may serve as crucial precursors for the production of renewable energy carriers, such as biofuel and biodiesel and they are mainly accumulated in terms of phospholipid membranes which are the composition of cell and thylakoid membranes1. Many reports have outlined the strength of genetic metabolic engineering approaches to enhance lipid production by genetically engineering the genes involved in lipid biosynthesis and its neighbouring pathways. For lipid biosynthesis in the cyanobacterium Synechocystis PCC6803, as shown in Fig. 1, the pathway starts from acetyl-CoA, which is converted to malonyl-CoA catalysed by acetyl-CoA carboxylase (ACC encoded by acc gene), and involves multiple steps of fatty acid synthesis II (FAS II) to obtain an intermediate fatty acyl-ACP, which is mainly used as a substrate for phospholipid synthesis and minor alkane production2. A genetically modified Synechocystis 6803 strain in which alkane production is blocked by deleting aar, encoding an acyl-ACP reductase that catalyses the conversion of fatty acyl-ACP to alkane, and together with an overexpression of aas, a gene encoding acyl-acyl carrier protein synthetase (AAS), had enhanced lipid level of approximately 1.8-fold3. The FFA recycling occurs via the activity of AAS, encoded by aas, to recycle the degraded membrane lipids by lipase A, encoded by lipA, to intracellular FFAs and which is then reused as a precursor for fatty acyl-ACP production4,5. In brief, in the linkage pathway with lipid synthesis, the Calvin-Benson-Bassham (CBB) cycle uses ATP and NADPH obtained from the light reaction to generate sugars6. Ribulose-1,5-bisphosphate carboxylase/oxygenase, commonly known as RuBisCO, is the key enzyme in the CBB cycle and plays a crucial rate-limiting role in this pathway. An engineered Synechocystis PCC 6803 strain overexpressing RuBisCO, encoded by RuBisCo gene operon (rbcLXS), exhibited increased RuBisCO activity, growth rate and biomass7. Not only the carbon source but also many carbon intermediates are produced within the CBB cycle and indirectly flow to other crucial metabolic processes; for example, the conversion of the intermediate 3-phosphoglycerate (3-PG) to pyruvate and subsequently to acetyl-CoA, which are used in central metabolic pathways8. Moreover, dihydroxyacetone phosphate (DHAP) which is one of intermediates in the CBB cycle can be converted to glycerol-3-phosphate (Gro3P) catalysed by glycerol-3-phosphate dehydrogenase (GPD encoded by glpD gene)6, which is further partly used as a glycerol backbone for lipid molecules. Recent research revealed the enhancement of Gro3P synthesis, which enhanced lipid production in Synechocystis PCC6803, by introducing the heterologous GPD1 gene (encoding glycerol-3-phosphate dehydrogenase) from Saccharomyces cerevisiae and the atf1 gene (encoding diacylglycerol acyltransferase, DGAT) from the oleaginous bacterium Rhodococcus opacus PD6309. However, Gro3P also generally functions as a substrate for other pathways including starch and glycogen biosynthesis, isoprene and carotenoid synthesis9,10,11,12.

Fatty acid biosynthesis and its neighbouring pathways in Synechocystis PCC 6803. Key enzyme-encoding genes in this study (represented in the circle) associated with lipid and fatty acid synthesis, the TCA cycle, the Calvin-Benson-Bassham (CBB) cycle, and free fatty acid (FFA) recycling including accABCD, a multi-subunit acetyl-CoA carboxylase gene; aas, acyl-ACP synthetase; glpD, glycerol-3-phosphate dehydrogenase; lipA, a lipolytic enzyme-encoding gene; phaA, β-ketothiolase; phaB, acetoacetyl-CoA reductase; plsX, plsY and plsC, putative phosphate acyl-transferases; the RuBisCo gene cluster, including rbcLSX, encoding RuBisCo large, small and chaperone subunits, respectively. The intermediates are: fatty acyl-ACP, fatty acyl-acyl carrier protein; Gro3P, glycerol-3-phosphate; 3PG, 3-phosphoglycerate.
In this study, we metabolically engineered Synechocystis 6803 strain via the integrated aspect on carbon fixation and lipid production by modifying genes related to fatty acid recycling and CBB cycle. There were three engineered strains constructed in this study including OG, OAG and OAGR. The OG strain was glpD-overexpressing strain which expectedly enhanced Gro3P product and was partly used as glycerol backbone in lipid molecules. The OAG strain contained double overexpression of aas_glpD which probably enhanced both glycerol backbone and fatty acyl-ACP substrate whereas triple-overexpression of aas_glpD_rbcLXS genes (OAGR) would synergistically induce RuBisCO function beside OAG. Our findings indicate that the apparent creation of genetically modified strains, in particular OAGR, was achieved to augment both intracellular lipid synthesis and free fatty acids secretion.
Results
Construct verification and growth curves of engineered Synechocystis PCC 6803 strains
After we successfully obtained all the engineered transformant cells (Table 1, Fig. 2), the strains were verified by monitoring gene location and segregation by PCR using many specific primers (Supplementary information, Table S1). Both wild type (WT) cells and the aas-overexpressing strain or OA, obtained from our previous study3, were used as hosts for further engineering in this study (Table 1). The OG and OAG strains were generated by the single homologous recombination into the native glpD gene (Fig. 2A,B), whereas OAGR was constructed by introducing glpD and rbcLXS gene fragments into the native RuBisCo gene operon (Fig. 2C). The gene integration was confirmed using PCR by each specific pair of primers (Supplementary information, Table S1) as shown in (a)–(d) of Fig. 2A–C. After that, cell growth of the engineered strains was subsequently monitored by comparing with Synechocystis PCC 6803 WT. The slight decrease of cell growth was observed in most engineered strains, except OG which apparently had increased cell growth after 6 days of cultivation when compared to WT (Fig. 3A). Doubling time of OG’s cell growth (4 days) was efficiently shortest among all strains studied whereas OAG (double aas_glpD-overexpressing strain) had longest doubling time. The OG strain apparently showed faster growth when compared to other strains. Moreover, intracellular pigments of chlorophyll a and carotenoids (Fig. 3B,C) of OG, OAG and OAGR strains showed the distinctly higher contents when compared to those of both WT and OA strains, in particular OG.

Genomic maps of engineered Synechocystis PCC 6803 strains, including the overexpression strains OG (A), OAG (B), and OAGR (C). The specific primers (Supplementary information, Table S1) are used for confirming the integration of each gene into Synechocystis genome. The OA strain3 was singly recombined with the aas gene. The OG and OAG strains were constructed by overexpressing the native glpd gene in WT and OA cells, respectively. OAGR was generated by overexpressing glpd and a RuBisCo cassette in the OA strain. Confirmation of integration was performed using PCR with genomic DNA from WT and engineered strains as the template. Lane M: GeneRuler DNA ladder (FERMENTAS). For (A) OG strain; Lane 1: negative control using WT as template (a–d), (a) Lanes 2–5: clone numbers 1 to 4 using Glpd_SF and Km_SR primers, (b) Lanes 2–6: clone numbers 1 to 5 using USpsbA2 and DSglpD primers, (c) Lanes 2–6: clone numbers 1 to 5 using USglpD and bb_cSR primers, and (d) Lanes 2–6: clone numbers 1 to 5 using bb_f1 and Glpd_SR primers. For (B) OAG strain; Lane 1: negative control using WT as template (a–d), (a) Lanes 2–6: clone numbers 1 to 5 using Glpd_SF and Km_SR primers, (b) Lanes 2–6: clone numbers 1 to 5 using USpsbA2 and DSglpD primers, (c) Lanes 2–6: clone numbers 1 to 5 using USglpD and bb_cSR primers, and (d) Lanes 2–6: clone numbers 1 to 5 using bb_f1 and Glpd_SR primers. For (C) OAGR strain; Lane 1: negative control using WT as template (a–d), (a) Lanes 2–6: Lanes 2–6: clone numbers 1 to 5 using USpsbA2 and RBC_SR2 primers, (b) Lanes 2–6: clone numbers 1 to 5 using GlpD_SF and DSrubisco primers, (c) Lanes 2–6: clone numbers 1 to 5 using bb_f1 and Glpd_SR primers, and (d) Lanes 2–6: clone numbers 1 to 5 using USrubisco and bb_cSR primers.

Growth curve (A), chlorophyll a content (B) and carotenoid content (C) of the Synechocystis PCC6803 wild-type (WT), OA, OG, OAG, and OAGR strains grown in BG11 medium for 19 days. The error bars represent standard deviations of means (mean ± S.D., n = 3). Doubling time of growth for each strain is shown in Table at right-handed side of (A).
Productions of intracellular lipids and extracellular free fatty acids (FFAs), and fatty acid (FA) compositions of engineered strains
At the start of cultivation (day 0), it was evident that all engineered strains contained significantly higher contents of intracellular lipids compared with WT cells (Fig. 4A). At day 5 of cultivation, the significant increases of intracellular lipid contents were observed in the OA, OAG and OAGR strains, but not in OG which exhibited similar content to that of WT with about 16% w/DCW. The highest content of intracellular lipids was noted at day 5 in OAGR strain, with approximately 35.9% w/DCW, followed by 32.2 and 32.1% w/DCW in OAG and OA, respectively. The certain decreases of lipid contents of all strains were observed at day 10 of cultivation, except OG strain which showed its constant level of intracellular lipid contents between day 5 and day 10 of cultivation. We also checked the intracellular lipid titer (mg/L) and production rate (mg/L/day) which were calculated and shown in Table 2. OA strain had the highest lipid titer and production rate at day 5 with 228.7 mg/L and 45.7 mg/L/day, respectively, whereas OG strain at day 10 had 358.3 mg/L and 35.8 mg/L/day, respectively (Table 2). As a result of faster growth of OG, its lipid content per cell mass (% w/DCW) was almost constant between day 5 and day 10 of cultivation (Fig. 4A).

Intracellular lipid content (A), extracellular free fatty acid content (B) and total contents of both intracellular lipids and extracellular free fatty acids (C) in Synechocystis PCC 6803 WT, OA, OG, OAG and OAGR strains grown in BG11 medium at the start of the experiment (day 0), day 5 and day 10 of cultivation. The error bars represent standard deviations of means (mean ± S.D., n = 3). Means with the same letter have nonsignificant differences at a significance level of P < 0.05.
Not only intracellular lipid synthesis but also free fatty acid (FFA) secretion contribute towards intracellular FFA balance upon toxic tolerance mechanism of cyanobacterial cells. Results of extracellular FFA content secreted into the medium are shown in Fig. 4B. At day 5, the Synechocystis WT strain secreted FFAs into BG11 medium at a level of approximately 6.1% w/DCW (Fig. 4B) with its high titer of about 7.3 mg/L (Table 3) when compared to the highest titer from OAGR (7.8 mg/L). The decreases of secreted FFAs levels from OA and OG strains were apparently noted when compared with that of WT (Fig. 4B), as well as their secreted titer (Table 3). Strikingly, OAG and OAGR strains were able to secrete significantly greater amounts of extracellular FFAs at day 5 than WT cells by about 7.3 and 9.6% w/DCW, respectively. In Table 3, the secreted FFA titers and production rates of OA, OG and OAG strains were lower than that of WT at day 5 but higher at day 10, except OG strain. The highest titer and production rate of the secreted FFAs occurred at day 5 and day 10 in OAGR and OA strains, respectively (Table 3). In Fig. 4C, we combined the results of both intracellular lipids and extracellular FFAs contents in order to reveal the capacity of cells based on their lipid synthesis and FFAs secretion, although FFA is only a component of lipid molecule. The highest capacity of triple overexpressing strain, OAGR, was evident on both intracellular lipid synthesis and FFA secretion, especially at day 5 of cultivation (Fig. 4C).
The fatty acid (FA) compositions from extracted cells (intracellular FAs) and supernatant (extracellular FAs) fractions of all strains were analysed by GC instrument. The results are shown as percentage of FA compositions (Fig. 5). The dominant FA in Synechocystis cells was palmitic acid (C16:0) in the range of 87–93% (Fig. 5A). The genetically modified strains in this study could increase C16:0 FA, especially OA and OAG strains. On the other hand, the decrease of stearic acid (C18:0) was markedly detected in all engineered strains when compared to that of WT whereas the increased ratio of linoleic acid (C18:2) were apparently noted in the OAGR strain. It was interesting that myristic acid (C14:0) was induced in the engineered strains, especially OG (Fig. 5A). In Fig. 5B, the FA composition in secreted FFAs fraction was depicted. The dominant C16:0 was also extracellularly found in all strains. The increased proportion of C18:0 by about 21–46% were secreted from all engineered strains when compared to that from WT (6%). Thus, the FFAs secretion from engineered Synechocystis cells mainly consisted of palmitic acid (C16:0) and stearic acid (C18:0) (Fig. 5B).

Intracellular (A) and extracellular (B) FA compositions (%) analysed by a GC instrument in Synechocystis PCC6803 WT and engineered strains grown in BG11 medium at day 10 of cultivation. The error bars represent standard deviations of means (mean ± S.D., n = 3).
Transcript levels of genes involved in CBB cycle and fatty acid biosynthetic pathway
The five day-cell culture of each strain was used to determine the transcript amounts of genes involved in CBB cycle, fatty acid and lipid biosynthesis and lipid degradation (Fig. 1) including accA, plsX, aas, lipA, rbcS, rbcL and glpD genes by RT-PCR using various specific pairs of primers (Supplementary information, Table S1). For genes related to fatty acid synthesis, the transcript levels of accA gene, encoding acetyl-CoA carboxylase subunit A, were obviously up-regulated in engineered strains, OA, OG, OAG and OAGR, when compared to WT (Fig. 6). Similarly, transcript levels of plsX gene, encoding putative acyltransferase for phospholipid synthesis13, were increased in engineered strains (Fig. 6B). When compared among four engineered strains, OAG gave lowest level of accA transcript whereas OA had lowest level of plsX transcript. In addition, the transcript levels of aas gene encoding acyl-ACP synthetase related to FFAs recycling, were highly induced in all engineered strains, even in OG strain when compared with WT. For phospholipid degradation, the increased transcript levels of lipA, encoding lipase A, were also observed, particularly in OG and OAGR strains, when compared to WT. The transcript levels of rbcL and rbcS, belonging to RuBisCo gene cassette, were significantly increased not only in OAGR strain but also in other engineered strains (Fig. 6B). Finally, transcript levels of glpD gene were higher in all engineered strains compared to WT.

The transcript levels (A) and relative intensity ratios (B) of the accA, plsX, aas, lipA, rbcS, rbcL, glpD and 16S rRNA genes of Synechocystis PCC 6803 WT, OA, OG, OAG, and OAGR strains grown in BG11 medium at day 5 of cultivation. The error bars represent standard deviations of means (mean ± S.D., n = 3). The cropped gels (in A) were taken from the different gels, and each cropped gel, between WT and OA bands, was taken from the same gel cutting out the non-related band of other sample as shown in Supplementary Information.
Discussion
In this study, we highlight our results from genetically modified strains of cyanobacterium Synechocystis PCC 6803 with enhanced capacity on their lipid production via the synergistic integration of carbon fixation reaction (or CBB cycle) and FFA recycling. Our previous reports demonstrated the increased lipid levels in engineered Synechocystis by modifying genes directly associated with fatty acid and phospholipid synthesis, including plsX, plsC, accDBCA, and FFA recycling including aas using metabolic engineering approach3,13. We previously suggested that carbon source supplementation, particularly acetate addition, could induce the intracellular lipid content up to 39.1% w/DCW in PlsC_PlsX-overexpressing Synechocystis cells13 (reviewed in Table 4). In this study, we then created the engineered strain with itself overproduced C-source via carbon fixation reaction by overexpressing RuBisCo gene operon (rbcL, rbcX and rbcS), thereby gaining more 3PG intermediate which partially changed to acetyl-CoA, the initial substrate required for fatty acid and phospholipid synthesis (Fig. 1). Moreover, recent overexpression of glpD gene encoding glycerol-3-phosphate dehydrogenase (GlpD), a crucial link between carbohydrate, pigment and lipid biosyntheses9,10 could increase glycerol backbone in terms of Gro3P which could partly contribute to phospholipid synthesis as well. On the other hand, Farmer and Liao (2001) reported that Gro3P could be also converted to pyruvate intermediate which was sequentially utilized for isoprene and carotenoid synthesis. Moreover, in our previous work, Synechocystis cells with overexpressing aas gene significantly increased their intracellular lipid level under normal growth condition3. To develop more efficient strains in this study, we recombined the glpD and/or RuBisCo with aas overexpression which consequently generated OG, OAG and OAGR strains. Our results demonstrated that glpD-overexpressing Synechocystis strain (or OG) had the highest increase of cell growth (or biomass) and intracellular pigments, including chlorophyll a and carotenoids (Fig. 3). The glpD overexpression of OG had higher induction effect on chlorophyll a content which tightly correlated with the light reaction of photosystems14. When cells gained higher CBB cycling by glpD overexpression, more ATP and NADPH generated from the light reaction would be needed. Thus, changes in chlorophyll a content may reflect changes in photosystem stoichiometry14. The time required to double population of cells or doubling time also confirmed that OG strain had fastest growth among all strains with the doubling time of 4 days. Our results suggested that the higher Gro3P product observed in OG strain might metabolically flow through glycolysis and TCA cycle which are crucial for generating energy for higher cell growth. The triple aas_glpD_RuBisCo overexpression (or OAGR) had 5.7 days of doubling time which slightly grew faster than WT (6 days of doubling time). However, the previous study suggested that overexpression of RuBisCO driven under psbA2 promoter could slightly increase the photosynthesis and biomass yield of Synechocystis PCC 68037. Only few studies have reported the use of Gro3P (glycerol-3-phosphate), a major product of the glpD-catalysed reaction, where Gro3P could ultimately flow to gluconeogenesis, glycolysis and lipid synthesis9. The strain overexpressing aas_glpD_Rubisco (OAGR strain) gave the highest level of intracellular lipid accumulation of approximately 35.9% w/DCW at day 5 of cultivation (Fig. 4), although its lipid production rate was 40.1 mg/L/day which was lower than that of OA with 45.7 mg/L/day due to its lowered biomass at day 5 (Table 2). Our results indicated that the intracellular lipid titer and production rate have a tight connection with cell biomass. Moreover, we proposed that Gro3P, the product of GPD activity, preferred to mainly flow to carbohydrate and pigment syntheses as supported by the increase of cell growth and intracellular pigment contents (Fig. 3). The reduced intracellular lipid content of the glpD-overexpressing strain (OG) (Fig. 4A) was reasonable due to the increased cell growth or biomass. This result was also supported by lipA transcript increase (Fig. 6), relating to phospholipid degradation into FFAs which were further utilized as energy for cell growth inside OG cells. We also demonstrated the high intracellular lipid content from our metabolic engineering design compared with previously reported works which focused on carbon supplementation (Table 4). The adjustment of nutrient composition in BG11 medium together with CO2 bubbling might be useful to further improve lipid production of our genetically-modified strains.
In this study, we also showed the results of free fatty acid (FFA) secretion (Fig. 3B). Recent reports revealed that the excessive level of intracellularly synthesized FFAs cause cell damage, i.e. unsaturated FAs can generate significant levels of reactive oxygen species (ROS) inside cells15 and FFAs could intercalate into both cell and thylakoid membranes which caused destabilization of both proteins and photosynthesis16,17,18. One of detoxification mechanisms is the FFAs secretion out of cells in order to reduce excessive FFA toxicity inside cells. Synechocystis cells have another mechanism to balance lipid metabolites or respond to changed environment by hydrolyzing phospholipid membrane into intracellular FFAs via the activity of lipase A enzyme encoded by lipA5. Our results demonstrated the increase of secreted FFAs content at day 5 of two engineered strains, OAG and OAGR, with up to 7.8 and 9.8% w/DCW, respectively compared with WT (6.0% w/DCW). The significant increase of both intracellular lipids and extracellular FFAs was observed in OAGR strain. We found that there was a variable correlation between the lipA transcript levels (Fig. 6) and extracellular FFA contents (Fig. 4B) among the engineered strains. For example, OAGR strain had high lipA transcript level and high extracellular FFA, whereas OG strain had high lipA transcript level but low extracellular FFA. This might suggest that the enhanced amount of intracellular FFAs via phospholipid degradation could be an internal regulatory mechanism of the utilization of FFA, i.e. being used intracellularly as energy source for cell growth including its recycling or being secreted into the medium. In this respect, OAGR strain was found to be most efficient for both lipid synthesis and FFA secretion (Fig. 4) amendable for commercial application. In addition, we analysed fatty acid composition of internal lipid and secreted FFAs fraction (Fig. 5). Palmitic acid (C16:0) was the dominant FA in Synechocystis cells which is in agreement with previous studies in wild type3,13,19. It is interesting that myristic acid (C14:0) was also found in all constructed strains at day 10. In a previous study, in vitro acyl-ACP synthetase (AAS) of bacterial Vibrio harveyi preferred C14:0 as one of the substrates for fatty acyl-ACP synthesis20. The high myristic acid levels were mainly found to be unique to Cyanothece sp., aided by the enzyme 1-acyl-sn-glycerol-3-phosphate acyltransferase with high specificity towards the 14:0-acyl-carrier protein21. Comparisons with genome databases such as Cyanobase revealed that this enzyme in Cyanothece PCC8801_1274 shares 59.6% identity with Sll1848 (PlsC) of Synechocystis PCC 6803. Coincidentally, our previous report demonstrated that Sll1848 overexpression led to increased lipid production in cells13. Moreover, our findings indicate the increased ratio of stearic acid (C18:0) in extracellular FFAs fraction from all engineered strains (Fig. 5B). In this respect, it should be noted that the structure of C18:0 is ordinarily more hydrophobic which can easily diffuse through cell membrane and thylakoid membranes. On the other hand, we also demonstrated the transcript levels of all the overexpression strains determined by RT-PCR (Fig. 6). Our results indicated that the induced transcript amounts of engineered strains were accA and plsX genes associated with fatty acid synthesis. The result of double glpD and aas overexpressing strain (or OAG) showing lower amount of accA transcript among engineered strains might indicate the feedback inhibition of fatty acyl-ACP to acetyl-CoA carboxylase (ACC) in fatty acid synthesis. However, triple overexpression by OAGR strain could increase an accA transcript level by about 2-fold compared to OAG strain suggesting that the OAGR strain could overcome feedback inhibition effect. Notably, our results also indicated the up-regulated transcript levels of lipA, encoding the lipolytic enzyme associated with phospholipid hydrolysis, in all engineered strains compared to WT. It was interesting that the rbcS transcript level was apparently higher than rbcL transcript in WT with low level of glpD gene expression (Fig. 6). All engineered strains significantly enhanced the expression of RuBisCo and glpD genes involved in CBB cycle.
In conclusion, our metabolically engineered Synechocystis strains were successfully constructed via integrative expression of aas, encoding acyl-ACP synthetase involved in FFA recycling, combined with RuBisCo and glpD genes in the Calvin-Benson-Bassham cycle. The genetically engineered Synechocystis strains in this study could be classified into three categories according to the desired purposes. First, the modified strain with best performance on internal lipid balance belongs to OAGR strain, as evident by highest lipid and FFA contents at day 5 of cultivation. Second, with regard to biotechnological productivity, OA is the best strain which prominently achieves highest lipid titer and production rate at day 5 of cultivation, Finally, for the purpose of increasing the biomass for lipids and other metabolites production, OG strain is the promising candidate. However, to further enhance the lipid productivity of selected genetically engineered strains, the stressed environment should be imposed in order to alter their metabolic flux and balance towards lipid synthesis.
Methods
Strains and culture conditions
The host Escherichia coli DH5α strain was propagated on Luria Bertani (LB) agar plates and in liquid broth containing 35 µg/mL kanamycin (Km) and 35 µg/mL chloramphenicol (Cm) at an optimum temperature of 37 °C. The unicellular cyanobacterium Synechocystis sp. PCC6803 (Synechocystis) strain was grown in BG11 medium at a culture temperature of 28 °C with continuous illumination of approximately 50 µmol photons/m2/s. The aas-overexpressing Synechocystis (OXAas) strain was obtained in our previous study3, hereafter OA in this study. Other engineered strains were OG, OAG and OAGR strains, all of which were constructed in this study (Table 1). The cells were routinely cultured in BG11 medium containing both 35 µg/mL kanamycin and 35 µg/mL chloramphenicol.
Construction of recombinant plasmids
All strains and plasmids constructed in this study are listed in Table 1. Initially, we modified the original pEERM expression vector22 by replacing the antibiotic Cmr cassette gene with the Kmr cassette gene to obtain the pEERM_Km vector, which was subsequently used as a cloning and expression vector. The recombinant pEERM_GlpD plasmid was constructed by inserting a homologous glpD gene fragment, amplified by PCR using the GlpD_F and GlpD_R primers (Supplementary information, Table S1) and genomic DNA of Synechocystis as a template, into the pEERM_Km vector between the XbaI and BcuI restriction sites. Additionally, the recombinant pEERM_GlpD_RubisCo (Table 1) was constructed by introducing a cassette fragment containing the rbcL, rbcX and rbcS genes, obtained from PCR using the RBC-F and RBC-R primers, into the pEERM_GlpD vector between the BcuI and PstI restriction sites. The pJAasCm vector (Table 1) was created by ligating the aas gene fragment amplified by PCR using the aas_F3 and aas_R3 primers (Supplementary information, Table S1) into the pJET1.2 blunt cloning vector whereas the antibiotic Cmr cassette gene fragment was amplified by the cm_F and cm_R primers (Supplementary information, Table S1) using the pEERM plasmid as the template. The Cmr fragment was subsequently inserted into the AvrII restriction site of the aas gene ligated into the pJET vector.
Natural transformation of recombinant plasmids into Synechocystis cells
To prepare host cells, Synechocystis PCC 6803 wild type (WT) and OA strains were initially grown in BG11 liquid medium until the optical density at 730 nm (OD730) reached between 0.3 and 0.5. 25 mL of cultured cells were harvested by centrifugation at 6,000 rpm (4,025 × g) for 10 min. Subsequently, the pelleted cells were washed and harvested again by centrifugation at the same speed for 10 min before being resuspended in 0.5 mL of fresh BG11 medium for cell condensation. To construct the OG strain, 1 µg of the recombinant pEERM_GlpD plasmid was added to the condensed WT cells and incubated at 28 °C for 6 h, and the tubes were inverted every 2 h. Next, the plasmid-cell mixture was spread on a 0.45-µm sterile nitrocellulose membrane placed over a BG11 agar plate overnight, and then, the membrane was transferred to a new BG11 agar plate containing 35 µg/mL kanamycin. Similarly, other recombinant plasmids were separately transformed into condensed host cells of WT and OA strains, and each mixture was spread on a 0.45-µm sterile nitrocellulose membrane placed over a BG11 agar plate overnight. Then, the membrane was transferred to BG11 agar containing the specific antibiotic type corresponding to the expression system, such as 35 µg/mL kanamycin alone or combined with 35 µg/mL chloramphenicol. After 2–3 weeks of incubation, the surviving colonies were picked and further checked for the corresponding gene location and segregation by PCR using specific pairs of primers (Supplementary information, Table S1).
Measurement of cell growth and pigment content
Synechocystis cell stock cultured in BG11 medium to mid-log phase of cell growth was harvested and subsequently diluted with fresh BG11 medium at an OD730 of approximately 0.1 before starting the cultivation experiment. Cell growth was periodically monitored by a spectrophotometer at OD730. For measurement of intracellular pigment content, including chlorophyll a (chl a) and carotenoids, 1 mL of cultured cells were harvested by centrifugation at 12,000 rpm and the pigments were extracted using the methods described by23,24. N,N-dimethylformamide (DMF), and the absorbance of the DMF extract were spectrophotometrically measured at 461, 625 and 664 nm. The values were then calculated and normalized to cell number to obtain final values corresponding to 1.0 × 108 cells. Dry cell weight (DCW) was measured by incubating the harvested cells at 60–70 °C until a constant dry weight was obtained.
Lipid extraction
The cultured cells started at OD730 of 0.1 cultured in 100 mL of fresh BG11 medium. Cultured cells (approximately 10–50 mL) of each strain were harvested by centrifugation at 6,000 rpm (4,025 × g) for 10 min at days 0 (start), 5 and 10 for lipid extraction. Lipids extracted from the pellet fraction represented intracellular lipids, whereas extracellular FFAs were obtained from the supernatant fraction. In this experiment, the lipid extraction method was slightly modified from the Bligh and Dyer method25. A 1-mL chloroform (CHCl3)/methanol (CH3OH) solution mixture with a ratio of 2:1 was added to harvested fractions, followed by incubation at 37 °C on a shaker for 2 h. After addition of 0.5 mL of 0.88% potassium chloride (KCl) and centrifugation at the same speed for 5 min, the aqueous phase was removed, whereas the lower organic phase was collected. Then, the lipid-dissolving solvent in the lower phase was evaporated at 70 °C.
Spectrophotometric method for determination of total lipid content
The total lipid content was determined by the potassium dichromate oxidation method26. K2Cr2O7 (0.18 M, 0.5 mL) and concentrated sulfuric acid were serially added into the lipid extract as described above. The reaction mixture was boiled at approximately 105 °C for 30 min. After cooling the mixture to room temperature, 0.5 mL of distilled water was added, and the absorbance at 600 nm (Abs600) was spectrophotometrically measured. Canola oil was used as a commercial standard control sample. The lipid content is represented as % w/DCW.
Analysis of fatty acid components
The fatty acid compositions were analysed using a gas chromatography (GC) instrument. The modified method according to27 was used to generate fatty acid methyl esters (FAMEs). A 1-mL mixture of 5% hydrochloric acid in MeOH solution was added to the tube containing extracted lipids. The reaction tube was further heated at 85 °C for 2 h and cooled to room temperature. Subsequently, distilled water (1 mL) was added, and the mixture was vortexed for a few seconds. After the addition of hexane solution (0.5 mL) into the reaction tube, centrifugation at 6,000 rpm (4,025 × g) was performed for 5 min to collect the hexane fraction in the GC vial tube. Additional hexane (0.5 mL) was added, and the mixture was centrifuged again to collect the hexane phase and pooled with the previous hexane extract. Compositions of fatty acids in the hexane phase were analysed by GC and interpreted by comparison with standard equations. The commercial standard of fatty acid mixtures (F.A.M.E., mixed C8-C24, MERCK KGaA, Germany) was prepared as a control. The calculated composition of fatty acids is presented as the percentage of each fatty acid component over the total amount of fatty acids.
Reverse transcription-polymerase chain reaction (RT-PCR)
TRIzol reagent (INVITROGEN, CA) was used to isolate total RNA from harvested cells. Before converting to cDNA using the SuperScript III First-Strand Synthesis Kit (INVITROGEN, CA), isolated total RNAs were treated with RNase-free DNAseI (FERMENTAS, MA) to remove contaminant genomic DNA. Then, the generated cDNAs were used as templates for PCR amplification of genes involved in the CBB cycle, lipid biosynthesis and some neighbouring pathways, including rbcL, rbcS, glpD, accA, aas, plsX, and lipA, with 16s rRNA as a reference, using the corresponding RT-PCR primers (Supplementary information, Table S1). The PCR conditions consisted of 95 °C for 3 minutes, followed by proper cycles for each gene at 95 °C for 30 seconds, primer melting temperature (Tm) for 30 seconds and 72 °C for 30 seconds, and then a final extension at 72 °C for 5 minutes. Proper cycles and Tm of each pair of primers are shown in Supplementary information (Table S1). The PCR products were verified by 1.2% (w/v) agarose gel electrophoresis. Quantification of band intensity was carried out using Syngene Gel Documentation (SYNGENE, Frederick, MD).
Data availability
The data that support the findings of this study are available within the article and its supplementary files or from the corresponding author upon reasonable request.
References
Quintana, N., Van Der Kooy, F., Van De Rhee, M. D., Voshol, G. P. & Verpoorte, R. Renewable energy from cyanobacteria: energy production optimization by metabolic pathway engineering. Applied Microbiology and Biotechnology 91, 471–490 (2011).
Wang, W., Liu, X. & Lu, X. Engineering cyanobacteria to improve photosynthetic production of alka(e)nes. Biotechnology for Biofuels 6, 69 (2013).
Eungrasamee, K., Miao, R., Incharoensakdi, A., Lindblad, P. & Jantaro, S. Improved lipid production via fatty acid biosynthesis and free fatty acid recycling in engineered Synechocystis sp. PCC 6803. Biotechnology for Biofuels 12, 8 (2019).
Kaczmarzyk, D. & Fulda, M. Fatty acid activation in cyanobacteria mediated by acyl-acyl carrier protein synthetase enables fatty acid recycling. Plant Physiology 152, 1598–1610 (2010).
Gao, Q., Tan, X. & Lu, X. Characterization of a key gene in membrane lipid cycle in Synechocystis sp. PCC6803. Chinese Journal of Biotechnology 28, 1473–1481 (2012).
Michelet, L. et al. Redox regulation of the Calvin–Benson cycle: something old, something new. Frontiers in Plant Science 4, 470 (2013).
Liang, F. & Lindblad, P. Synechocystis PCC 6803 overexpressing RuBisCO grow faster with increased photosynthesis. Metabolic Engineering Communications 4, 29–36 (2017).
Hellier, P., Purton, S. & Ladommatos, N. Molecular structure of photosynthetic microbial biofuels for improved engine combustion and emissions characteristics. Frontiers in Bioengineering and Biotechnology 3, 49 (2015).
Wang, X., Xiong, X., Sa, N., Roje, S. & Chen, S. Metabolic engineering of enhanced glycerol-3-phosphate synthesis to increase lipid production in Synechocystis sp. PCC 6803. Applied Microbiology and Biotechnology 100, 6091–6101 (2016).
Farmer, W. R. & Liao, J. C. Precursor balancing for metabolic engineering of lycopene production in Escherichia coli. Biotechnology Progress 17, 57–61 (2001).
Xue, Y. & He, Q. Cyanobacteria as cell factories to produce plant secondary metabolites. Frontiers in Bioengineering and Biotechnology 3, 57 (2015).
Liu, Z., Tian, L., Chen, Y. & Mou, H. Efficient extracellular production of κ-carrageenase in Escherichia coli: Effects of wild-type signal sequence and process conditions on extracellular secretion. Journal of Biotechnology 185, 8–14 (2014).
Towijit, U., Songruk, N., Lindblad, P., Incharoensakdi, A. & Jantaro, S. Co-overexpression of native phospholipid-biosynthetic genes plsX and plsC enhances lipid production in Synechocystis sp. PCC 6803. Scientific Reports 8, 13510 (2018).
Kirst, H., Formighieri, C. & Melis, A. Maximizing photosynthetic efficiency and culture productivity in cyanobacteria upon minimizing the phycobilisome light-harvesting antenna size. Biochimica et Biophysica Acta 1837, 1653–1664 (2014).
Dunlop, M. J., Keasling, J. D. & Mukhopadhyay, A. A model for improving microbial biofuel production using a synthetic feedback loop. Systems and Synthetic Biology 4, 95–104 (2010).
Desbois, A. P. & Smith, V. J. Antibacterial free fatty acids: activities, mechanisms of action and biotechnological potential. Applied Microbiology and Biotechnology 85, 162942 (2010).
Ruffing, A. M. RNA-Seq analysis and targeted mutagenesis for improved free fatty acid production in an engineered cyanobacterium. Biotechnology for biofuels 6, 113 (2013).
Ruffing, A. M. Improved free fatty acid production in cyanobacteria with Synechococcus sp. PCC 7002 as host. Frontiers in Bioengineering and Biotechnology 2, 17 (2014).
Merritt, M. V., Rosenstein, S. P., Rachel, C. L., Chou, H. S. & Allen, M. M. A comparison of the major lipid classes and fatty acid composition of marine unicellular cyanobacteria with freshwater species. Archives of Microbiology 155, 107–113 (1991).
Byers, D. M. & Holmes, C. G. A soluble fatty acyl–acyl carrier protein synthetase from the bioluminescent bacterium Vibrio harveyi. Biochemistry and Cell Biology 68, 1045–1051 (1990).
Saito, M. et al. High myristic acid content in the cyanobacterium Cyanothece sp. PCC 8801 results from substrate specificity of lysophosphatidic acid acyltransferase. Biochimica et Biophysica Acta - Molecular and Cell Biology of Lipids 1863, 939–947 (2018).
Englund, E., Andersen-Ranberg, J., Miao, R., Hamberger, B. & Lindberg, P. Metabolic engineering of Synechocystis sp. PCC 6803 for production of the plant diterpenoid manoyl oxide. ACS Synthetic Biology 4, 1270–1278 (2015).
Chamovitz, D., Sandmann, G. & Hirschberg, J. Molecular and biochemical characterization of herbicide-resistant mutants of cyanobacteria reveals that phytoene desaturation is a rate-limiting step in carotenoid biosynthesis. Journal of Biological Chemistry 268, 17348–17353 (1993).
Moran, R. Formulae for determination of chlorophyllous pigments extracted with N,N-dimethylformamide. Plant Physiology 69, 1376–1381 (1982).
Bligh, E. G. & Dyer, W. J. A rapid method of total lipid extraction and purification. Canadian Journal of Biochemistry and Physiology 37, 911–917 (1959).
Fales, M. F. Evaluation of a spectrophotometric method for determination of total fecal lipid. Clinical Chemistry 17, 1103–1108 (1971).
O’Fallon, J. V., Busboom, J. R., Nelson, M. L. & Gaskins, C. T. A direct method for fatty acid methyl ester synthesis: application to wet meat tissues, oils, and feedstuffs. Journal of Animal Science 85, 1511–1521 (2007).
Runguphan, W. & Keasling, J. D. Metabolic engineering of Saccharomyces cerevisiae for production of fatty acid-derived biofuels and chemicals. Metabolic Engineering 21, 103–113 (2014).
Greer, M. S. et al. Engineering increased triacylglycerol accumulation in Saccharomyces cerevisiae using a modified type 1 plant diacylglycerol acyltransferase. Applied Microbiology and Biotechnology 99, 2243–2253 (2015).
He, Q. et al. Oleaginicity of the yeast strain Saccharomyces cerevisiae D5A. Biotechnology for Biofuels 11, 258 (2018).
Wang, J., Xu, R., Wang, R., Haque, M. E. & Liu, A. Overexpression of ACC gene from oleaginous yeast Lipomyces starkeyi enhanced the lipid accumulation in Saccharomyces cerevisiae with increased levels of glycerol 3-phosphate substrates. Bioscience, Biotechnology, and Biochemistry 80, 1214–1222 (2016).
La Russa, M. et al. Functional analysis of three type-2 DGAT homologue genes for triacylglycerol production in the green microalga Chlamydomonas reinhardtii. Journal of Biotechnology 162, 13–20 (2012).
Kang, N. K. et al. Increased lipid production by heterologous expression of AtWRI1 transcription factor in Nannochloropsis salina. Biotechnology for Biofuels 10, 231 (2017).
Shields-Menard, S. A. et al. Lipid accumulation by Rhodococcus rhodochrous grown on glucose. Journal of Industrial Microbiology and Biotechnology 42, 693–699 (2015).
Davis, M. S., Solbiati, J. & Cronan, J. E. Jr. Overproduction of acetyl-CoA carboxylase activity increases the rate of fatty acid biosynthesis in Escherchia coli. Journal of Biological Chemistry 275, 28593–28598 (2000).
Modiri, S. et al. Lipid production and mixotrophic growth features of cyanobacterial strains isolated from various aquatic sites. Microbiology 161, 662–673 (2015).
Acknowledgements
This research was funded by the Ratchadapisek Sompoch Endowment Fund (2016), Chulalongkorn University (CU-59-018-FW) to S.J. and the Development and Promotion of Science and Technology Talents Project (DPST)’s scholarship for postgraduate tuition and expenses to K.E.
Author information
Authors and Affiliations
Contributions
K.E. was responsible for study conception, experimenter, data collection and analysis, manuscript preparation; A.I. and P.L. for study conception and manuscript revision; S.J. for study conception and design, critical revision and manuscript writing, and final approval of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Eungrasamee, K., Incharoensakdi, A., Lindblad, P. et al. Synechocystis sp. PCC 6803 overexpressing genes involved in CBB cycle and free fatty acid cycling enhances the significant levels of intracellular lipids and secreted free fatty acids. Sci Rep 10, 4515 (2020). https://doi.org/10.1038/s41598-020-61100-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-61100-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
