
Abstract
A rigidification strategy was applied to the preclinical candidate donecopride, an acetylcholinesterase inhibitor possessing 5-HT4R agonist activity. Inspired by promising bioactive benzisoxazole compounds, we have conducted a pharmacomodulation study to generate a novel series of multitarget directed ligands. The chemical synthesis of the ligand was optimized and compounds were evaluated in vitro against each target and in cellulo. Structure-activity relationship was supported by docking analysis in human acetylcholinesterase binding site. Among the synthesized compounds, we have identified a novel hybrid 32a (3-[2-[1-(cyclohexylmethyl)-4-piperidyl]ethyl]-4-methoxy-1,2-benzoxazole) able to display nanomolar acetylcholinesterase inhibitory effects and nanomolar Ki for 5-HT4R.
Similar content being viewed by others
Introduction
As people live longer, the number of people with dementia in low- and middle-income countries and estimates of prevalence raised rapidly and are constantly being revised upwards. In 2018, 50 million people are living with dementia worldwide and this number will increase to 152 million by 20501. Currently ranked in the first places leading causes of death for older people, the most common dementia, Alzheimer’s disease (AD), is a progressive and irreversible brain disorder characterized by a slowly decline in memory and thinking skills up to the inability to carry out the simplest everyday tasks2. The histological diagnosis of AD is based on the presence in abnormally high quantities of two types of lesions in the central nervous system: the senile plaques formed by aggregation of β-amyloid protein (Aβ) and the neurofibrillary tangles due to hyperphosphorylation of tau protein3.
For more than 20 years, therapeutic approaches, focused on the neuronal dysfunction, only allowed to develop treatment with symptomatic benefits for patients, by regulating neurotransmitters4. Except memantine, an NMDA receptor antagonist approved to regulate the glutamatergic activity, the three other drugs authorized by the FDA (donepezil, rivastigmine and galanthamine) are acetylcholinesterase (AChE) inhibitors. Usually used to treat mild to moderate forms of AD, AChE inhibitors (AChEIs) reduce, by inhibiting the catalytic active site (CAS) of the enzyme, the excessive degradation of acetylcholine (ACh), a neurotransmitter involved in many physiological processes including memory and learning and thus restore cholinergic transmission. However, these drugs only work for a limited time and do not change the underlying disease process.
The multifactorial origin of AD has led researchers to develop a new approach that could exert a disease-modifying effect, aimed at slowing down, stopping or reversing the progression of AD, by direct actions on pathophysiological pathways as early as possible5. While disease modifying strategies have not yet allowed any marketing authorization despite many preclinical and clinical trials (ineffectiveness, side effects, lack of selectivity)6 the development of a selective compound against a single AD target does not seem to be the best-suited strategy to the complex AD puzzle.
In order to target several molecular causes implicated in the pathogenesis of AD disease, a new concept emerged about 10 years ago: the development of Multi-Target Directed Ligands (MTDLs)7,8. This innovative and promising drug discovery approach allows the design of ligands targeting, from a single active compound, several pharmacological targets involved in the pathogenesis of a same disease and showing a more synergistic effect. Compared to traditional pharmacological approaches (multiple-medication therapy or multiple-compound medication) used for multifactorial diseases, this concept has advantages such as a simplified clinical development (pharmacokinetic study and optimization of ADME-Tox properties) and a lower risk of drug-drug interactions or poor patient compliance avoided. Based on the benefit that could be the synergy of several therapeutic effects obtained from different pharmacological targets on clinical efficacy, Morphy et al. have nevertheless reported the difficulty of adjusting the ratio of activities towards the various targets7.
Many MTDLs have been developed for the inhibition of AChE, and more particularly dual-binding site (DBS) AChE inhibitors targeting simultaneously the two sites of the enzyme, the CAS and the peripheral anionic site (PAS)9,10. Indeed, it has been shown that the PAS could form a toxic complex with Aβ favoring the formation of fibrils and consequently their aggregation into senile plaques11,12,13.
The interaction with both sites allows the restoration of the cholinergic deficit by blocking the CAS activity and, in addition to preventing the access of ACh to the active site, the inhibition of PAS disrupts amyloid aggregation. Other MTDLs have been developed by adding another activity to the AChE inhibition (BACE1 or MAOs inhibitors, metal chelators, antioxidants…)14,15 or by combining different rational therapeutic activities without targeting the enzyme16.
Our laboratory has published data on several chemical families of MTDLs useful for treating AD17,18,19, Among them, donecopride, based on a structural compromise between donepezil, an AChEI drug, and RS67333, a serotonin subtype 4 receptor (5-HT4R) agonist, seems to be a promising drug candidate with excellent in vitro activities (hAChE IC50 = 16 nM and 5-HT4R Ki = 8.5 nM, 48% of control agonist response) and in vivo effects (improvement of memory performances on the object recognition test in mice)20. Donecopride, the first MTDL described in the literature associating these both complementary activities, is actually engaged in preclinical trials.
Located in the CNS, 5-HT4Rs are involved in cognitive functions (learning, memory…) and their stimulation leads to an improvement in the release of neurotransmitters and an increase in their concentration in brain areas21,22,23. The partial agonist RS67333 showed an improvement of cognitive performances in animals24. The increased release of ACh in combination with decreased breakdown due to inhibition of AchE is particularly interesting to counteract memory deficits as in AD. Additionally, activation of 5-HT4Rs promotes the non-amyloidogenic cleavage of amyloid precursor protein (APP), producing the soluble and neurotrophic fragment sAPPα to the detriment of Aβ production25. We have earlier demonstrated that the co-modulation of those two targets (AChE inhibition and 5-HT4R agonism with two reference compounds) appears synergistic in in vivo models of memory deficit26. The published results obtained with donecopride demonstrated the potential of this strategy. We would also like to explore novel heterocyclic scaffold in order to verify if this combination of activities is transposable to novel chemical series that could combine a symptomatic action (restoration of the cholinergic activity) and a disease-modifying effect (promotion of sAPPα, inhibition of the AChE-induced Aβ aggregation) that seems to be a relevant approach to treat AD.
Villalobos et al. have reported the development of a series of N-benzylpiperidine benzisoxazoles as potent and selective AChEI27. While keeping a benzylated piperidine, the benzisoxazole ring was investigated as a suitable bioisostere to replace the benzoylamino scaffold present in a class of AChE inhibitors previously described28. All benzisoxazoles synthesized displayed submicromolar hAChE IC50s. The most promising N-acetyl derivative [B] (Fig. 1) showed an excellent AChE inhibition activity with an IC50 of 2.8 nM and a favorable profile in vivo (a dose-dependent elevation of ACh in mouse and a reversion of amnesia in a mouse passive avoidance model). According to the docking realized for this AChEI, the compound seems able to interact with the PAS of the enzyme. More recently, Brodney et al. have identified 5-HT4R partial agonists with a similar benzisoxazole scaffold29. By exploring various alkoxyl substituents in position C3 of the benzisoxazole ring and two different substituents on the piperidine ring, the resulting compounds displayed a nanomolar affinity towards 5-HT4R. Many ADME parameters (clearance from human liver microsomes, passive permeability, cyclic AMP production, neuropharmacokinetic parameters…) have been studied and two compounds, among them the benzisoxazole [A] (Fig. 1) with a 5-HT4R Ki of 9.6 nM, were chosen to continue in preclinical trials.

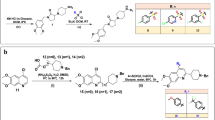
Rational design of novel MTDLs with acetylcholinesterase inhibition and serotoninergic 5-HT4 receptors activity, guided by previous works in the literature.
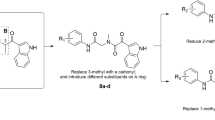
On the basis of these encouraging results for the development of AChE inhibitors on one hand and 5-HT4R agonists on the other, we decided to synthesize new benzisoxazole MTDLs while keeping a structure close to donecopride (Fig. 1). Based on a structural compromise between the two chemical series already described, this work should allow us to rigidify the structure of the latter to study the impact of this new heterocycle on the two pharmacological targets.
Results
Chemistry
Many chemical reactions have been reported in literature concerning the formation of the 1,2-benzoxazole ring, substituted or not in position C3. To summarize some of them, we can quote non-exhaustively the concomitant formation of a C–C bond and an O–C bond by a [3 + 2] cycloaddition of nitrile oxides and arynes30, the formation of a O–N bond from readily accessible ortho-hydroxyaryl N–H ketimines through a N–Cl imine31 or the formation of a N=C bond via an intramolecular transoximation32.
We first focused on the synthesis of compounds 11–16, substituted by a C–O bond in position C3 and bearing a hydrogen or a methoxy group in position C4, from an intermediate N,2-dihydroxybenzamide. The latter can be obtained in particular according from the Angeli-Rimini reaction between an aldehyde and N-hydroxybenzenesulfonamide. In our case, the synthesis of hydroxamic acids 5 and 6 began by an esterification reaction of benzoic acids 1–2 under classical acidic conditions, followed by treatment of esters 3–4 with hydroxylamine in the presence of sodium hydroxide33. Cyclization of 5–6 with 1,1′-carbonyldiimidazole in anhydrous THF under reflux for 3 h led to build the benzisoxazole ring. The precipitation of the crude in water by addition of HCl allowed to afford derivatives 7–8 after suction filtration. The compound 7 was obtained in 94% yield and its structure was confirmed by X-ray diffraction of obtained crystals (Fig. 2). In the second case, the compound 8 was obtained in mixture with the compound 8′ having a 1,3-benzoxazole ring (see Supporting Information). This reaction was repeated several times and the expected product 8 with a 1,2-benzoxazole ring has nevertheless still been obtained as the major compound. The formation of the byproduct 8′ seems to be explained by the Lossen rearrangement, already described in the literature and allowing the conversion of an activated hydroxamic acid to isocyanate under the action of a base or a thermal activation34. The isocyanate can then easily react with an alcohol-containing compound. A reaction, according to the classical conditions described by Mitsunobu et al., then allows the conversion of the primary alcohols of the cyclized products 7–8 into ether links35. Compound 7 and the mixture 8–8′ (known ratio) were reacted with a hydroxylated piperidine chain in the presence of PPh3 and DEAD in dry THF to led to the derivative 9 in 44% yield and to the mixture 10–10′ in 79% yield. The separation of the both 1,2- and 1,3-benzoxazole 10–10′ was then carried out after this step (see Supporting Information). The piperidines 9–10 were deprotected under acidic conditions and we introduced several chains by an N-alkylation reaction using halogenated derivatives and a base according various conditions, leading to the desired compounds 11–16 in 22–45% yields over two steps (Fig. 2).

Synthesis of C3 substituted-benzisoxazole by a C–O bond with a hydrogen or a methoxy group in C4. Reagents and conditions: (a) conc. H2SO4, MeOH, reflux, 48 h, 92–94% (b) NH2OH.HCl, NaOH, H2O/dioxane (3:1), 1 h, rt then 15 h, 40 °C, 65–93% (c) CDI, dry THF, reflux, 3 h, 74–94% (d) tert-butyl 4-(hydroxymethyl)piperidine-1-carboxylate, DEAD, PPh3, dry THF, reflux, 19 h, 44–65% (e) TFA, CH2Cl2, rt, 1 h (f) bromomethylcyclohexane, K2CO3, DMF, 110 °C, 3 h, 22–45% (2 steps) (g) bromomethylbenzene, Et3N, CH2Cl2, rt, 15 h, 26–86% (2 steps) (h) 1-iodo-2-methyl-propane, Et3N, EtOH, reflux, 48 h, 23–36% (2 steps).
We then decided to evaluate the influence of a methylene linker in position C3 instead of a C-O bond following a similar chemical strategy as that developed by Villalobos et al., namely the formation of the C-C bond by deprotonation of the 3-methyl-1,2-benzoxazole derivative using a lithiated base and subsequent reaction with an electrophile27. Synthesis of the expected methylated compound 19 began by a reaction between the o-bromoacetophenone and hydroxylamine in the presence of sodium acetate to obtain the mixture of oximes 18a–18b (E/Z, 2:1) in 90% yield. A cyclization of these 2-bromophenyl ketoxime, engaged without separation of the both isomers according to the conditions reported by Udd and co-workers, allowed to form quickly and at room temperature the 3-methyl-1,2-benzoxazole 1936. The overall yield was 27%, but the copper-catalyzed cyclization of o-brominated aromatic oximes was reported as being effective only for oximes of (Z) configuration (65% yield for the only Z isomer). Deprotonation of methylated compound 19 occurred at −78 °C in anhydrous THF using a lithium diisopropylamide solution and was followed by the addition of a (iodomethyl)piperidine chain to obtain the derivative 20. Finally, the cleavage of the tert-butoxycarbonyl group was performed using trifluoroacetic acid and the deprotected piperidines was subsequently reacted with halogenated derivatives to afford the target compounds 21–23 in 15–50% yields over two steps (Fig. 3).

Synthesis of C3 substituted-benzisoxazole by a C-C bond. Reagents and conditions: (a) NH2OH.HCl, NaOAc, H2O/EtOH (3:2), 1 h, rt then 2 h, reflux, 90% (mixture of isomers E/Z 2:1) (b) t-BuONa, cat. DMEDA, cat. CuI, dry THF, 1 h, rt, 27% (c) LDA 1.0 M sol., dry THF, −78 °C then tert-butyl 4-(iodomethyl)piperidine-1-carboxylate, 1 h, −78 °C, 22% (d) TFA, CH2Cl2, rt, 1 h (e) bromomethylcyclohexane, K2CO3, DMF, 110 °C, 3 h, 50% (2 steps) (f) bromomethylbenzene, Et3N, CH2Cl2, rt, 15 h, 38% (2 steps) (g) 1-iodo-2-methyl-propane, Et3N, DMF, 110 °C, 3 h, 15% (2 steps).
Our last synthesized chemical series consisted in the study of the influence of the C4 substitution by various alkoxyls, while maintaining a C–C bond in C3. The mixture of oximes 25a–25b was prepared in quantitative yield (Z/E 1:4) using a condensation reaction between the dihydroxyacetophenone and hydroxylamine. After separation of the both isomers, the ketoxime 25b was firstly reacted with PPh3 and DDQ in dry THF at room temperature for 1 h but we obtained the undesired 2-methyl-1,3-benzoxazol-4-ol 2737. Its structure was confirmed by X-ray diffraction of obtained crystals (see Supporting Information), The formation of 27 seems to be explained by the Beckmann rearrangement, allowing oximes to rearrange into substituted amides. The ketoxime 25b was finally transformed into the corresponding activated oxime 26 by treatment with acetic anhydride for 1 h in 83% yield38. The acetylated intermediate was cyclized in pyridine to form the mixture of 1,3- and 1,2-benzoxazole 27–28 (3:7). At this stage, we introduced various alkyl groups (cyclo-, linear or branched alkyls) or benzyl on the hydroxyl group of the latter mixture 27–28 to obtain 1,3-benzoxazole 29a–e and 1,2-benzoxazole 30a–e, which were separated. The desired compounds 32a–e were synthesized, under the same conditions as previously described, in a four-step sequence through the lithiation reaction of methylated compounds 30a–e, the subsequent addition of the (iodomethyl)piperidine chains, the deprotection of the Boc groups before alkylating the piperidines by a methylcyclohexyl group (Fig. 4). Additionally, the compound 33 was obtained by O-debenzylation of the derivative 32e using hydrobromic acid in acetic acid in 85% yield.

Synthesis of C3 substituted-benzisoxazole by a C-C bond with various ether groups or a hydroxyl group in C4. Reagents and conditions: (a) NH2OH.HCl, NaOAc, H2O/EtOH (3:2), 1 h, rt then 3 h, reflux, quantitative yield (mixture of isomers E/Z 4:1) (b) Ac2O, 1 h, rt, 83% (c) pyridine, 1 h, reflux, 71% (mixture 27/28 3:7) (d) halogenated derivatives, K2CO3, rt, 15–48 h or reflux, 3 h, 32–98% (e) LDA 1.0 M sol., dry THF, −78 °C then tert-butyl 4-(iodomethyl)piperidine-1-carboxylate, 1 h, −78 °C, 21–47% (f) TFA, CH2Cl2, rt, 1 h (g) bromomethylcyclohexane, K2CO3, DMF, 110 °C, 3 h, 23–79% (2 steps) (h) HBr sol. (33 wt.% in AcOH), 50 °C, 3 h, 85%.
In vitro evaluation
Compounds 11–16, 21–23, 32a–e and 33 were evaluated in vitro for their capacity to inhibit hAChE using the spectrometric Ellman method39 and to bind to human 5-HT4R (h5-HT4R) in competition studies40 with the tritiated ligand [3H]–GR113808, a specific and highly potent 5-HT4R antagonist (Table 1). The 5-HT4R pharmacological profile was also determined for some compounds by quantification of cAMP production (Fig. 5).

Determination of the 5-HT4 pharmacological profiles for compounds 16, 32a and 33 by quantification of cAMP production. (A) Assay to determine the agonism activity (with serotonin and the partial agonist RS-67333 as references). (B) Assay to determine the antagonism activity (with the antagonist GR113808 as reference). Data presented are the mean of three independent experiments performed in duplicate.
Discussion
Correlating well with the results reported by Brodney et al.29 the first unsubstituted 1,2-benzoxazoles 11–13 with an ether link displayed potent in vitro affinity towards 5-HT4R (similar to donecopride with a Ki of 9.6 nM for the benzisoxazole analogue 11) but showed a loss of inhibition of the enzyme (Table 1).
Villalobos et al. have reported that, in comparison with unsubstituted compounds, an increase of AChE inhibition was observed by substitution of the benzisoxazole ring with electron-donating and some electron-withdrawing groups27. In accordance with these comments and in order to design donecopride-inspired compounds, the substitution of the benzisoxazole ring by a methoxy group in position C4 led to compounds 14 and 15 presenting a slight activity with a range of submicromolar inhibition of AChE (respectively 441 nM and 939 nM). The isobutyl-substituted piperidine derivative 16 did not show any inhibition of the enzyme. This isobutyl group probably cannot interact with AChE via the hydrophobic interaction conventionally observed with the tryptophan residue in the anionic subsite of the active site (residue Trp86 for hAChE). The introduction of the methoxy group also increased the in vitro capacity of derivatives 14–16 to bind to human 5-HT4R with an affinity of less than 5 nM.
Thanks to the pharmacomodulation of donecopride, we demonstrated that the modulation of the linker chain between the aromatic ring and the piperidine has an important impact on both pharmacological activities. Indeed, an amide or ester bond causes loss of AChE inhibition, while improving the affinity for 5-HT4R in comparison with two-carbon methylene bridge. Docking work has revealed a difference in the interaction of an amide or ester compound with the enzyme due to the rigidity induced by these structures (loss of one of the interactions)41.
The synthesis of compounds 21–22, with a two-carbon methylene link, seems to confirm this hypothesis because the compounds showed, in comparison with the ether analogues 11–13, much better AChE inhibition activity with respectively IC50s of 240 nM and 36 nM. The modification of an ether bond between the benzisoxazole and piperidine rings to a two-carbon methylene bond allows to increase flexibility and freedom of rotation of our structures, and to implement an AChE inhibition property. However, these structures presented in return a decrease in affinity towards 5-HT4R.
In order to better understand this difference docking studies of compounds 11 and 21 were performed into a human AChE structure co-crystallised with donepezil (PDB ED: 4EY742, Fig. 6A) using the GOLD software43,44. Firstly, compound 11 and 21 were built using Discovery Studio45 and protonated on piperidine ring following the ChemAxon software prediction (http://www.chemaxon.com/).

(A) Donepezil co-crystallized in the hAChE (PDB ED: 4EY7). Docking poses of the compound 21 (B) and the compound 11 (C) in hAChE binding sites. The compounds and the side chains of the binding site residues are in stick and the protein in ribbon representation. This figure was made with PYMOL (DeLano Scientific, 2002, San Carlo, USA).
During the docking of compound 11 and 21 into human AChE a water molecule interacting with protonated piperidine ring of donepezil was conserved (residue number 931). In the selected best scoring pose for compound 21 (Fig. 6B, ChemPLP Score = 101.62) its orientation was closed to the donepezil one. Indeed, compound 21 reproduced well the donepezil interactions: (i) the charged nitrogen of the piperidine ring is oriented in a position suitable for interacting with the water molecule slotting between Tyr337 and Tyr341, (ii) the nitrogen atom of benzisoxazole ring was close to NH of the Phe295 backbone and formed with it a hydrogen bond and in the same time the benzisoxazole ring was positioned in a parallel way with respect to the Trp286 indole ring of the PAS at a 3.1 Å distance to favor the π-stacking interaction. (iii) the cyclohexane ring occupied the donepezil benzyl ring place in neighboring of Trp86 and when the cyclohexane ring was replaced by benzyl one (compound 22) the AChE inhibition activities increased got closer to the donepezil one.
However, the best docked pose of compound 11 (ChemPLP Score = 98.21) does not allow us to lighten its worst AChE activity compared to the compound 21. The three attachment points observed for compound 21 were well reproduced in the compound 11 docking pose (see Fig. 6C). We could however postulate that the replacement of the methylene linker present in compound 21 by an oxygen atom in compound 11 could increase its rigidity due to its mesomeric effect as earlier described in Donecopride analog41. This rigidity might affect its binding within human AChE binding site.
Finally, the last chemical series was synthesized with both the two-carbon methylene link and various alkoxyls in position C4. The compounds 32a–32e demonstrated in most of the cases a correlation between the steric effect of the alkyl group and the results of both pharmacological activities. Except for the compound 32c without AChE inhibition, the increasing bulk of the alkoxyl group (O-benzyl 32e > O-cyclopentyl 32d > O-isobutyl 32c > O-ethyl 32b > O-methyl 32a) results in less % 5-HT4R affinity and AChE inhibition are interesting. The methoxy 32a and the alcohol 33 were the most promising of all benzisoxazoles synthesized in this work, with respectively IC50 values of 63.5 nM and 97.3 nM towards AChE and Ki values of 59 nM and 37 nM for the 5-HT4R.
On the basis of these results, the two benzisoxazoles 32a and 33 were selected to determine the 5-HT4R pharmacological profile of our compounds. In these assays both behaved as antagonists (Fig. 5) with IC50 of 97.2 ± 17.2 nM and 883.0 ± 597.8 nM, respectively. This profile is not in accordance with our objective. We determined also the 5-HT4R pharmacological profile of the compound 16 with an ether link and the compound also behaved as a highly potent antagonist (IC50 of 5.6 ± 2.2 nM).
In order to better understand their profile, docking studies were performed in a homology model of the 5-HT4 receptor. The docking of compound 32a was compared to Donecopride41, and appear similar which could explain their affinities for the 5-HT4R (Fig. 7). For both compounds the basic piperidine nitrogen interacts with Asp100 on the transmembrane helix 3 (consistent with the constraint used during docking) and the aromatic rings are oriented towards the transmembrane helix 5 (TM5) (in yellow in Fig. 7). However two differences can be underlined between both compounds poses: (i) lack of an H-bond interaction for compound 32a with Ser197 and (ii) lack of an electrostatic interaction for compound 32a with Thr104. One of these two interactions could therefore be responsible for 5-HT4R agonism/antogonism profile.

Compound 32a (A) and Donecopride (B) positioned in the 5-HT4R binding sites from docking studies. The compound and the side chains of the binding site residues are in stick and the protein in ribbon representation. This figure was made with PYMOL (DeLano Scientific, 2002, San Carlo, USA).
On the basis of this docking study, we could postulate that further modulation of the benzisoxazole moiety, either by introducing novel substituent or by modulation of the methoxy group will be needed to obtain a 5-HT4R agonist. Such modulation should reproduce one or the two missing H-bonds with Ser197 or Thr104 to reproduce the agonist profile of Donecopride41 or compound [A]29.
Conclusion
To briefly summarize, all benzisoxazoles synthesized allowed to establish structure-activity relationships and to study the impact of a pharmacomodulation of each part of the structure on the pharmacological results. In general, compounds with an ether link promote the affinity for 5-HT4R, whereas compounds with a two-carbon methylene link promote the inhibition of AChE. Several compounds have shown good activities in vitro on both targets and constitute a real basis for further works, in particular the compounds 32a and 33. It is necessary to combine all pharmacomodulations that have been performed to achieve the design of compounds meeting the selected criteria to belong to this MTDL family.
Methods
Chemistry
All commercially available compounds were used without further purification. Melting points were determined on a Köfler apparatus. Analytical thin-layer chromatography (TLC) was performed on silica gel 60 F254 on aluminium plates (Merck) and visualized with UV light (254 nm). Flash chromatography was conducted on a VWR SPOT II Essential instrument with silica gel 60 (40–63 µm). Column’s size and flow rate were used according to manufacturer’s recommendation. NMR spectra were recorded at 400 or 500 MHz (Bruker Avance III 400/500 MHz) for 1H NMR and at 100 or 125 MHz for 13C NMR in chloroform-d, methanol-d4 or DMSO-d6 with chemical shift (δ) given in parts per million (ppm) relative to TMS as internal standard and recorded at 295 K. The following abbreviations are used to describe peak splitting patterns when appropriate: br = broad, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, dd = doublet of doublet, dt = doublet of triplet. Coupling constants J are reported in hertz units (Hz). Infrared spectra (IR) were obtained on a PERKIN-ELMER FT-IR spectrometer and are reported in terms of frequency of absorption (cm−1) using KBr discs. High-resolution mass spectra (HRMS) were obtained by electronic impact (HRMS/EI), or by electrospray (HRMS/ESI) on a Bruker maXis mass spectrometer. LC-MS (ESI) analyses were realized with Waters Alliance 2695 as separating module using the following gradients: A (95%)/B (5%) to A (5%)/B (95%) in 4.00 min. This ratio was hold during 1.50 min before return to initial conditions in 0.50 min. Initial conditions were then maintained for 2.00 min (A = H2O, B = CH3CN; each containing HCOOH: 0.1%; column XBridge C18 2.5 µm/4.6 × 50 mm; flow rate 0.8 mL/min). MS were obtained on a SQ detector by positive ESI. Mass spectrum data are reported as m/z. X-ray diffraction experiments were performed with graphite–monochromatized Mo Kα radiation on a Bruker-Nonius Kappa CCD area detector diffractometer either at 298 K or at 150 K IUPAC nomenclature was used for all compounds. For the description of NMR spectra, the numbering used for hydrogens can be different than the IUPAC numbering. Compounds, that are not fully characterized, may already have been described in the literature, according to the references cited. Yields refer to chromatographically and spectroscopically (1H NMR) homogeneous materials and are mostly unoptimized. NMR spectroscopy (1H) was used to determine the proportions of mixture41.
Representative procedure for the synthesis of 11–16, 21–23 and 32a-e
To a stirred solution of tert-butyl piperidine-1-carboxylate derivative (1.0 eq.) in CH2Cl2 (20 mL/mmol) was added TFA (2 mL/mmol). The resulting mixture was stirred at room temperature for 1 h. Removal of the solvent under vacuum afforded the crude product, which was directly engaged in the next step. The residue obtained (1.0 eq.) was dissolved in the appropriate solvent (MeOH, DCM or EtOH, 10 mL/mmol), an alkyl-halogenated derivative (1.1 eq.) and a base (2.0–10.0 eq.) were then added. The resulting mixture was stirred at the appropriate temperature (room temperature, 80 °C or 110 °C) for 3–48 h, then concentrated in vacuo. Ethyl acetate was added, the organic layer was washed several times with brine, dried over MgSO4 and concentrated in vacuo. The crude was purified by chromatography on silica gel column and concentrated under reduced pressure to afford the corresponding alkylated compound41.
3-[[1-(cyclohexylmethyl)-4-piperidyl]methoxy]-1,2-benzoxazole (11)
The compound was prepared from tert-butyl 4-(1,2-benzoxazol-3-yloxymethyl)piperidine-1-carboxylate 9 (100 mg, 0.30 mmol, 1.0 eq.), bromomethylcyclohexane (47 µL, 0.33 mmol, 1.1 eq.) and K2CO3 (415 mg, 3.0 mmol, 10.0 eq.) in DMF according to the representative procedure (E) and stirring the reaction for 3 h at 110 °C. The crude was purified by flash chromatography on silica gel (cyclohexane/EtOAc, gradient 100:0 to 80:20) to give 11 as a yellow oil (22 mg, 22% yield over two steps); 1H NMR (CDCl3-d, 400 MHz) δ 7.65 (dt, 3J = 7.9 Hz, 4J = 5J = 1.0 Hz, 1H), 7.52 (ddd, 3J = 8.4 Hz, 3J = 7.0 Hz, 4J = 1.2 Hz, 1H), 7.43 (dt, 3J = 8.5 Hz, 4J = 5J = 0.8 Hz, 1H), 7.26 (m, 1H), 4.29 (d, 3J = 6.6 Hz, 2H), 2.92 (m, 2H), 2.12 (d, 3J = 7.0 Hz, 2H), 1.94–1.89 (m, 3H), 1.84–1.62 (m, 7H), 1.50–1.44 (m, 3H), 1.27–1.14 (m, 3H), 0.88 (m, 2H); 13C NMR (CDCl3-d, 100 MHz) δ 166.7, 163.9, 130.4, 122.9, 120.9, 114.4, 110.2, 74.8, 66.2, 53.9 (2 C), 35.8, 35.3, 32.1 (2 C), 28.8 (2 C), 26.8, 26.2 (2 C); LC-MS (ESI) tR = 3.65 min; m/z [M + H]+ 329.59; HRMS/ESI: m/z calcd. for C20H29N2O2 [M + H]+ 329.2224, found 329.2224; IR (neat, cm−1) ν 2925, 2848, 2801, 2763, 1614, 1538, 1449, 1361, 1237, 1158, 989, 922, 751.
Docking studies (hAChE)
For each docked compound a preliminary calculation on its protonation state at pH 7.4 was carried out using standard tools of the ChemAxon Package (Marvin 16.1.4.0, 2016, ChemAxon (http://www.chemaxon.com/) and the majority protonated microspecies at this pH was built by Discovery Studio software (Discovery Studio Modeling Environment, release 3.5, San Diego, CA: 2012) for docking studies.
The crystallographic coordinates of human acetylcholinesterase used for docking studies were obtained from X-ray structure of the donepezil/AChE complex (PDB ID 4EY7, a structure refined to 2.35 Å with an R factor of 17.7%)41.
The docking of the compounds into the hAChE was carried out with the GOLD program (v5.6.3) using the default parameters43,44. This program applies a genetic algorithm to explore conformational spaces and ligand binding modes. To evaluate the proposed ligand positions, the ChemPLP fitness function was applied in these docking studies. The binding site in the hAChE model was defined as a 7 Å sphere from the co-crystallized ligand donepezil and a water molecule interacting with protonated piperidine ring of donepezil was conserved during the docking (residue number 931).
Docking studies (5-HT4R)
In this study, the 3D model of the human 5-HT4R built previously by homology sequence approach46 was used. This model was generated using the crystal structure (PBD: 2RH1) of the human β2 adrenergic receptor-T4 lysozyme fusion protein complexed with the carazolol47 as a template. The docking of the 32a compound and donecopride into the generated model was carried out with the GOLD program (v5.7.3) using the default parameters43,44 and the proposed ligand positions were evaluated by the ChemPLP fitness function. The binding site in the 5-HT4R model was defined as a 10 Å sphere centred on the aspartic acid residue Asp100. As the mutagenesis studies have shown that the interaction between the positively ionisable amine of ligands and Asp100 of 5-HT4R is crucial for ligand binding, a hydrogen bond constraint between positively ionisable amine ligand and OD atom of Asp100 was used during the docking48. Furthermore, special attention was paid during the docking procedure to the following amino acids in the binding site, which were kept flexible: Arg96, Asp100, Thr104, Tyr192, Ser197 and Trp294.
Pharmacological characterization of drugs on human 5-HT4R
The method was validated from saturation studies: six concentrations of [3H]GR113808 were used to give final concentrations of 0.0625–2 nM, and nonspecific binding of [3H]GR113808 was defined in the presence of 30 μM serotonin to determine the Kd and the Bmax. The competition studies used membrane preparations made from proprietary stable recombinant cell lines expressing the 5-HT4(b) receptor to ensure high-level of GPCR surface expression (HTS110M, Millipore). Membranes (2.5 µg protein) were incubated in duplicate at 25 °C for 60 min in the absence or the presence of 10–6 or 10–8M of each drug and 0.2 nM [3H]-GR 113808 (VT 240, ViTrax) in 25 mM Tris buffer (pH 7.4, 25 °C). At the end of the incubation, homogenates were filtered through Whatman GF/C filters (Alpha Biotech) pre-soaked with 0.5% polyethylenimine using a Brandel cell harvester. Filters were subsequently washed three times with 4 mL of ice-cold 25 mM Tris buffer (pH 7.4, 4 °C). Non-specific binding was evaluated in parallel in the presence of 30 μM serotonin.
For some of these compounds, affinity constants were calculated from five-point inhibition curves using the EBDA-Ligand software and expressed as Ki ± SD41.
In vitro tests of AChE biological activity
Inhibitory capacity of compounds on AChE biological activity was evaluated through the use of the spectrometric method of Ellman39. Acetylthiocholine and 5,5-dithiobis-(2-nitrobenzoic) acid (DTNB) were purchased from Sigma Aldrich. AChE from human erythrocytes (buffered aqueous solution, ≥500 units/mg protein (BCA), Sigma Aldrich) was diluted in 20 mM HEPES buffer pH 8, 0.1% Triton X-100 such as to have enzyme solution with 0.25 unit/mL enzyme activity. In the procedure, 100 μL of 0.3 mM DTNB dissolved in phosphate buffer pH 7.4 were added into the 96 wells plate followed by 50 μL of test compound solution and 50 μL of enzyme (0.05 U final). After 5 min of preincubation at 25 °C, the reaction was then initiated by the injection of 50 μL of 10 mM acetylthiocholine iodide solution. The hydrolysis of acetylthiocholine was monitored by the formation of yellow 5-thio-2-nitrobenzoate anion as the result of the reaction of DTNB with thiocholine, released by the enzymatic hydrolysis of acetylthiocholine, at a wavelength of 412 nm using a 96-well microplate plate reader (TECAN Infinite M200, Lyon, France). Test compounds were dissolved in analytical grade DMSO. Donepezil was used as a reference standard. The rate of absorbance increase at 412 nm was followed every minute for 10 min. Assays were performed with a blank containing all components except acetylthiocholine, in order to account for non-enzymatic reaction. The reaction slopes were compared and the percent inhibition due to the presence of test compounds was calculated by the following expression: 100 − (vi/v0 × 100) where vi is the rate calculated in the presence of inhibitor and v0 is the enzyme activity.
First screening of AChE activity was carried out at a 10−6 or 10−5 M concentration of compounds under study. For the compounds with significant inhibition (≥50%), IC50 values were determined graphically by plotting the % inhibition versus the logarithm of six inhibitor concentrations in the assay solution using the Origin software.
Determination of cAMP production
COS-7 cells were purchased from ATCC (ATCC CRL-1651; LGC STANDARTS, Molsheim, France). T were grown in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% dialyzed fetal calf serum (dFCS) and antibiotics. Cells were transiently transfected with plasmid encoding HA-tagged human 5-HT4 receptor, then seeded in 96-well plates (35,000 cells/well). 24 hrs after transfection, cells were washed once with 200 µl of HBS (20 mM HEPES; 150 mM NaCl; 4.2 mM KCl; 0.9 mM CaCl2; 0.5 mM MgCl2; 0.1% glucose; 0.1% BSA) and, after HBS removal, exposed to the indicated concentrations of 5-HT4R ligands in the presence of 0.1 mM of the phosphodiesterase inhibitor RO-20-1724, at 37 °C in 100 µl of HBS. After 10 min, cells were then lysed by addition of the same volume of Triton-X100 (0.1%) during 30 min at 37 °C. The competition assay in antagonist mode was performed as follows: after one wash in 200 µl of HBS, cells were exposed to 5-HT4R ligands at twice the indicated concentration, at 37 °C in 50 µl of HBS. After 7 min, serotonin (final concentration of 5.10−7M) was added in 50 µl of HBS in the presence of the phosphodiesterase inhibitor RO-20-1724 (final concentration of 0.1 mM). After 10 min, cells were then lysed by addition of the same volume of Triton-X100 (0.1%) during 30 min at 37 °C. Quantification of cAMP production was performed by HTRF by using the cAMP Dynamic kit (Cisbio Bioassays) according to the manufacturer’s instructions
References
World Alzheimer Report. The state of the art of dementia research: New frontiers, https://www.alz.co.uk/research/world-report-2018 (2018)
Stelzmann, R. A., Schnitzlein, H. N. & Murtagh, F. R. An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin. Anat. 8, 429–431, https://doi.org/10.1002/ca.980080612 (1995).
Bird, T. D. Genetic aspects of Alzheimer disease. Genet. Med. 10, 231–239, https://doi.org/10.1097/GIM.0b013e31816b64dc (2008).
Melnikova, I. Therapies for Alzheimer’s disease. Nat. Rev. Drug. Discov. 6, 341–342, https://doi.org/10.1038/nrd2314 (2007).
Cummings, J. L., Morstorf, T. & Zhong, K. Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimer’s Res. Ther. 6, 37, https://doi.org/10.1186/alzrt269 (2014).
Mangialasche, F., Solomon, A., Winblad, B., Mecocci, P. & Kivipelto, M. Alzheimer’s disease: clinical trials and drug development. Lancet Neurol. 9, 702–716, https://doi.org/10.1016/S1474-4422(10)70119-8 (2010).
Morphy, R. & Rankovic, Z. Designed multiple ligands. An emerging drug discovery paradigm. J. Med. Chem. 48, 6523–6543, https://doi.org/10.1021/jm058225d (2005).
Cavalli, A. et al. Multi-target-directed ligands to combat neurodegenerative diseases. J. Med. Chem. 51, 347–372, https://doi.org/10.1021/jm7009364 (2008).
Lewis, W. G. et al. Click chemistry in situ: acetylcholinesterase as a reaction vessel for the selective assembly of a femtomolar inhibitor from an array of building blocks. Angew Chem Int Ed 41, 1053–1057, 10.1002/1521-3773(20020315)41:6<1053::AID-ANIE1053>3.0.CO;2-4 (2002).
Garcia-Palomero, E. et al. Potent beta-amyloid modulators. Neurodegener. Dis. 5, 153–156, https://doi.org/10.1159/000113688 (2008).
Inestrosa, N. C., Sagal, J. P. & Colombres, M. Acetylcholinesterase interaction with Alzheimer amyloid beta. Subcell. Biochem. 38, 299–317, https://doi.org/10.1007/0-387-23226-5_15 (2005).
Alvarez, A. et al. Stable complexes involving acetylcholinesterase and amyloid-beta peptide change the biochemical properties of the enzyme and increase the neurotoxicity of Alzheimer’s fibrils. J. Neurosci. 18, 3213–3223, https://doi.org/10.1523/JNEUROSCI.18-09-03213.1998 (1998).
Reyes, A. E. et al. Acetylcholinesterase-Abeta complexes are more toxic than Abeta fibrils in rat hippocampus: effect on rat beta-amyloid aggregation, laminin expression, reactive astrocytosis, and neuronal cell loss. Am. J. Pathol. 164, 2163–2174, https://doi.org/10.1016/S0002-9440(10)63774-1 (2004).
Agis-Torres, A., Sollhuber, M., Fernandez, M. & Sanchez-Montero, J. M. Multi-Target-Directed Ligands and other Therapeutic Strategies in the Search of a Real Solution for Alzheimer’s Disease. Curr. Neuropharmacol. 12, 2–36, https://doi.org/10.2174/1570159X113116660047 (2014).
Kowal, N. M. et al. Novel Approach for the Search for Chemical Scaffolds with Activity at Both Acetylcholinesterase and the α7 Nicotinic Acetylcholine Receptor: A Perspective on Scaffolds with Dual Activity for the Treatment of Neurodegenerative Disorders. Molecules 24, 446, https://doi.org/10.3390/molecules24030446 (2019).
Lalut, J., Rochais, C. & Dallemagne, P. Multiple Ligands in Neurodegenerative Diseases, Drug Selectivity – An Evolving Concept in Drug Discovery, Book series “Methods and Principles in Medicinal Chemistry”, Wiley-VCH Publishing House. https://doi.org/10.1002/9783527674381.ch16 (2017).
Lecoutey, C. et al. Synthesis of dual AChE/5-HT4 receptor multi-target directed ligands. Med. Chem. Commun. 3, 627–634, https://doi.org/10.1039/c2md20063e (2012).
Jourdan, J.-P. et al. Novel benzylidenephenylpyrrolizinones with pleiotropic activities potentially useful in Alzheimer’s disease treatment. Eur. J. Med. Chem. 114, 365–379, https://doi.org/10.1016/j.ejmech.2016.03.023 (2016).
Lalut, J. et al. Novel Multitarget-Directed Ligands Targeting Acetylcholinesterase and σ1 Receptors as Lead Compounds for Treatment of Alzheimer’s Disease: Synthesis, Evaluation, and Structural Characterization of their Complexes with Acetylcholinesterase. Eur. J. Med. Chem. 162, 234–248, https://doi.org/10.1016/j.ejmech.2018.10.064 (2019).
Lecoutey, C. et al. Design of donecopride, a dual serotonin subtype 4 receptor agonist/acetylcholinesterase inhibitor with potential interest for Alzheimer’s disease treatment. Proc. Natl Acad. Sci. US 111, E3825–E3830, https://doi.org/10.1073/pnas.1410315111 (2014).
Bockaert, J., Claeysen, S., Compan, V. & Dumuis, A. 5-HT(4) receptors: history, molecular pharmacology and brain functions. Neuropharmacol. 55, 922–931, https://doi.org/10.1016/j.neuropharm.2008.05.013 (2008).
Matsumoto, M. et al. Evidence for involvement of central 5-HT(4) receptors in cholinergic function associated with cognitive processes: Behavioral, electrophysiological, and neurochemical studies. J. Pharmacol. Exp. Ther. 296, 676–682 (2001).
Consolo, S., Arnaboldi, S., Giorgi, S., Russi, G. & Ladinsky, H. 5-HT4 receptor stimulation facilitates acetylcholine release in rat frontal cortex. Neuroreport 5, 1230–1232, https://doi.org/10.1097/00001756-199406020-00018 (1994).
Lamirault, L. & Simon, H. Enhancement of place and object recognition memory in young adult and old rats by RS67333, a partial agonist of 5-HT4 receptors. Neuropharmacol. 41, 844–853, https://doi.org/10.1016/S0028-3908(01)00123-X (2001).
Russo, O. et al. Design, synthesis, and biological evaluation of new 5-HT4 receptor agonists: application as amyloid cascade modulators and potential therapeutic utility in Alzheimer’s disease. J. Med. Chem. 52, 2214–2225, https://doi.org/10.1021/jm801327q (2009).
Freret, T. Synergistic effect of acetylcholinesterase inhibition (donepezil) and 5-HT4 receptor activation (RS67333) on object recognition in mice. A new hope to design a treatment for Alzheimer’s disease. Behav. Brain Res. 230, 304–308, https://doi.org/10.1016/j.bbr.2012.02.012 (2012).
Villalobos, A. et al. Novel Benzisoxazole Derivatives as Potent and Selective Inhibitors of Acetylcholinesterase. J. Med. Chem. 37, 2721–2734, https://doi.org/10.1021/jm00043a012 (1994).
Sugimoto, H. et al. Novel Piperidine Derivatives. Synthesis and Anti-Acetylcholinesterase Activity of 1-Benzyl-4-[2-(N-benzoylamino)ethyl]piperidine Derivatives. J. Med. Chem. 33, 1880–1887, https://doi.org/10.1021/jm00169a008 (1990).
Brodney, M. A. et al. Identification of Multiple 5-HT4 Partial Agonist Clinical Candidates for the Treatment of Alzheimer’s Disease. J. Med. Chem. 55, 9240–9254, https://doi.org/10.1021/jm300953p (2012).
Dubrovskiy, A. V. & Larock, R. C. Synthesis of Benzisoxazoles by the [3 + 2] Cycloaddition of in situ Generated Nitrile Oxides and Arynes. Org. Lett. 12, 1180–1183, https://doi.org/10.1021/ol902921s (2010).
Chen, C.-y et al. A Divergent and Selective Synthesis of Isomeric Benzoxazoles from a Single N–Cl Imine. Org. Lett. 13, 6300–6303, https://doi.org/10.1021/ol202844c (2011).
Shutske, G. M. A new synthesis of 3-phenyl-1,2-benzisoxazoles: sterically constrained 3-phenyl-1,2-benzisoxazoles by intramolecular carbon:nitrogen bond formation at a hindered carbonyl group. J. Org. Chem. 49, 180–183, https://doi.org/10.1021/jo00175a042 (1984).
Liu, W. et al. Benzimidazolones: a new class of selective peroxisome proliferator-activated receptor γ (PPARγ) modulators. J. Med. Chem. 54, 8541–8554, https://doi.org/10.1021/jm201061j (2011).
Hoare, D. G. et al. The reaction of hydroxamic acids with water-soluble carbodiimides. A lossen rearrangement. J. Am. Chem. Soc. 90, 1638–1643, https://doi.org/10.1021/ja01008a040 (1968).
Mitsunobu, O. & Yamada, M. Preparation of Esters of Carboxylic and Phosphoric Acid via Quaternary Phosphonium Salts. Bull. Chem. Soc. Jpn. 40, 2380–2382, https://doi.org/10.1246/bcsj.40.2380 (1967).
Udd, S. et al. Copper-catalyzed cyclization of Z-oximes into 3-methyl-1,2-benzisoxazoles. Tetrahedron Lett. 51, 1030–1033, https://doi.org/10.1016/j.tetlet.2009.12.070 (2010).
Iranpoor, N. et al. A novel method for the highly efficient synthesis of 1,2-benzisoxazoles under neutral conditions using the Ph3P/DDQ system. Tetrahedron Lett. 47, 8247–8250, https://doi.org/10.1016/j.tetlet.2006.09.120 (2006).
Alvaro, G. et al. Dérivés d’hydantoïne utiles en tant qu’inhibiteurs de kv3. WO 2012/076877 A1 (2012).
Ellman, G. L. et al. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 7, 88–95, https://doi.org/10.1016/0006-2952(61)90145-9 (1961).
Grossman, C. J. et al. Development of a radioligand binding assay for 5-HT4 receptors in guinea-pig and rat brain. Br. J. Pharmacol. 109, 618–624, https://doi.org/10.1111/j.1476-5381.1993.tb13617.x (1993).
Rochais, C. et al. Novel Multitarget-Directed Ligands (MTDLs) with Acetylcholinesterase (AChE) Inhibitory and Serotonergic Subtype 4 Receptor (5-HT4R) Agonist Activities As Potential Agents against Alzheimer’s Disease: The Design of Donecopride. J. Med. Chem. 58, 3172–3187, https://doi.org/10.1021/acs.jmedchem.5b00115 (2015).
Cheung, J. et al. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 55, 10282–10286, https://doi.org/10.1021/jm300871x (2012).
Jones, G., Willett, P. & Glen, R. C. Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J. Mol. Biol. 245, 43–53, https://doi.org/10.1016/S0022-2836(95)80037-9 (1995).
Jones, G. et al. Development and Validation of a Genetic Algorithm for Flexible Docking. J. Mol. Biol. 267, 727–748, https://doi.org/10.1006/jmbi.1996.0897 (1997).
Discovery Studio Modeling Environment, release 4.5, San Diego, CA: (2012).
Dubost, E. et al. Synthesis and Structure-Affinity Relationships of Selective High-Affinity 5-HT4 Receptor Antagonists: Application to the Design of New Potential Single Photon Emission Computed Tomography Tracers. J. Med. Chem. 55, 9693–9707, https://doi.org/10.1021/jm300943r (2012).
Cherezov, V. et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Sci. 318, 1258–1265, https://doi.org/10.1126/science.1150577 (2007).
Rivail, L. et al. New insights into the human 5-HT4 receptor binding site:Exploration of a hydrophobic pocket. Br. J. Pharmacol. 143, 361–370, https://doi.org/10.1038/sj.bjp.0705950 (2004).
Acknowledgements
This work was supported by funding from the French Agence Nationale de la Recherche Projects MALAD ANR-12-JS007–0012–01. The authors gratefully acknowledge the Conseil Régional de Normandie, as well as the European Community (FEDER) for their contribution to the CERMN’s analytical platform. European COST action CA15135 (Multi-Target Paradigm for Innovative Ligand Identification in the Drug Discovery Process, MuTaLig) supports this article. This work was also supported by funding from the Fondation Alzheimer (AAP2015 Project TRIAD 016). Part of this work was performed using computing resources of CRIANN (Normandy, France) as well as the European Community (FEDER) for the molecular modeling software.
Author information
Authors and Affiliations
Contributions
S.C., P.D. and C.R. designed the research and the manuscript was written through contributions of J.L., S.C., J.S.d.O.S. and C.R. J.L. performed the synthesis and H.P., A.D., C.L. and R.L. performed the experiments. J.S.d.O.S. performed the docking experiments. All authors have given approval to the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lalut, J., Payan, H., Davis, A. et al. Rational design of novel benzisoxazole derivatives with acetylcholinesterase inhibitory and serotoninergic 5-HT4 receptors activities for the treatment of Alzheimer’s disease. Sci Rep 10, 3014 (2020). https://doi.org/10.1038/s41598-020-59805-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-59805-7
This article is cited by
-
Characterization of β-secretase inhibitory extracts from sea cucumber (Stichopus japonicus) hydrolysis with their cellular level mechanism in SH-SY5Y cells
European Food Research and Technology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
