
Abstract
Highly porous self-assembled nanostructured Ni@C and NiO@C were synthesized via calcination of a Ni-based metal–organic framework. The morphology, structure, and composition of as synthesized Ni@C and NiO@C were characterized by SEM, FIB-SEM, TEM, and XRD. The electro-catalytic activity of the Ni@C and NiO@C catalysts towards urea oxidation was investigated using cyclic voltammetry. It was found that the Ni@C had a higher residual carbon content and a higher specific surface area than NiO@C, thus exhibiting an enhanced electrochemical performance for urea oxidation. A direct urea fuel cell with Ni@C as an anode catalyst featured an excellent maximum power density of 13.8 mW cm−2 with 0.33 M urea solution in 1 M KOH as fuel and humidified air as oxidant at 50 °C, additionally showing excellent stability during continuous 20-h operation. Thus, this work showed that the highly porous carbon-supported Ni catalysts derived from Ni-based metal–organic framework can be used for urea oxidation and as an efficient anode material for urea fuel cells.
Similar content being viewed by others


Introduction
The non-toxicity, affordability, and ease of storage/transport of urea make direct urea fuel cells (DUFCs) promising power sources1,2. The synthesis of urea features the reaction of ammonia with carbon dioxide, thus simultaneously achieving carbon dioxide fixation and the conversion of toxic ammonia into a non-toxic product2. Moreover, DUFCs can be operated using urine or urea-rich wastewater as fuels. Although the high energy density (16.9 MJ L−1) and high solubility of urea allow its use as a hydrogen carrier in solid oxide fuel cells3, its direct utilization for electrical energy production is believed to bring about further system simplification and increase the energy conversion efficiency4. In a urea/O2 type DUFC, urea is electro-oxidized at the anode (Eq. 1), while oxygen is reduced at the cathode (Eq. 2):
Although the theoretical open circuit voltage (OCV) of a urea/O2 fuel cell equals 1.147 V at room temperature which is lower than that of a H2/O2 fuel cell (1.23 V)5, the theoretical efficiency of the former cell is ~20% higher than that of the latter6 due to the positive entropy change of the overall reaction (Eq. 3). In addition, DUFCs can be operated using anion exchange membranes and catalysts based on non-precious metal such as Ni that exhibit good activity in alkaline environments3. However, the practical implementation of DUFCs is hindered by their relatively low power density of 5–10 mW cm−2 compared to H2 and CH3OH fueled anion exchange membrane fuel cells which have typical maximum power densities of 500–600 and 100–300 mW cm2, respectively3,7,8. Hence, most research efforts are directed at the development of suitable anode catalysts to speed up the sluggish kinetics of the six-electron-transfer urea oxidation reaction (UOR). Currently, non-precious Ni is considered to be the most efficient anode catalyst9,10 in DUFCs. The activity and stability of Ni-based catalysts have been improved by doping with various transient metals such as Co, Mo, Cr, Mn, and Fe11,12,13,14. In addition, nanostructured Ni catalysts (e.g., those featuring nanowires10,15, nanosheets9,16, and nanoparticles17,18) and/or composition with carbon-based nanostructures including carbon nanotubes19,20, graphene17, and aerogels21 are reported to exhibit increased numbers of active sites, electrical connections, porosity, and stability.
Recently, the highly tunable porosity, large surface area, and open metal sites of metal-organic frameworks (MOFs) have attracted much attention, allowing MOFs to be viewed as excellent electrochemically functional materials22. For instance, we have previously demonstrated the excellent catalytic activity of a Ni-based MOF (Ni-MOF) for urea oxidation20. However, the electrical conductivity of Ni-MOFs is generally poor, requiring hybridization with highly conductive compounds such as carbon nanotubes to obtain high urea oxidation currents20. Furthermore, the implementation of MOFs as electrochemical catalysts is restricted by their weak mechanical stability23,24,25. Ni-MOF has been pyrolyzed to obtain NiO@C for lithium storage. The NiO@C was reported to exhibit a hierarchical core-shell structure and feature a large surface area and high porosity26 due to the intrinsic pore structure of the MOF precursor27. Additionally, an iron-based MOF has been utilized to prepare a spindle-like porous α-Fe2O3 electrocatalyst28.
In this study, we pyrolized Ni-MOF under air and N2 environment to obtain novel NiO@C and Ni@C catalysts, respectively, for UOR. The structural, morphological, and electrochemical properties of the prepared NiO@C and Ni@C catalysts were characterized. The performance of DUFCs (urea/O2 fuel cells) comprising the NiO@C and Ni@C catalysts as anode materials was evaluated.
Results and Discussion
Characterization of Ni-MOF, Ni@C, and NiO@C
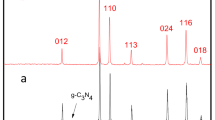
The crystallographic structure and phase purity of the Ni-MOF precursor, Ni@C, and NiO@C powders were analyzed by XRD (Fig. 1a). Ni-MOF exhibited no appreciable crystalline phase peaks, which indicated its amorphous nature and was in agreement with previous reports20,26. In contrast, the patterns of Ni@C and NiO@C showed peaks of metallic Ni [(111), (200), (220); JCPDF No. 65-0380]29 and NiO [(111), (220), (220), (311), (222), (225); JCPDF No. 65-2901]26, confirming the successful synthesis of the above catalysts. Moreover, the absence of any impurity peaks in the above patterns confirmed that the thermal decomposition of Ni-MOF was complete and afforded phase-pure Ni and NiO. In addition, the co-presence of carbon in Ni@C and NiO@C was confirmed by the observation of a peak at 2θ = 23.2°30. The presence of residual carbon was thought to account for the self-assembled hierarchical structure of Ni@C and NiO@C, as is further discussed later in the text. The carbon content of Ni@C (calcined in N2) and NiO@C (calcined in air) were 27.0 and 0.2 wt%, respectively, as measured by an elemental analyzer (Table 1). The difference in carbon content of Ni@C and NiO@C was close to the difference in remaining weight of each sample as shown in TG analysis (Suppl. Fig. S1) due to the removal of carbon by CO2 formation during the calcination in air environment. The XPS spectra of Ni-MOF, NiO@C and Ni@C are presented in Fig. 1b, indicating the presence of Ni, carbon and oxygen. The deconvulated core level spectra of the different materials shows two distinct peaks corresponding to Ni 2p3/2 and Ni 2p1/2 (Suppl. Fig. S2). In case of Ni-MOF, the peak at 855.6 eV with satellite peak at 860.9 eV was assigned to the Ni 2p3/2, and the peak at 873.0 eV with satellite peak at 880.0 eV was assigned to the Ni 2p1/2, which are typical peaks for the Ni. In case of NiO@C and Ni@C the Ni 2p3/2 is centered on 852.2 eV and 853.3 eV possible suggesting the existence of metallic Ni. Besides, the appearance of a shoulder peak at 855.33 eV for NiO@C and Ni@C suggests that some oxides of the Ni are also present in the structure.

(a) XRD patterns of Ni-MOF, Ni@C, and NiO@C and (b) XPS spectra of Ni-MOF, NiO@C, and Ni@C.
Figure 2 shows SEM and cross-sectional FIB-SEM images of as-prepared Ni-MOF, Ni@C, and NiO@C particles, revealing that those of Ni-MOF were spherical (Fig. 2a-1,a-2) with a porous inside (Fig. 2a-3). Similarly, Ni@C and NiO@C also featured spherical particles with a more porous structure, which reflected the decomposition of organic compounds in the Ni-MOF precursor. NiO@C particles, obtained by calcination in air, exhibited a ball-in-ball structure with a crevice between the spherical core and the shell (Fig. 2b), as observed previously26, probably because fast mass transfer and high weight loss during calcination in air resulted in core shrinkage and the formation of a gap between the core and the shell. In contrast, Ni@C, obtained by calcination in nitrogen, retained the structure of the Ni-MOF precursor, featuring more abundant and uniformly distributed pores (Fig. 2c). The SEM images also showed that each catalyst particle comprised nano-sized subunits forming nano-sized pores. In addition, the elemental maps of both the catalysts revealed uniform distributions of Ni and C, and the elemental spectra showed that the Ni/C ratio was close to that observed by an elemental analyzer (Suppl. Fig. S3).

SEM (1, 2) and cross-sectional FIB-SEM (3) images of Ni-MOF (a), NiO@C (b), and Ni@C (c).
The structures of Ni@C and NiO@C subunits were characterized by TEM. Figure 3 clearly shows that Ni@C and NiO@C particles comprised clusters of individual uniformly distributed nanoparticles with average sizes of ~50 nm and clusters of randomly distributed nanoparticles with sizes of 10–50 nm, respectively. Interestingly, the subunit particles of Ni@C featured two distinct regions (Fig. 3a-2), i.e., a Ni-abundant core (darker area) and a carbon-abundant outer layer (lighter area)27. However, no clear distinct regions were observed in NiO@C subunit particles, because NiO@C contained only 0.2 wt% carbon. Therefore, the uniform distribution of fine Ni particles in Ni@C was attributed to the presence of carbon therein. Selected area electron diffraction (SAED) patterns (insets of Fig. 3a-3,b-3) confirmed the presence of NiO (JCPDF No. 65-2901)26 and cubic-structured Fm-3m Ni (JCPDF 65-0380)29 in NiO@C and Ni@C, respectively, in good agreement with XRD results.

TEM (1, 2) and HRTEM (3) images of Ni@C (a) and NiO@C (b). Insets show SAED pattern.
The structural differences of Ni-MOF, Ni@C, and NiO@C resulted in different specific surface areas and porosities as shown in Table 1. Calcination of Ni-MOF considerably increased BET specific surface area and porosity, depending on calcination conditions. Ni@C, which was calcined in N2, had higher carbon content and higher BET surface area than NiO@C, which was calcined in air. The result might indicate that the carbon content affected the particle morphology and specific surface area of calcined products. The average pore sizes of Ni-MOF, Ni@C, and NiO@C particles were calculated from the Barrett-Joyner-Halenda (BJH) pore size distribution curve as 8–12 nm (Suppl. Fig. S4), which were indicative of mesoporosity.
Electrocatalytic properties of Ni-MOF, Ni@C, and NiO@C
The catalytic activities of Ni-MOF, Ni@C, and NiO@C for urea electro-oxidation were examined by cyclic voltammetry (CV) measurements (Fig. 4). All CV curves of Ni-based catalysts in the absence of urea (Fig. 4a) exhibited a pair of redox peaks at 0.5–0.6 and 0.15–0.35 V vs. Ag/AgCl, which corresponded to anodic and cathodic peak potentials, respectively, and indicated the previously reported inter-conversion of Ni2+/Ni3+ in alkaline medium31,32. In the presence of 0.1 mM urea in 0.1 M KOH, the Ni-based catalysts (Fig. 4b) exhibited redox peaks (with increased oxidation peak intensity and decreased reduction peak intensity) at the similar potentials as those observed in 0.1 M KOH in the absence of urea, which confirmed the activity of these catalysts for urea oxidation occurring via Ni2+/Ni3+ redox pathway; i.e., Ni3+OOH was formed by electrochemical oxidation of Ni2+(OH)2 and it catalyzed chemical oxidation of the adsorbed urea while being reduced to Ni2+OH, called an indirect electrochemical-chemical (EC) mechanism13,33. The result, therefore, implied that Ni3+OOH, a high valence of Ni oxide34,35, was the active site for UOR. As seen in Fig. 4, redox peak potentials were shifted by the presence of urea. Although further study should be needed for the clear explanation on the potential shift, it might be due to the reactions involving the adsorbed urea. Among the catalysts tested, Ni@C exhibited the highest peak current density of 30.3 mA cm–2, whereas values of 22.0 and 25.8 mA cm−2 were obtained for Ni-MOF and NiO@C, respectively. The results indicated that Ni@C and NiO@C derived from Ni-MOF exhibited enhanced catalytic activities for UOR compared to that of their precursor, which was mainly ascribed to their improved surface area and porosity. Furthermore, the highest electrochemical activity of Ni@C was ascribed to its high BET surface area and the co-presence of carbon, which could increase electron conductivity26, as evidenced by the resistivity measurement. As shown in Fig. 5, Ni@C powder pellet, which contained highest residual carbon, exhibited lowest resistivity and thus highest catalytic activity. The positive role of carbon on the catalytic performance via the enhanced conductivity was also reported previously36.

CV curves of Ni-MOF (a), NiO@C (b), and Ni@C (c) recorded in the absence and presence of 0.1 M urea in 0.1 M KOH at scan rate of 10 mV s−1.

Electrical resistivity of Ni-MOF, NiO@C, and Ni@C pellets as measured by a four-point probe method.
The electrochemical UOR on the Ni@C catalyst was appeared to be an irreversible diffusion-controlled process because the peak current density for the urea oxidation increased linearly with the square root scan rate (Suppl. Fig. S5) as observed previously33,34. In addition, the effect of urea and KOH concentration revealed saturation kinetics of UOR on the Ni@C catalyst (Suppl. Figs. S6 and S7).
Electrochemical active surface areas (EASAs) were calculated from the CV data (Fig. 4) as described elsewhere21,37. The calculation procedure and other electrochemical parameters extracted from CV data are presented in Suppl. Table S1. The EASAs of Ni-MOF, NiO@C, and Ni@C were 45.3, 55.6, and 95.1 m2 g−1, respectively, and agreed with the results of BET surface area measurements. The highest EASA of Ni@C was ascribed to the good dispersion of fine Ni active sites owing to its uniform and highly porous structure. Chronoamperometry measurements revealed that all catalysts exhibited a stable urea oxidation current for 3600 s (Suppl. Fig. S8) with the highest current observed for Ni@C being in agreement with the results of CV measurements.
Performances of DUFCs with Ni-MOF, Ni@C, and NiO@C anode catalysts
DUFCs (urea/O2 type) were fabricated using Ni-MOF, Ni@C, and NiO@C catalysts as anode materials. The I-V polarization and power density curves of these cells recorded in 0.1 M urea/1.0 M KOH at 50 °C are shown in Fig. 6, and the maximum power densities (MPDs) of cells comprising Ni-MOF, NiO@C, and Ni@C anode catalysts were obtained as 2.91, 3.21, and 6.99 mW cm−2, respectively. The best fuel cell performance was observed for the Ni@C anode catalyst, which was mainly ascribed to its higher BET surface area and EASA, as discussed earlier.

Performances DUFCs with various anode materials (Ni-MOF, NiO@C, and Ni@C) in 0.1 M urea and 1.0 M KOH at 50 °C.
To optimize fuel cell operating conditions, the effects of urea concentration and operating temperature were studied for DUFC with Ni@C anode. As shown in Fig. 7, as the urea concentration increased in the range of 0.1–0.33 M, the MPD reached a maximum and then decreased as the urea concentration further increased, which probably reflected the deactivation and/or blockage of the catalyst at such high concentrations. At a urea concentration of 0.5 M, a transition zone (i.e., decrease of current density) was observed in the polarization curve, indicating catalyst site blockage33,38. The formation of the transition zone was previously attributed to the sluggish kinetics and flooding of catalyst pores by water and products of urea electro-oxidation, which prevented urea molecules from reaching the catalyst sites39,40.

Performances of the DUFC with Ni@C as anode at different urea concentrations in 1.0 M KOH at 50 °C.
The performance of DUFC was also significantly affected by operating temperature, e.g., the MPD increased approximately threefold as the temperature increased from 25 to 50 °C (Fig. 8a), mainly because of the concomitant increase of electrochemical redox reaction rates and the ion conductivity of the AEM41,42. It should be noted that a further increase of operating temperature to values above 50 °C would enhance cell performance because CV measurements obtained for Ni@C at temperatures of 25–80 °C revealed that the peak current for urea oxidation increased continuously with temperature as shown in Suppl. Fig. S9. However, the AEM used in this study was not stable at higher temperatures, which reflected the necessity of designing a heat-stable AEM to achieve high MPD. Under optimum conditions studied (0.33 M urea/1.0 M KOH at 50 °C), the MPD of the DUFC with Ni@C anode equaled 13.8 mW cm−2 with an open circuit of 0.93 V, which was higher than those reported previously (Suppl. Table S2)4,5,6,11,15,19,29,43,44,45,46,47. The excellent performance of the Ni@C anode was mainly attributed to its high specific surface area and mesoporous structure, which provided a high number of active Ni-catalyst sites and allowed fast mass transfer of urea and products in the anode. A high MPD of 31.5 mW cm−2 in urea/H2O2 type fuel cell at 70 °C using 0.5 M urea/7.0 M KOH as anolyte, and 2 M H2O2 and 2 M H2SO4 as catholyte has been reported4. However, the operation with H2O2 and H2SO4 as catholyte chemicals should require a higher cost in comparison to the humidified air.

(a) Effect of operation temperature on the performance of DUFC with Ni@C anode in 0.33 M urea and 1.0 M KOH (Inset is peak oxidation current density vs. Temperature) and (b) stability test of the DUFC with Ni@C anode at a constant current density of 20 mA cm−2 in 0.33 M and 1.0 M KOH at 50 °C.
The stability of the DUFC with Ni@C anode was further examined under a constant current density of 20 mA cm−2 in 0.33 M urea/1.0 M KOH at 50 °C (Fig. 8b). Notably, the cell voltage was retained at ~0.6 V during 15 h of operation, slightly decreasing and remaining constant until 20 h.
Conclusion
Highly porous nanostructured Ni@C and NiO@C composites synthesized by simple calcination of a Ni-based MOF exhibited higher specific surface areas and EASAs than their Ni-MOF precursor and thus featured excellent electro-catalytic activity for urea electro-oxidation. A DUFC containing Ni@C as an anode catalyst achieved a maximum power density of 13.8 mW cm−2 in 330 mM urea/1 M KOH at 50 °C, outperforming previously reported DUFCs. Moreover, the above fuel cell exhibited a stable performance during 20 h of continuous operation. In view of the above, it was concluded that the simply prepared nanostructured Ni@C and NiO@C catalysts are suitable anode materials for DUFCs.
Methods
Syntheses of Ni-MOF, Ni@C, and NiO@C
Ni@C and NiO@C were synthesized using Ni-MOF as a template precursor. To prepare Ni-MOF ([Ni3(btc)2], btc = benzene-1,3,5-tricarboxylate), a solution of Ni(NO3)3 · 6H2O in methanol was mixed with an appropriate amount of benzene-1,3,5-tricarboxylic acid, stirred for 1 h at room temperature, and autoclaved at 150 °C for 24 h. After reaction completion, the suspension was centrifuged, and the solid product was repeatedly washed with methanol and dried in a vacuum oven at 60 °C for 24 h. The thus obtained Ni-MOF precursor powder was calcined at 400 °C (5 °C min−1) for 4 h in nitrogen or air to synthesize Ni@C or NiO@C, respectively.
Preparation of electrodes and urea/O2 single cell
As prepared Ni-MOF, Ni@C, and NiO@C catalysts were mixed with Vulcan carbon (XC-72) at 30 wt%, and the obtained mixture was added into 0.05 wt% Nafion solution in isopropanol and sonicated for 2 h. The resulting anode catalyst inks were coated onto a nickel foam (pore diameter 0.25 mm, MTI, USA) electrode to achieve a catalyst loading of 3.4 mg cm−2. A commercial Pt/C (40 wt%, E-TEK)-coated carbon paper with a Pt loading of 0.6 mg cm−2 was used as a cathode. An anion exchange membrane (AEM; Fumasep FAA-3-PK-130, Germany) was used as a polymer electrolyte separating anode and cathode compartments, as described elsewhere48. The active electrode area equaled 5.0 cm2. A single-cell bipolar plate was prepared using graphite with serpentine flow channels. Urea solutions were pumped into the anode side by a peristaltic pump at 3 mL min–1, and humidified air was supplied to the cathode by passing air through room-temperature water.
Analysis
Crystal structures were analyzed by X-ray diffraction (XRD; Rigaku DMAX 2200, Japan) at a scan rate of 5° min−1 using Cu Kα radiation (λ = 1.506 Å). Sample morphology and structure were probed by scanning electron microscopy (SEM; Hitachi S-4700, Japan), focused ion-beam SEM (FIB-SEM; Hitachi, Japan), and transmission electron microscopy (TEM; Tecnai G2 F30 S-Twin, USA). Thermogravimetric (TG) analysis was performed using a TG analyzer (SDT Q600, TA, USA). Carbon contents were measured by an elemental analyzer (Vario MICRO cube, Germany). Specific surface area and porosity were determined based on nitrogen adsorption-desorption measurements performed using a Micromeritics ASAP 2020 apparatus according to the Brunauer-Emmett-Teller (BET) theory. Electrical resistivity of the catalyst powder compressed into pellets under 1500 bar at 25 °C in 30 minutes was determined by four-probe resistivity method (CMT-SR2000N, South Korea). The electrochemical properties of catalysts were analyzed by a potentiostat-galvanostat (VSP, Bio-Logic Science Instruments, France) using a conventional three-electrode system with Pt wire and Ag/AgCl (3 M NaCl) as counter and reference electrodes, respectively. The working electrode was prepared by coating catalyst inks (as described above) on carbon fiber (5 cm2) to achieve a catalyst loading of 1.0 mg cm−2. Fuel cell performance was evaluated utilizing the potentiostat-galvanostat interfaced with EC-lab 11.01 data acquisition software.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Rollinson, A. N., Jones, J., Dupont, V. & Twigg, M. V. Urea as a hydrogen carrier: a perspective on its potential for safe, sustainable and long-term energy supply. Energy Environ. Sci. 4, 1216–1224 (2011).
Radenahmad, N. et al. Proton-conducting electrolytes for direct methanol and direct urea fuel cells - A state-of-the-art review. Renew. Sust. Energ. Rev. 57, 1347–1358 (2016).
Xu, W., Wu, Z. & Tao, S. Urea-Based Fuel Cells and Electrocatalysts for Urea Oxidation. Energy Technol. 4, 1329–1337 (2016).
Guo, F. et al. Enhancement of direct urea-hydrogen peroxide fuel cell performance by three-dimensional porous nickel-cobalt anode. J. Power Sources 307, 697–704 (2016).
Serban, E. C. et al. Urea/hydrogen peroxide fuel cell. Dig. J. Nanomater. Bios. 9, 1647–1654 (2014).
Lan, R., Tao, S. & Irvine, J. T. S. A direct urea fuel cell – power from fertiliser and waste. Energy Environ. Sci. 3, 438–441 (2010).
Guerrero Moreno, N., Cisneros Molina, M., Gervasio, D. & Pérez Robles, J. F. Approaches to polymer electrolyte membrane fuel cells (PEMFCs) and their cost. Renew. Sust. Energ. Rev. 52, 897–906 (2015).
Joghee, P., Malik, J. N., Pylypenko, S. & O’Hayre, R. A review on direct methanol fuel cells – In the perspective of energy and sustainability. MRS Energ. Sust. 2, 1–31 (2015).
Zhu, X. et al. Metallic Nickel Hydroxide Nanosheets Give Superior Electrocatalytic Oxidation of Urea for Fuel Cells. Angew. Chem. Int. Ed. 55, 12465–12469 (2016).
Yan, W., Wang, D., Diaz, L. A. & Botte, G. G. Nickel nanowires as effective catalysts for urea electro-oxidation. Electrochim. Acta. 134, 266–271 (2014).
Xu, W., Zhang, H., Li, G. & Wu, Z. Nickel-cobalt bimetallic anode catalysts for direct urea fuel cell. Sci. Rep. 4, 5863 (2014).
Shi, W., Ding, R., Li, X., Xu, Q. & Liu, E. Enhanced performance and electrocatalytic kinetics of Ni-Mo/graphene nanocatalysts towards alkaline urea oxidation reaction. Electrochim. Acta. 242, 247–259 (2017).
Singh, K. R. & Schechter, A. Electroactivity of Urea Oxidation on NiCr Catalysts in Alkaline Electrolyte. ChemCatChem. 9, 3374–3379 (2017).
Xu, W., Du, D., Lan, R., Humphreys, J. & Wu, Z. Highly active Ni–Fe double hydroxides as anode catalysts for electrooxidation of urea. New J. Chem. 41, 4190–4196 (2017).
Guo, F., Cheng, K., Ye, K., Wang, G. & Cao, D. Preparation of nickel-cobalt nanowire arrays anode electro-catalyst and its application in direct urea/hydrogen peroxide fuel cell. Electrochim. Acta. 199, 290–296 (2016).
Wu, M. S., Lin, G. W. & Yang, R. S. Hydrothermal growth of vertically-aligned ordered mesoporous nickel oxide nanosheets on three-dimensional nickel framework for electrocatalytic oxidation of urea in alkaline medium. J. Power Sources 272, 711–718 (2014).
Barakat, N. A. M. et al. Nickel nanoparticles-decorated graphene as highly effective and stable electrocatalyst for urea electrooxidation. J. Mol. Catal. A: Chem. 421, 83–91 (2016).
Barakat, N. A. M., El-Newehy, M. H., Yasin, A. S., Ghouri, Z. K. & Al-Deyab, S. S. Ni&Mn nanoparticles-decorated carbon nanofibers as effective electrocatalyst for urea oxidation. Appl. Catal. A. 510, 180–188 (2016).
Kakati, N., Maiti, J., Lee, K. S., Viswanathan, B. & Yoon, Y. S. Hollow sodium nickel fluoride nanocubes deposited MWCNT as an efficient electrocatalyst for urea oxidation. Electrochim. Acta. 240, 175–185 (2017).
Tran, T. Q. N., Das, G. & Yoon, H. H. Nickel-metal organic framework/MWCNT composite electrode for non-enzymatic urea detection. Sens. Actuators, B. 243, 78–83 (2017).
Das, G., Tesfaye, R. M., Won, Y. & Yoon, H. H. NiO-Fe2O3 based graphene aerogel as urea electrooxidation catalyst. Electrochim. Acta. 237, 171–176 (2017).
Kreno, L. E. et al. Metal-Organic Framework Materials as Chemical Sensors. Chem. Rev. 112, 1105–1125 (2012).
Zhang, Y. et al. Facile synthesis of a Cu-based MOF confined in macroporous carbon hybrid material with enhanced electrocatalytic ability. Chem. Commun. 49, 6885–6887 (2013).
Yang, J., Xiong, P., Zheng, C., Qiu, H. & Wei, M. Metal-organic frameworks: a new promising class of materials for a high performance supercapacitor electrode. J. Mater. Chem. A. 2, 16640–16644 (2014).
Yang, J., Zhao, F. & Zeng, B. One-step synthesis of a copper-based metal–organic framework–graphene nanocomposite with enhanced electrocatalytic activity. RSC Adv. 5, 22060–22065 (2015).
Liu, L. et al. Self-assembled hierarchical yolk-shell structured NiO@C from metal-organic frameworks with outstanding performance for lithium storage. Chem. Commun. 50, 94859488 (2014).
Zou, F. et al. Controllable self-catalytic fabrication of carbon nanomaterials mediated by a nickel metal organic framework. Environ. Sci.: Nano. 4, 46 (2017).
Xu, X., Cao, R., Jeong, S. & Cho, J. Spindle-like mesoporous Fe2O3 anode material prepared from MOF template for high-rate lithium batteries. Nano Lett. 12, 4988–4991 (2012).
Lan, R. & Tao, S. Preparation of nano-sized nickel as anode catalyst for direct urea and urine fuel cells. J. Power Sources 196, 5021–5026 (2011).
Wang, Z. B., Li, C. Z., Gu, D. M. & Yin, G. P. Carbon riveted PtRu/C catalyst from glucose in-situ carbonization through hydrothermal method for direct methanol fuel cell. J. Power Sources 238, 283–289 (2013).
Sattar, M. A. & Conway, B. E. Electrochemistry of the nickel-oxide electrode-VI. Surface oxidation of nickel anodes in alkaline solution,. Electrochim. Acta. 14, 695–710 (1969).
Fleischmann, M., Korinek, K. & Pletcher, D. The oxidation of organic compounds at a nickel anode in alkaline solition. J. Electroanal. Chem. 31, 39–49 (1971).
Vedharathinam, V. & Botte, G. G. Understanding the electro-catalytic oxidation mechanism of urea on nickel electrodes in alkaline medium. Electrochim. Acta. 81, 292–300 (2012).
Yang, D., Yang, L., Zhong, V., Yu, X. & Feng, L. Urea electro-oxidation efficiently catalyzed by nickel-molybdenumoxide nanorods. Electrochim. Acta. 295, 524–531 (2019).
Yang, D. et al. Nanostructured Ni2P‐C as an Efficient Catalyst for Urea Electrooxidation. ChemElectroChem 5, 659–664 (2018).
Liu, H., Liu, Z. & Feng, L. Bonding state synergy of the NiF2/Ni2P hybrid with the co-existence of covalent and ionic bonds and the application of this hybrid as a robust catalyst for the energy-relevant electrooxidation of water and urea. Nanoscale 11, 16017 (2019).
Yan, W., Wang, D. & Botte, G. G. Electrochemical decomposition of urea with Ni-based catalysts. Appl. Catal. B. 127, 221–226 (2012).
Ye, K. et al. Highly porous nickel@carbon sponge as a novel type of three-dimensional anode with low cost for high catalytic performance of urea electro-oxidation in alkaline medium. J. Power Sources 283, 408–415 (2015).
Basu, D. & Basu, S. Synthesis, characterization and application of platinum based bi-metallic catalysts for direct glucose alkaline fuel cell. Electrochim. Acta. 56, 6106–6113 (2011).
Basu, D. & Basu, S. Performance studies of Pd-Pt and Pt-Pd-Au catalyst for electro-oxidation of glucose in direct glucose fuel cell. Int. J. Hydrogen Energy 37, 4678–4684 (2012).
Dekel, D. R. Review of cell performance in anion exchange membrane fuel cells. J. Power Sources 375, 158–169 (2018).
Wu, Y. et al. Novel silica/poly(2,6-dimethyl-1,4-phenylene oxide) hybrid anion-exchange membranes for alkaline fuel cells: Effect of silica content and the single cell performance. J. Power Sources 195, 3069–3076 (2010).
Nagao, M., Kobayashi, K. & Hibino, T. A Direct Urine Fuel Cell Operated at Intermediate Temperatures. Chem. Lett. 44, 363–365 (2015).
Xu, W., Zhang, H., Li, G. & Wu, Z. A urine/Cr(VI) fuel cell - Electrical power from processing heavy metal and human urine. J. Electroanal. Chem. 764, 38–44 (2016).
Fan, Z., Kwon, Y., Yang, X., Xu, W. & Wu, Z. In-situ production of hydrogen peroxide as oxidant for direct urea fuel cell. Energy Procedia. 105, 1858–1863 (2017).
Yousef, A., El-Newehy, M. H., Al-Deyab, S. S. & Barakat, N. A. M. Facile synthesis of Ni-decorated multi-layers graphene sheets as effective anode for direct urea fuel cells. Arab. J. Chem. 10, 811–822 (2017).
Zhang, H., Wang, Y., Wu, Z. & Leung, D. Y. C. A direct urea micro fluidic fuel cell with flow-through Ni-supported- carbon- nanotube-coated sponge as porous electrode. J. Power Sources 363, 61–69 (2017).
Tesfaye, R. M., Gautam Das, G., Park, B. J., Kim, J. & Yoon, H. H. Ni-Co bimetal decorated carbon nanotube aerogel as an efficient anode catalyst in urea fuel cells. Sci. Rep. 9, 479 (2019).
Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2017R1AB4002083) and by the Korea Institute of Energy Technology Evaluation and Planning (KETEP) and the Ministry of Trade, Industry & Energy (MOTIE) of the Republic of Korea (No. 20194030202290).
Author information
Authors and Affiliations
Contributions
T.Q.N. Tran, T.N. Duong, and W.H. Yun carried out the experiments and wrote the main manuscript text. B.J. Park designed the experimental work. H.H. Yoon directed the research and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tran, T.Q.N., Park, B.J., Yun, W.H. et al. Metal–organic framework–derived Ni@C and NiO@C as anode catalysts for urea fuel cells. Sci Rep 10, 278 (2020). https://doi.org/10.1038/s41598-019-57139-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-57139-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
