
Abstract
Brain tumours that are refractory to treatment have a poor prognosis and constitute a major challenge in offering effective treatment strategies. By targeting molecular alterations, precision cancer medicine may be a viable option for the treatment of brain tumours. In this retrospective analysis of our PCM platform, we describe the molecular profiling of primary brain tumours from 50 patients. Tumour samples of the patients were examined by a 161-gene next-generation sequencing panel, immunohistochemistry, and fluorescence in situ hybridization (FISH). We identified 103 molecular aberrations in 36 (72%) of the 50 patients. The predominant mutations were TP53 (14.6%), IDH1 (9.7%) and PIK3CA (6.8%). No mutations were detected in 14 (28%) of the 50 patients. IHC demonstrated frequent overexpression of EGFR and mTOR, in 38 (76%) and 35 (70%) patients, respectively. Overexpression of PDGFRa and PDGFRb were less common and detected in 16 and four patients, respectively. For 35 patients a targeted therapy was recommended. In our database, the majority of patients displayed mutations, against which targeted therapy could be offered. Based on our observations, PCM may be a feasible novel treatment approach in neuro-oncology.
Similar content being viewed by others

Introduction
Effective alternative therapies in adult patients with primary brain tumours relapsing after standard therapy is often lacking. For most neuro-oncological patients, there is no standard treatment recommendation after failure of first-line therapy. This is also true for patients with a glioblastoma, which constitutes a large proportion of patients treated by neuro-oncologists. Glioblastoma multiforme (GBM) is the most frequent malignant brain tumour, making up over 50% of all gliomas and 16% of all primary brain tumours1. In addition, for patients with relapsed diffuse or anaplastic astrocytomas or oligodendrogliomas, there are no standard alternative therapeutic recommendations, and for patients with rare brain tumours and for the few patients with relapsed or progressive meningiomas, further neurosurgical or radiotherapeutic treatment options may be very limited or absent.
The cancer database GLOBOCAN 2018 revealed that roughly 300,000 patients (1.6% of all new cancer cases) were diagnosed with a brain tumour, and 241,000 patients (2.5% of all cancer deaths) died of this disease2. According to GLOBOCAN 2018, brain tumours are the 19th most common cancer2. But despite their relatively low frequency, when compared with other malignant diseases, brain tumours cause a disproportionate amount of morbidity and mortality, partly because of the critical location of the tumour mass3. The management of advanced and relapsed brain tumours poses a great challenge to physicians and healthcare providers and requires co-operation between and management by a multidisciplinary team. There is no consensus on treatment pathways in recurrent or progressive brain tumours, and the existing therapeutic options are limited4,5.
In recent years, there has been an effort to progressively individualize therapy options in specific cancers. In a few particular cancers, treatment with immunotherapeutics or tyrosine kinase inhibitors, tailored to the individual, are possible, for example, trastuzumab in human epidermal growth factor receptor 2 (HER2)-positive breast cancer or gastric cancer, imatinib in philadelphia chromosome positive chronic myelogenous leukaemia (Ph + CML) or in KIT+ gastrointestinal stromal tumour (GIST), pazopanib and sunitinib in advanced renal cell carcinoma (RCC), or BRAF-directed therapy with vemurafenib or dabrafenib/trametinib in melanoma6,7,8.
Emerging techniques, such as profiling tumour molecular alterations and mutations, identifying molecular targets amenable to specific treatments, and developing drug treatments specific to an individual patient, provide the potential for novel and effective therapies. The ground-breaking pilot trial by Daniel von Hoff have ushered in a new era of medicine. This approach is known by a number of different names, including individualized, stratified, tailored or precision cancer medicine (PCM)9. The main rationale of PCM is to match a therapeutic agent to its corresponding molecular target, to allow a precise treatment tailored to a specific patient. It aims to achieve a better and more sustained response than more generic treatments, without damaging healthy cells and tissues10. We conducted a retrospective subgroup analysis of all 50 patients with primary brain tumour (PBT) that had been enrolled and profiled in our special PCM platform of molecular oncological diagnostics and therapy (MONDTI) of the comprehensive cancer centre of the Medical University of Vienna (CCC-MUV). We sought to map the molecular profiles (MP) of highly advanced, mainly relapsed PBT and to specifically target the found molecular alterations.
Materials and Methods
Patients and design of the precision medicine platform
Patients with progressive and mainly recurrent PBT who had progressed to all standard treatment options were eligible for inclusion in MONDTI, provided archival tissue samples were available. Patients had to have an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1. MONDTI is not a clinical trial, but intends to provide the possibility of a targeted therapy to patients where no active anti-tumoural treatment is available.
The study was performed according to the International Conference on Harmonization E6 requirements for Good Clinical Practice and with the ethical principles described in the Declaration of Helsinki. Patients had to provide informed consent before inclusion in MONDTI. Furthermore, the Institutional Ethics Committee of the Medical University of Vienna has also approved this subanalysis (Nr. 1039/2017).
Tissue samples
Formalin-fixed, paraffin-embedded tissue sections from patients with advanced PBT that were resistant to all standard therapy options were sent to or retrieved from the archive of the Department of Neuropathology and sent to the Department of Pathology, Medical University Vienna, Vienna, Austria.
Cancer gene panel sequencing
DNA was obtained from paraffin-embedded tumour tissue blocks with a QIAamp Tissue KitTM (Qiagen, Hilden, Germany), and 10 ng DNA per sample was employed for sequencing. The DNA library was generated by multiplex polymerase chain reaction with the 161-gene next-generation sequencing panel of Oncomine Comprehensive Assay v3 (Thermo Fisher Scientific, Waltham, MA, USA).
Immunohistochemistry (IHC)
IHC was done utilizing 2-μm-thin tissue sections. The following antibodies were used: ALK (clone 1A4; Zytomed, Berlin, Germany), cluster of differentiation 30 (CD30) (clone BerH2; Dako, Vienna, Austria), CD20 (clone L26; Dako), epidermal growth factor receptor (EGFR) (clone 3C6; Ventana), oestrogen receptor (ER) (clone SP1; Ventana), HER2 (clone 4B5; Ventana), HER3 (clone SP71; Abcam), KIT (clone 9.7; Ventana), MET (clone SP44; Ventana), phosphorylated- mechanistic target of rapamycin (p-mTOR) (clone 49F9; Cell Signaling Technology, Danvers, MA, USA), platelet-derived growth factor receptor (PDGFRA) (rabbit polyclonal; Thermo Fisher Scientific), PDGFRB (clone 28E1; Cell Signaling Technology), programmed death-ligand 1 (PD-L1) (clone E1L3N; Cell Signaling Technology), progesterone receptor (PR) (clone 1E2; Ventana), phosphatase and tensin homolog (PTEN) (clone Y184; Abcam), and ROS1 (clone D4D6; Cell Signaling Technology). An immunohistochemical score was calculated by multiplying the percentage of positive cells with their corresponding staining intensity (0 = negative, 1 = weak, 2 = moderate, 3 = strong), as described here: (maximum 300) = (% negative × 0) + (% weak × 1) + (% moderate × 2) + (% strong × 3).
The cut offs employed for each immunohistochemical stain used for treatment recommendation as follows: Oestrogen receptor and progesteron receptor: ≥10% positive tumour cells of any staining intensity, HER2: positive, Score 2+ or 3+ and additionally presence of an HER2 gene amplification with an HER2:centromer 17 ratio ≥2, KIT: IHC score ≥100 and presence of a pathogenic/likely pathogenic KIT mutation, p-mTOR: IHC score ≥100 and loss of PTEN expression or presence of a pathogenic/likely pathogenic PTEN mutation, PDGFRA and PDGFRB: IHC score of ≥100, PD-L1: tumour proportion score ≥1.
Fluorescence in situ hybridization
FISH was done with 4-μm-thick formalin-fixed, paraffin-embedded tissue samples. The following fluorescent probes were utilized: ALK (2p23.1; Abbott, Abbott Park, IL, USA), RET (10q11; Kreatech, Berlin, Germany), PTEN (10q23.31)/Centromere 10, and ROS1 (ZytoVision, Bremerhaven, Germany). Two hundred cell nuclei per tumour were assessed. The cut-off level for an aberrant ALK, RET, and ROS1 FISH was ≥15% of cells with a split-apart signal. The PTEN FISH was considered positive for PTEN gene loss with ≥30% of cells with only one or no PTEN signals. A chromosome 10 centromere FISH probe served as a control for ploidy of chromosome 10.
Multidisciplinary boards (molecular tumour boards for PCM)
After thorough examination of the molecular profile of each tumour sample by a qualified and competent molecular pathologist, the results and findings were reviewed in a multidisciplinary tumour boards (MTB) that were held every other week. Members of the board included molecular pathologists, radiologists, clinical oncologists, biostatisticians, and basic scientists. The MTB recommended the targeted therapy based on the specific molecular profile of each patient. The targeted therapies included tyrosine kinase inhibitors, checkpoint inhibitors (e.g. anti- PD-L1 monoclonal antibodies), and growth factor receptor antibodies with or without endocrine therapy. The treatment recommendations by the MTB were prioritized dependent on the level of evidence from high to low according to phase III to phase I trials.
In cases where more than one druggable molecular aberration was identified, the MTB recommended a therapy regimen to target as many molecular aberrations as possible, with special consideration to toxicity profile of each antitumoural agent and their potential interactions. Since all patients were given all available standard treatment options for their cancer disease prior to their inclusion in our PCM platform, nearly all targeted agents were suggested as off-label use. If the tumour profile and the clinical characteristics of a patient met the requirements of a clinical trial for targeted therapies that was conducted in our cancer centre, patients were preferentially asked if they wanted to participate in this trial.
Descriptive statistics
For data description, we used measures of central tendency including the mean and median. We also used the method of frequency distribution to delineate the characteristics of the PBT patients.
Results
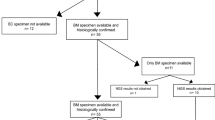
Fifty patients diagnosed with a primary brain tumour were included in this subgroup analysis from the cohort of the PCM project MONDTI, that has so far profiled 550 patients with various highly advanced cancer types. In this analysis, all patients were Caucasians. There were 26 men and 24 women, diagnosed with a total of 24 different types of PBT. The median age at first diagnosis was 39 years, range 10 to 71, and the median age at the time when the molecular profiling was performed was 45 years, range 10 to 72 (Table 1). The tumour tissue was obtained during surgical intervention.
Thirty patients had a diagnosis of glioma. The analysis also included rare brain tumours, including papillary tumour of the pineal region (PTPR), pineal parenchymal tumour of intermediate differentiation (PPTID), gliofibroma, and a very rare, as yet unclassified, type of glioma with loss of H3 K27me3 and absence of H3 K27 mutation that has been verified by IHC and sequencing, and described only in extremely rare cases.
There was a time span of between six months and two years between diagnosis and when molecular profiling was performed. By the time of molecular profiling, 42 patients had suffered relapses of their malignant disease and had received a median of two courses of drug therapy (range 1–4). All patients had undergone at least one surgical intervention.
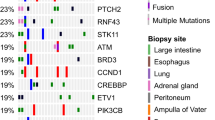
In total, we identified 103 molecular aberrations in 36 patients. The predominant mutations were TP53 (14.6%), IDH1 (9.7%) and PIK3CA (6.8%). No mutations were detected in 14 patients. Some patients were found to have more than one mutation. The three patients diagnosed with an anaplastic oligodendroglioma had a total of 37 mutations. In contrast, ten patients with GBM were found to have a total of 27 aberrations. In four of the 30 patients with glioma, the MGMT promoter was methylated. IDH1 was mutated in ten patients. IDH2 was mutated in one patient with secondary GBM. Interestingly, BRCA1 was mutated in one patient diagnosed with GBM, and BRCA2 mutation was shown in one sample of anaplastic oligodendroglioma. A genetic alteration of EGFR was observed in three patients; however, none of them was EGFRvIII-positive. See Tables 2 and 3 for a detailed analysis of all the reported mutations. Apart from TP53, IDH1, PIK3CA, ATM, BRAF and CDKN2A, mutations in other genes were detected in only one to two cases each.
IHC detected EGFR overexpression in 38 patients, with a high median score of 180. Seventeen patients had an EGFR score between 200 and 300. The overexpression of mTOR was detected in 35 patients, and was lower than was EGFR overexpression, with a median score of the former of 65. PTEN expression was revealed in exactly half of the patients, with a median score of 100. Less common and of lower levels were overexpression of PDGFRa and PDGFRb, which were found in 15 and eight patients, respectively. Ten patients with PDGFRa overexpression and two patients with PDGFRb had a high-grade glioma, including GBM, anaplastic oligoastrocytoma, anaplastic oligodendroglioma and anaplastic pleomorphic xanthoastrocytoma (PXA). Tumour expression of PD-L1 was seen in six tumour samples, including two cases of GBM, and in one case each of anaplastic oligodendroglioma, malignant prolactinoma, anaplastic oligodendroglioma and anaplastic hemangiopericytoma. The progesterone receptor was expressed in five glioma patients, and expression of the oestrogen receptor was found in only one woman, who had a malignant prolactinoma. Overexpression of KIT was reported in four patients. Three of these patients had a concomitant PDGFRa overexpression. FISH analysis revealed a loss of PTEN in three patients.
In over two-thirds (35) of the 50 patients, a targeted therapy was suggested, based on the identified genetic mutations. The most frequently recommended specific treatments were imatinib (n = 12), sunitinib (n = 5) and pembrolizumab (n = 5). For five patients, a more general treatment recommendation was produced: EGFR inhibitor (n = 2), AKT inhibitor (n = 1), FGFR inhibitor (n = 1), PDGFRa inhibitor (n = 1). For two patients, erlotinib was considered. Everolimus, cetuximab, dabrafenib/trametinib, olaparib, pazopanib and vismodegib were each proposed in one case each. Table 4 describes the rationale behind the specific therapy suggestions. In two cases, sunitinib or erlotinib were recommended, after the patients had received a course of imatinib but had subsequently become refractory to imatinib treatment. Except for two patients, all patients with GBM (n = 8) were given a therapy suggestion. For all patients with anaplastic oligodendroglioma, anaplastic astrocytoma, meningioma, oligoastrocytoma or PXA, an experimental therapy option was suggested.
Discussion
In this report, we show that the information of individual genomic alterations in patients with primary brain tumours that are refractory to standard treatment has been translated into specific therapeutic recommendations. Notably in this study, the histological type was diverse, and patients with rare brain tumours were also included and investigated.
In this retrospective single-centre analysis, we present the molecular profiling (MP) of all 50 patients with PBT from the MONDTI cohort. Their disease was therapy-refractory and highly advanced. Tumour tissue was obtained from all patients and characterized for their MP. Subsequently, the genomic information of the patients was discussed in a multidisciplinary tumour board (MTB) for PCM to evaluate the possibility of a genomic-based treatment that was independent of the tumour’s histological classification (tissue-agnostic treatment). A treatment recommendation was derived for 35 patients from the MTB. The drugs were carefully selected to form an individualized treatment, taking into account the patient’s clinical and treatment history and concomitant therapies and comorbidities.
Tumour samples commonly displayed mutations in TP53, IDH1 and PIK3CA, and revealed a high median EGFR score. Common therapeutic agents recommended were imatinib, sunitinib and pembrolizumab. The detected mutations and IHC scores observed in gliomas in this analysis are in keeping with previous studies11,12,13. The analysis presented in this study shows that molecular profiling from tumour samples of patients and subsequent identification of therapeutic options with highly advanced PBT appears feasible and safe, and consistent with previous work. While outcome data are not presented in this study, previous work has shown that the drug treatments used here may be effective in improving clinical outcomes.
A similar study used molecular profiling to offer an individualized therapy only in patients with recurrent or advanced glioblastoma. They enrolled a limited number of 20 patients of whom seven actually received the recommended treatment. The investigators reported the feasibility of treatment plan guided by the identified molecular profiling14.
A pilot trial evaluated the feasibility and potential of precision medicine in young patients below 25 years with newly diagnosed diffuse intrinsic pontine glioma. Based on whole exome sequencing and RNA sequencing, a personalized therapy concept was developed for 15 patients15.
Another trial performed exome and transcriptome sequencing in 91 young patients with therapy refractory or relapsed cancer and integrated and incorporated the information into clinical management. In roughly half of the patients (46%), druggable molecular alterations were found and were considered in the treatment plan16.
Imatinib and sunitinib were offered in this current study as a treatment when overexpression of PDGFRa/b or KIT was identified. Haberler et al. have shown previously that, in a cohort of 101 glioblastoma patients, the expression of anti-PDGFRa and b, c-kit, c-abl and arg protein that can be targeted by imatinib17. According to a phase II trial in patients with recurrent gliomas, treatment with imatinib is well tolerated, with a limited antitumour activity18. Hassler et al. also tested the antitumour activity of imatinib in 24 recurrent glioblastoma patients with at least one tyrosine kinase expression that can be targeted by imatinib. Six patients survived over one year, and twelve patients achieved stable disease (SD). Median progression-free survival was 3 months and median overall survival was 6 months19. Based on preclinical data, sunitinib appears to impede brain tumour progression and reduce tumour-induced neurodegeneration in the microenvironment20. In this study, imatinib was preferred over sunitinib or erlotinib due to its relatively favourable and manageable safety profile and longer experience of its use.
Pazopanib has previously been used in a patient with hemangioblastoma and von Hippel-Lindau (VHL) disease. An important study published in The Lancet investigated the efficacy of pazopanib in 31 patients with VHL and reported an objective response in 13 patients (42%). However, treatment-related serious adverse events included one case each of appendicitis and gastritis, and one patient had a fatal central nervous system bleeding21.
Notably, we identified a BRCA1/2 and an ATM mutation that are rational targets for Poly [ADP-ribose] polymerase 1 (PARP) inhibitors, for example olaparib22. It has been shown that olaparib penetrates the blood-brain barrier (BBB) and therefore easily accesses the tumour site, and it may enhance the cytotoxic effects of ionising radiation and temozolomide. Halford et al. treated 35 patients with olaparib and temozolomide and achieved six-month progression-free survival (PFS) rates in all patients, with a favourable safety profile23.
BRAF V600E mutation has been reported in a young woman with anaplastic PXA, who was offered dabrafenib/trametinib as tailored therapy. This drug combination has achieved Food and Drug Administration (FDA) approval for the treatment of melanoma displaying the BRAF V600E mutation. Our observation is in line with a retrospective study by Burger et al., who noted SD in three patients with recurrent, BRAF V600E-mutated malignant glioma after receiving dabrafenib24.
Pembrolizumab has been suggested in patients with tumour expression of PD-L1 or with a hypermutability. In a small study of 25 patients with refractory high-grade glioma, pembrolizumab was administered as salvage treatment and achieved a median overall survival of four months25. In May 2017, the FDA granted accelerated approval to pembrolizumab for the first tissue-agnostic indication in patients with unresectable or metastatic, microsatellite instability-high (MSI-H) or mismatch repair-deficient (dMMR) solid tumours that have progressed following prior treatment and which have no satisfactory alternative treatment options26.
For one male patient with recurrent medulloblastoma and SMO mutation, vismodegib was considered as targeted therapy in our analysis. The antitumour activity of vismodegib acting as a sonic hedgehog (SHH) pathway inhibitor has previously been confirmed by Robinson et al.27.
In patients with chordoma, PTPR and diffuse midline glioma imatinib or sunitinib were recommended due to expression of PDGFR28,29.
Despite the efforts and the investigated agents in brain tumours, progressive recurrent PBT have a very poor prognosis and there are only few FDA-approved therapeutic agents for brain tumours currently recommended or in use.
It is a difficult and complex task to classify and prioritize the plethora of reported genetic alterations and epigenetic changes of brain tumours, in order to identify molecular targets and to choose adequate tailored therapeutic measures. Most of the identified alterations are therefore currently undefined regarding their role in the pathogenesis of brain tumours and their therapeutic consequences and implications30.
With the exception of TP53, IDH1, PIK3CA, ATM, BRAF and CDKN2A, mutations in all other genes were detected only one to two times in the current study. This finding is consistent with the well-described extreme and complex intratumoural heterogeneity which occurs within the same tumour tissue; vascularization, proliferation, tumour mutational burden (TMB) and subclones are all known to be highly variable. The pattern of genetic and epigenetic aberrations changes both spatially and temporally. The tumour biology at metastatic sites is different from the primary site and differs again at the time point of relapse. In addition, it is known that the therapy itself can influence and inform the clonal tumour evolution, by creating new driver mutations in subclones that become insensitive to drugs30,31.
Another relevant issue is the drug delivery to the brain tumour. Tailored drugs applied in neuro-oncology must be capable of bypassing the physiological constraint of the BBB to reach the tumour32.
Taken together, the extremely complex tumour biology, the spatial and temporal heterogeneity in PBT genetics, and the difficulty of bypassing the BBB pose unique challenges for the management of brain tumours and in drug development. Further research is required to better comprehend the tumour biology.
One limitation of this subgroup analysis is that neurotrophin receptor kinases (TRK) fusion genes were not assessed and evaluated. TRK fusion genes are important oncogenic drivers in high-grade glioma that have a therapeutic consequence, as they can be targeted by specific inhibitors such as larotrectinib or entrectinib33,34. Another limitation is that the tumour tissue was obtained during surgical intervention and there was subsequently a time delay of between six months and two years until molecular profiling was performed. This limitation is important, since the tumour biology differs at the time of relapse or progression following the failure of initial therapy. Thus, the antitumour activity of targeted agents that are aimed at mutations present at the time of diagnosis may be reduced due to the changed molecular landscape of the tumour. For future analysis and studies, it would be of major clinical relevance to obtain genetic information of the tumour at the time of relapse or progression e.g. via liquid biopsy of cerebrospinal fluid to recommend the targeted therapy according to the current molecular characteristics of the tumour35,36,37,38.
Despite the limited number of patients included in this subgroup analysis, the results are promising. PCM clearly has the potential to inform treatment of PBT by expanding the therapeutic repertoire. Currently, PCM in neuro-oncology is in its infancy but has the potential to become more widely used in cancer drug development and therapy planning and strategy11,31.
References
Ostrom, Q. T. et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2011–2015. Neuro. Oncol. 20, iv1–iv86 (2018).
Bray, F. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424 (2018).
Macdonald, D. R., Kiebert, G., Prados, M., Yung, A. & Olson, J. Benefit of temozolomide compared to procarbazine in treatment of glioblastoma multiforme at first relapse: effect on neurological functioning, performance status, and health related quality of life. Cancer Invest. 23, 138–144 (2005).
Weller, M. et al. EANO guideline for the diagnosis and treatment of anaplastic gliomas and glioblastoma. Lancet Oncol. 15, e395–e403 (2014).
Fernandes, C. et al. Current standards of care in glioblastoma therapy in Glioblastoma (ed. De Vleeschouwer, S.) (2017).
Hauschild, A. et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 380, 358–365 (2012).
Bang, Y. J. et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 376, 687–697 (2010).
Kantarjian, H. et al. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N. Engl. J. Med. 346, 645–652 (2002).
Von Hoff, D. D. et al. Pilot study using molecular profiling of patients’ tumors to find potential targets and select treatments for their refractory cancers. J. Clin. Oncol. 28, 4877–4883 (2010).
König, I. R., Fuchs, O., Hansen, G., von Mutius, E. & Kopp, M. V. What is precision medicine? Eur. Respir. J. 50, 4 (2017).
Xiu, J. et al. Multi-platform molecular profiling of a large cohort of glioblastomas reveals potential therapeutic strategies. Oncotarget. 7, 21556–21569 (2016).
Park, S. H. et al. Molecular testing of brain tumor. J. Pathol. Transl. Med. 51, 205–223 (2017).
Olar, A. & Aldape, K. D. Using the molecular classification of glioblastoma to inform personalized treatment. J. Pathol. 232, 165–177 (2014).
Prados, M. et al. Precision medicine in recurrent glioblastoma: a feasibility trial conducted by the Ivy Foundation Early Phase Clinical Trials Consortium. J. Clin. Oncol. 34, 2031 (2016).
Mueller, S. et al. A pilot precision medicine trial for children with diffuse intrinsic pontine glioma-PNOC003: a report from the Pacific Pediatric Neuro-Oncology Consortium. Int. J. Cancer. 145, 1889–1901 (2019).
Mody, R. J. et al. Integrative clinical sequencing in the management of refractory or relapsed cancer in youth. JAMA. 314, 913–925 (2015).
Haberler, C. et al. Immunohistochemical analysis of platelet-derived growth factor receptor-alpha, -beta, c-kit, c-abl, and arg proteins in glioblastoma: possible implications for patient selection for imatinib mesylate therapy. J. Neurooncol. 76, 105–109 (2006).
Raymond, E. et al. Phase II study of imatinib in patients with recurrent gliomas of various histologies: a European Organisation for Research and Treatment of Cancer Brain Tumor Group Study. J. Clin. Oncol. 26, 4659–4665 (2008).
Hassler, M. R. et al. Response to imatinib as a function of target kinase expression in recurrent glioblastoma. Springerplus. 3, 111 (2014).
Hatipoglu, G. et al. Sunitinib impedes brain tumor progression and reduces tumor-induced neurodegeneration in the microenvironment. Cancer Sci. 106, 160–170 (2015).
Jonasch, E. et al. Pazopanib in patients with von Hippel-Lindau disease: a single-arm, single-centre, phase 2 trial. Lancet Oncol. 19, 1351–1359 (2018).
Chen, H. Z., Bonneville, R. & Roychowdhury, S. Implementing precision cancer medicine in the genomic era. Semin. Cancer Biol. 55, 16–27 (2019).
Halford, S. E. R. et al. Results of the OPARATIC trial: A phase I dose escalation study of olaparib in combination with temozolomide (TMZ) in patients with relapsed glioblastoma (GBM). J. Clin. Oncol. 35, 2022 (2017).
Burger, M. C. et al. Dabrafenib in patients with recurrent, BRAF V600E mutated malignant glioma and leptomeningeal disease. Oncol. Rep. 38, 3291–3296 (2017).
Reiss, S. N., Yerram, P., Modelevsky, L. & Grommes, C. Retrospective review of safety and efficacy of programmed cell death-1 inhibitors in refractory high grade gliomas. J. Immunother. Cancer. 5, 99 (2017).
Marcus, L. et al. FDA Approval Summary: Pembrolizumab for the Treatment of Microsatellite Instability-High Solid Tumors. Clin Cancer Res. 25, p. 3753–3758.
Robinson, G. W. et al. Vismodegib exerts targeted efficacy against recurrent sonic hedgehog-subgroup medulloblastoma: results from phase II pediatric brain tumor consortium studies PBTC-025B and PBTC-032. J. Clin. Oncol. 33, 2646–2654 (2015).
Iqbal, N. & Iqbal, N. Imatinib: a breakthrough of targeted therapy in cancer. Chemother. Res. Pract. 2014, 357027 (2014).
Nazarenko, I. et al. PDGF and PDGF receptors in glioma. Ups. J. Med. Sci. 117, 99–112 (2012).
Prados, M. D. et al. Toward precision medicine in glioblastoma: the promise and the challenges. Neuro. Oncol. 17, 1051–1063 (2015).
Klughammer, J. et al. The DNA methylation landscape of glioblastoma disease progression shows extensive heterogeneity in time and space. Nat. Med. 24, 1611–1624 (2018).
Pardridge, W. M. The blood-brain barrier: bottleneck in brain drug development. NeuroRx. 2, 3–14 (2005).
Ziegler, D. S. et al. Brief report: potent clinical and radiological response to larotrectinib in TRK fusion-driven high-grade glioma. Br. J. Cancer. 119, 693–696 (2018).
Xu, T. et al. Gene fusion in malignant glioma: an emerging target for next-generation personalized treatment. Transl. Oncol. 11, 609–618 (2018).
Saenz-Antoñanzas, A. et al. Liquid biopsy in glioblastoma: opportunities, applications and challenges. Cancers (Basel). 11, 950 (2019).
Klekner, Á. et al. Significance of liquid biopsy in glioblastoma - a review. J. Biotechnol. 298, 82–87 (2019).
Shankar, G. M., Balaj, L., Stott, S. L., Nahed, B. & Carter, B. S. Liquid biopsy for brain tumors. Expert Rev. Mol. Diagn. 17, 943–947 (2017).
Miller, A. M. et al. Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature. 565, 654–658 (2019).
Author information
Authors and Affiliations
Contributions
Conception, design: H.T., C.M. and G.P. Manuscript writing: H.T. and G.P. Collection and assembly of data: H.T. Provision of study materials or patients: L.M., C.M., M.P., J.H., J.F. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
M. Preusser has received honoraria for lectures, consultation or advisory board participation from the following for-profit companies: Bayer, Bristol-Myers Squibb, Novartis, Gerson Lehrman Group (GLG), CMC Contrast, GlaxoSmithKline, Mundipharma, Roche, Astra Zeneca, AbbVie, Lilly, Medahead, Daiichi Sankyo, Merck Sharp & Dome. G.W. Prager received speaker’s fees from Bayer, Roche, Merck-Serono, Amgen, Servier, Celgene, Shire, MSD, Lilly, and Sanofi-Aventis. M.K. received travel support from Merck, Bayer, Bristol-Myers Squibb, and Roche. All other authors have nothing to declare.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Taghizadeh, H., Müllauer, L., Furtner, J. et al. Applied Precision Cancer Medicine in Neuro-Oncology. Sci Rep 9, 20139 (2019). https://doi.org/10.1038/s41598-019-56473-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-56473-0
This article is cited by
-
Radiomics for characterization of the glioma immune microenvironment
npj Precision Oncology (2023)
-
The new era of bio-molecular imaging with O-(2-18F-fluoroethyl)-L-tyrosine (18F-FET) in neurosurgery of gliomas
Clinical and Translational Imaging (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
