
Abstract
Regulatory T cells (Tregs) are a double-edged regulator of the immune system. Aberrations of Tregs correlate with pathogenesis of inflammatory, autoimmune and neoplastic disorders. Phenotypically and functionally distinct subsets of Tregs have been identified in humans and mice on the basis of their extensive portfolios of monoclonal antibodies (mAb) against Treg surface antigens. As an important veterinary species, dogs are increasingly recognised as an excellent model for many human diseases. However, insightful study of canine Tregs has been restrained by the limited availability of mAb. We therefore set out to characterise CD4+CD25high T cells isolated ex vivo from healthy dogs and showed that they possess a regulatory phenotype, function, and transcriptomic signature that resembles those of human and murine Tregs. By launching a cross-species comparison, we unveiled a conserved transcriptomic signature of Tregs and identified that transcript hip1 may have implications in Treg function.
Similar content being viewed by others


Introduction
Regulatory T cells (Tregs) are dominant regulators of immune responses against self, pathogenic and commensal antigens in the periphery1. As key players in the maintenance of immune health, aberrations of Tregs have pathogenic implications in a number of inflammatory, autoimmune and neoplastic diseases, making them a compelling biomarker and immunotherapeutic target2,3,4,5,6,7. Tregs are heterogeneous in the periphery8,9. Despite the discovery of various Treg subtypes such as type 1 regulatory T (Tr1)10,11, CD8+ 12,13, CD4+CD25−LAG3+ 14,15, γδ TCR+16,17 and invariant natural killer T (iNKT)18,19 regulatory cells, CD4+FoxP3+ Tregs remain as the principal target of investigation in humans and mice20,21. CD4+CD25+FoxP3+ T cells in mice are suppressive20,22, whereas accumulating evidence suggests that CD4+FoxP3+ T cells in humans are heterogeneous in phenotype and function23,24,25. In addition to the extensive portfolio of surface markers for human Tregs, including CD25, CD127, CD45RA, ICOS and HLA-DR, sialyl lewis x (CD15s) identifies terminally differentiated effector Tregs26.
Although murine models for a number of pathobiological and immunotherapeutic studies are firmly established, large animal models are increasingly gaining traction. Of these, the dog recapitulates human autoimmune and neoplastic diseases remarkably well. Such diseases are spontaneous in canine patients, which have a competent immune system, and clinical presentations, treatment modalities and living environments shared with their human counterparts27,28,29. However, in-depth study of canine Tregs has been hampered by the limited availability of monoclonal antibodies (mAb) against surface antigens. Apart from the cross-reactive clones validated for canine intracellular FoxP3 (clone FJK-16s)30,31 and Helios (clone 226 F)31, anti-CD25 (clone P4A10) is the only known mAb labelling the extracellular surface of canine Tregs32. CD4+CD25+/high T cells are enriched for suppressive FoxP3+ T cells in humans and mice33,34,35. Our previous work has shown that canine CD4+CD25high T cells induced in vitro are regulatory31, but studies examining these cells ex vivo are limited in number and scope36,37,38. We therefore set out to characterise canine CD4+CD25high T cells isolated ex vivo, hypothesising that they possess regulatory phenotype and function. Furthermore, we investigated the transcriptomic phenotype of Tregs in dogs and compared it with those of humans and mice on the basis of published transcriptomic data, revealing a broadly conserved Treg signature across these species and consensus transcripts encoding molecules not hitherto associated with Tregs.
Materials and Methods
Sample collection
This study was approved by the Clinical Research Ethical Review Board (URN 2016 1592) of the Royal Veterinary College (RVC) in the United Kingdom. Eleven healthy dogs, defined by the absence of clinical signs and a normal physical examination undertaken by a veterinarian or veterinary nurse, were recruited at the RVC. Peripheral blood samples were collected from the jugular or lateral saphenous vein in sterile fashion by a veterinarian or veterinary nurse under the Animals (Scientific Procedures) Act 1986, following informed written consent by the owners of the dogs.
Isolation of peripheral blood mononuclear cells
Mononuclear cells were isolated from the peripheral blood by density gradient centrifugation, using Histopaque®-1077 (Sigma-Aldrich, Dorset, UK). Blood was diluted by an equal volume of phosphate-buffered saline (PBS; Sigma-Aldrich) with 2% v/v fetal bovine serum (FBS; Thermo Fisher Scientific, Waltham, MA, USA). The diluted blood was then layered onto an equal volume of Histopaque, before centrifugation at 400 g for 30 minutes at room temperature with minimal acceleration and braking. The purified peripheral blood mononuclear cells (PBMCs) were washed twice in PBS with 10% v/v FBS by centrifuging at 600 g for five minutes at 4 °C. After washing, cells were re-suspended in PBS with 10% v/v FBS, and counted using a haemocytometer before flow cytometric analysis. Dead cells were excluded by trypan blue staining.
Flow cytometry
Freshly isolated PBMCs were analysed by flow cytometry using mAb against canine-specific or cross-reactive antigens (all from Thermo Fisher Scientific). Extracellular labelling was performed by incubating PBMCs for 30 minutes at 4 °C with a mixture of FITC-conjugated anti-dog CD45 (clone YKIX716.13), PerCP-eFluor® 710-conjugated anti-dog CD5 (clone YKIX322.3), PE-Cy7-conjugated anti-dog CD4 (clone YKIX302.9), eFluor® 450-conjugated anti-dog CD8 (clone YCATE55.9) and PE-conjugated anti-dog CD25 (clone P4A10). After washing twice with PBS, cells were incubated in eBioscience™ FoxP3/transcription factor fixation/permeabilisation buffer (Thermo Fisher Scientific) according to the manufacturer’s instructions, then labelled with APC-conjugated anti-mouse/rat FoxP3 (clone FJK-16s) for 30 minutes at 4 °C. After washing with 1x permeabilisation buffer, cells were re-suspended in 200 μL PBS before being acquired on a FACSCanto II flow cytometer (Becton-Dickinson (BD); Franklin Lakes, NJ, USA). Flow cytometric data were analysed using FlowJo® software, version 7.6 (Tree Star, Ashland, OR, USA). Positive events were gated according to corresponding isotype or fluorescence minus one (FMO) controls.
Fluorescence-activated cell sorting
Fluorescence-activated cell sorting (FACS™) was used to sort PBMCs into subpopulations for subsequent experiments. Freshly isolated PBMCs were labelled by a mixture of PerCP-eFluor® 710-conjugated anti-dog CD5 (clone YKIX322.3), PE-Cy7-conjugated anti-dog CD4 (clone YKIX302.9), PE-conjugated anti-dog CD25 (clone P4A10) and Alexa Fluor® 700-conjugated anti-mouse CD11b for 30 minutes at 4 °C. After washing twice with PBS, cells were stained with 4′,6-diamidino-2-phenylindole (DAPI; BioLegend, San Diego, CA, USA) at room temperature for 10 minutes prior to sorting on BD FACSAria™ II. CD4+CD25high and CD4+CD25− T cells were isolated from CD5+CD11b− cells, and autologous antigen-presenting cells (APCs) were identified as CD5−CD11b+. For functional assays, CD4+CD25high T cells were defined as the 5% of CD4+ T cells showing the highest CD25 expression, whereas CD4+CD25− T cells were defined as the 20% of CD4+ T cells showing the lowest CD25 expression. For transcriptomic assays, CD4+CD25high T cells were defined as the 1% of CD4+ T cells showing the highest CD25 expression, whereas CD4+CD25− T cells were defined as before.
In vitro suppression assay
CD4+CD25high and CD4+CD25− T cells sorted from the peripheral blood of healthy dogs were immediately re-suspended in complete culture medium (RPMI-1640 complemented with 10% v/v FBS, 10 mM HEPES, 100 μg/mL streptomycin, 100 U/mL penicillin and 0.5 mM β-mercaptoethanol; all reagents from Sigma-Aldrich). The responder T (Tresp) cell population (CD4+CD25−) was stained with CellTrace™ violet proliferation dye according to the manufacturer’s instructions (Thermo Fisher Scientific), and seeded into a 96-well plate at a density of 1–5 × 104 cells per well. The suppressor cell population (CD4+CD25high) was co-cultured with Tresp cells at a ratio (Treg:Tresp) of 1:1 and/or 1:2. A population of autologous CD5−CD11b+ monocytes at a proportion of 1/5 of that of Tresp cells were also seeded into each well, as APCs. The mixed cell culture contained a total volume of 200 μL with 2.5 μg/mL concanavalin A (ConA) (Sigma-Aldrich) and was incubated for 96 hours at 37 °C, with 5% CO2. Three control groups were set up in the same fashion, including un-stimulated Tresp alone, stimulated Tresp alone and CD4+CD25− co-cultured with Tresp.
RNA extraction
CD4+CD25high and CD4+CD25− T cells sorted from the peripheral blood of five healthy dogs were immediately re-suspended in RNA Bee (AMS Biotechnology, Abingdon, UK) at a density of 2 × 106 cells/mL. Two hundred microlitres of chloroform (Sigma-Aldrich) per millilitre of RNA Bee suspension were added, before thorough admixture, transfer to a 2 mL MaXtract High Density tube (QIAGEN, Hilden, Germany), and incubation on ice for three minutes. The tube was then centrifuged at 12,000 g for 15 minutes at 4 °C. After centrifugation, the upper aqueous layer was carefully transferred to a 1.5 mL DNase/RNase-free Eppendorf Tube® (Eppendorf, Stevenage, UK), before being mixed completely with an equal volume of 100% ethanol (Sigma-Aldrich). The mixture was then transferred into a Zymo-Spin™ IC column on top of a collection tube and centrifuged according to the manufacturer’s instructions (Direct-zol™ RNA MicroPrep Kit, Zymo Research, Irvine, CA, USA). All samples were treated with DNase I during extraction; the final product was eluted in 6–10 μL of DNase/RNase-free water.
Library construction and sequencing
SMARTer® Universal Low Input RNA Kit (Clontech, California, USA) was used to construct the complementary (c) DNA library at the Oxford Genomics Centre, University of Oxford (Oxford, UK). RNA was converted to cDNA using Oligo (dT) primers and adapters, followed by PCR amplification. The cDNA library was then sheared into short fragments using a Covaris S220 Focused-Ultrasonicator (Thermo Fisher Scientific) for subsequent random shotgun Illumina sequencing. The 75-bp, paired-end sequencing was performed on the prepared DNA libraries, using the HiSeq. 4000 System (Illumina, San Diego, CA, USA) at the Oxford Genomics Centre. Samples were loaded onto the clustered sequencing Flow Cell, which was then primed with sequencing by synthesis (SBS) reagents and hybridised by Read 1 and Read 2 primers. The run was recorded by HCS 3.4.0 (Illumina).
Read processing and expression quantification
Sequencing reads were trimmed using Skewer (version 0.1.125) to remove the adapter and anchor sequences added during library construction and sequencing. Trimmed transcript reads were mapped to the canine genome, CanFam3.1 (Ensembl Genes, release 91), using HISAT2 (version 2.0.0-beta). The uniquely mapped read pairs were quantified using featureCounts (version 1.5.0), and annotated using the same canine genomic data. Mapping metrics were generated using Picard Tools (version 1.92). The metrics and variants for assessing read distribution, biotype distribution and mapped transcripts were generated using R packages (version 3.4.2) with in-house scripts. Read counts were all converted to transcripts per million (TPM) to normalise sequencing depth and gene lengths.
Differential expression analysis
Transcripts differentially expressed between canine CD4+CD25high and CD4+CD25− T cells were identified using Bioconductor package edgeR (Bioconductor version 3.6), with fold change (FC) values and statistical significance, the latter of which was represented by false discovery rate (FDR). R version 3.4.2 was used to conduct principal component analysis (PCA) and volcano plots.
Ingenuity pathway analysis
Differentially expressed transcripts (FDR < 0.05) with FC and FDR values were input into the software Ingenuity Pathway Analysis (IPA; Ingenuity Systems Inc., Redwood City, CA, USA) to identify biological pathways affected by the altered expression of these transcripts (|Z| score ≥ 2).
Reverse transcription and quantitative PCR
Purified total RNA was converted to cDNA by performing reverse transcription (RT), using the Precision nanoScript™ 2 Reverse Transcription Kit (Primerdesign, Southampton, UK). One reaction of 20 μL volume in total contained RNA template (up to 2 μg), combined Oligo (dT) and random nonamer primers, nanoScript™ 2 Buffer, dNTP mix, nanoScript™ 2 enzyme and RNase/DNase free water. The reaction included an annealing step of 65 °C for five minutes, then immediate cooling on ice, followed by an extension step at room temperature for five minutes and 42 °C for 20 minutes, then 75 °C for 10 minutes. The abundance of transcripts of interest was then measured by quantitative (q) PCR, using cDNA as reaction template, according to the manufacturer’s instructions. Primers specific to each transcript were all from the Taqman® Gene Expression Assays (GEAs) (Thermo Fisher Scientific), targeting fam129a (Cf02724989_m1), lmna (Cf02678125_g1), cadm1 (Cf02645230_m1), anxa2 (Cf02734571_gH), ctsz (Cf02661948_m1), actn4 (Cf02689744_g1), csf1 (Cf01094425_m1), hip1 (Cf02698307_m1), galm (Cf02648153_m1), pou2f2 (Cf00922171_g1), frmd4b (Cf02646908_m1), il2ra (Cf02623133_m1), foxp3 (Cf02741700_m1) and ikzf2 (Cf00915981_m1). Two reference transcripts, ubc encoding CG11624-PA, isoform A and sdha encoding succinate dehydrogenase flavoprotein subunit, were selected following validation by means of the Primerdesign Dog geNorm™ Kit. The relative expression of the target transcript was calculated using Pfaffl’s model39 as below:
E represents E value; TAR, target transcript; REF, reference transcript; Control, CD4+CD25− cells; Sample, CD4+CD25high cells. The relative expression ratio calculated by this equation indicated the FC of the target transcript abundance in the sample population when compared to that of the control population.
Interspecies comparisons
To compare the transcriptomic profiles of canine CD4+CD25high T cells across species with those of human and murine Tregs, published resources were used. The selected human and murine studies22,40 used different analytical methods from those in this study, but were the most comprehensive in the literature and conducted on freshly isolated Tregs in comparison to CD4+CD25− T cells. Raw transcriptomic data of the published human and murine studies were analysed following the same pipeline as for canine CD4+CD25high T cells, with respective genomic information. The data were processed using the web-based bioinformatics platform Galaxy41. Similarity scores were calculated using R OrderedList42 (version 1.48.0), to determine the number of shared transcripts between two species in the first n consensus transcripts, which were ordered by differential expression FC values. A similarity score was yielded, in which transcripts received higher weight the closer they were to the top or bottom end of the ordered list. Similarity scores for n = 100, 150, 200, 300, 400, 500 and 750 transcripts were reported, respectively. Statistical significance was assessed for each of the similarity scores, by comparing with a null distribution generated by randomly scrambling the order of the transcripts.
Statistical analysis
Summary data are shown as mean ± standard error of the mean (SEM). Statistical analysis was performed using GraphPad Prism version 7 (GraphPad Software, La Jolla, CA, USA).
Results
Freshly isolated canine CD4+CD25high T cells are enriched for FoxP3
To test the hypothesis that freshly isolated canine CD4+CD25+ T cells have a regulatory phenotype, PBMCs of 11 healthy dogs were labelled with a mAb panel incorporating all markers of canine Tregs to date. When the CD25 gate was moved upwards to incorporate increasing CD25 expression per cell, from the highest 5% to the highest 0.2%, the proportion of FoxP3+ cells significantly increased from 36.89 ± 2.79% to 74.07 ± 4.81%, suggesting that ex vivo CD4+CD25high T cells were enriched for FoxP3 (Fig. 1a,b).

CD4+CD25high T cells isolated ex vivo are enriched for FoxP3. (a) Representative flow cytometric plots showing that proportional expression of FoxP3 increased with increasing CD25 expression by CD4+ T cells from the highest 5% to the highest 0.5% of one healthy dog (all CD4+CD25+ T cells in this figure were analysed as CD45+CD5+CD8-CD4+CD25+, following a cascaded gating strategy). (b) Scatter dot plot summarising the increasing proportional expression of FoxP3 (mean ± SEM) among CD4+ T cells of 11 healthy dogs, with increasing CD25 expression from the highest 5% to the highest 0.2%. (c) Summary scatter dot plot comparing the higher proportional expression of FoxP3 in top 1% of CD25high cells, in which gating was determined by the corresponding isotype control (iso) or biological negative control (bio; CD25−). No significant difference was found in CD25high cells between the two gating methods. Statistical significance in (b,c) was analysed by one-way ANOVA, followed by Dunn’s multiple comparisons test (****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05).
The top 1% of CD4+CD25+ T cells were selected for subsequent phenotypic characterisation, balancing the enrichment for FoxP3 (61.59 ± 4.76%) with the need to isolate sufficient numbers. The proportional expression of FoxP3 in CD4+CD25high T cells was compared to CD4+CD25− cells of the same dogs, the latter selected by gating the 20% of CD4+ T cells showing the lowest CD25 expression. FoxP3+ cells in the CD25high fraction were gated in two ways, making a comparison with either the corresponding isotype control or the paired CD25− population (a negative biological control). The two gating methods yielded similar results: CD25high T cells had significantly greater FoxP3 expression than CD25− T cells from the same dogs (Fig. 1c).
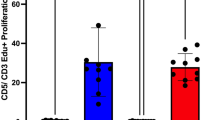
Freshly isolated canine CD4+CD25high T cells are suppressive in vitro
Freshly isolated CD4+CD25high T cells suppressed conventional CD4+CD25− T cell proliferation, as indicated by reduced cell divisions at a ratio of 1:1 or 1:2 in the presence of autologous monocytes (CD5-CD11b+) and ConA (Fig. 2). Our findings therefore confirmed the suppressive function of ex vivo canine CD4+CD25high T cells. Given their regulatory phenotype and function, we then hypothesised that canine CD4+CD25high T cells have a transcriptomic profile characteristic of Tregs.

CD4+CD25high T cells isolated ex vivo are suppressive in vitro. (a) Representative flow cytometric plots showing the proliferation of pre-labelled Tresp cells analysed in flow cytometry after a 96-hour incubation. Suppressor and responder cells were co-cultured at the ratio of 1:1. Tresp T cells of the four groups were gated following the same cascaded strategy: from live cells, to lymphocytes, to Tresp cells, followed by measurement via proportional proliferation. (b) Summary bar charts showing the proliferation of Tresp cells post 96-hour incubation (mean ± SEM), measured by means of proportional proliferation, at both 1:1 and 1:2 suppressor: responder ratios (five independent experiments). Statistical significance was analysed by one-way ANOVA, followed by Holm-Sidak’s multiple comparisons test. (c) Summary bar charts showing the percent suppression mediated by the suppressor population, normalised to parallel stimulated Tresp cells ((proliferating % of Tresp only − proliferating % of co-cultured Tresp)/(proliferating % of Tresp only) × 100 (five independent experiments; mean ± SEM). Statistical significance was determined by means of a paired t test (****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05).
Canine CD4+CD25high T cells possess the transcriptomic signature of Tregs
We conducted RNA-seq on freshly isolated CD4+CD25high and CD4+CD25− T cells. PCA analysis revealed distinct expression signatures of the two cell types (Fig. 3a). A volcano plot confirmed the distinction and suggested a Treg-like phenotype of CD25high T cells, which preferentially expressed nearly all of the known Treg-specific transcripts, such as il2ra, foxp3, ikzf2, ctla4, il10, lgals3, tigit, nrp1, lag3, icam1 and tnfrsf1843,44 (Fig. 3b).

CD4+CD25high T cells possess the transcriptomic signature of Tregs. (a) Genome-wide expression data of 9,476 transcripts of five CD4+CD25high and paired CD4+CD25− T cell samples isolated ex vivo from five healthy dogs were plotted by PCA, with the principle component 1 (PC1) of 29.8% and PC2 of 17.9%. (b) Expression data of differentially expressed transcripts of the same five CD4+CD25high versus paired CD4+CD25− T cell samples as in (a), revealed by volcano plot. Threshold line in red indicates FDR = 0.05, and each dot represents one transcript. Transcripts above threshold were differentially expressed, with Tregs-specific transcripts annotated with symbols. For better visualisation, transcript symbols were designated in upper case. The transcript il2ra was also designated with coordinates, owing to its striking values for FC and statistical significance, both off scales. (c) Stacked bar charts showing z-scores of enriched biological pathways identified by IPA, with red colour representing activated status. The dashed line highlights a z-score of 2; absolute values ≥ 2 indicate high consistency of expression direction between the input transcripts and IPA knowledge database. All highlighted pathways were statistically significant (p < 0.05).
Ingenuity pathway analysis of canine CD4+CD25high T cells
Pathway analysis further consolidated functional annotations of the CD25high T cell expression signature in comparison to CD25− T cells, which identified three pathways associated with development and function of Tregs to be activated, namely phospholipase C signalling, p38-mitogen activated protein kinase (MAPK) signalling and cell cycle regulation (Fig. 3c).
A Treg-specific expression signature is conserved in humans, mice and dogs
We compared Treg-specific transcriptomic signatures between species using similarity scores, which revealed a resemblance of canine CD4+CD25high T cells to both human and murine Tregs for the top 100 most differentially expressed transcripts (Fig. 4a). Of interest, human and murine Tregs showed no significant similarity (Supplementary Fig. S1). Thirty-one transcripts highly enriched in Tregs (FC > 2) were consensus in all three species (Fig. 4b). Among them, six transcripts encode the Treg signature molecules il2ra, foxp3, il10, ikzf2, lgals3, and tigit43,44. Thirteen transcripts, namely ccr8, ccr4, il2rb, trib1, rgs1, itgb1, ccl20, s100a4, prdm1, fas, ptger2, gata3 and ikzf4, are associated with development and function of Tregs44,45,46,47,48,49,50. The remaining 12 transcripts have not been associated with Tregs previously (Fig. 4c). Preferential expression of 11 transcripts not hitherto related to Tregs was confirmed by RT-qPCR, together with il2ra, foxp3 and ikzf2 as positive controls; primers for canine ptprj were unavailable at the time of this study, precluding confirmation of this transcript (Fig. 4d). All of the 14 transcripts examined by RT-qPCR showed greater expression in canine CD4+CD25high T cells compared to CD4+CD25− T cells, with FC values comparable to those detected by RNA-seq (Fig. 4e).

A Treg-specific transcriptomic signature is conserved in humans, mice and dogs. (a) Similarity score analysis measured the resemblance between differentially expressed transcripts of canine CD4+CD25high T cells with those of human and murine Tregs, on the basis of 772 consensus transcripts. Similarity score was calculated using the ranked top 100, 150, 200, 300, 400, 500 and 750 transcripts, respectively, with an accompanying p value. The dashed line indicates p = 0.05. (b) Venn diagram showing highly enriched transcripts (with more than two-fold preferential expression) consensus between canine CD4+CD25high T cells and, human and murine Tregs. (c) Stacked bar charts showing the 31 consensus transcripts conserved in all three species, with corresponding FC values in log2 format. Transcripts selected for RT-qPCR validation are highlighted in orange. (d) Scatter plots showing relative expression FC values of transcripts validated by RT-qPCR, plotted in log2 format. The line indicates median value of the three or four sample replicates. (e) Stacked bar charts showing expression FC values of transcripts preferentially expressed by canine CD4+CD25high T cells compatible between RNA-seq and RT-qPCR detection, plotted in log2 format.
Discussion
We have shown that canine CD4+CD25high T cells isolated ex vivo have the transcriptomic signature of Tregs, reconciling with their regulatory phenotype and function. Moreover, the transcriptomic signature of canine CD4+CD25high T cells resembled those of human and murine Tregs, consistent with our view that they represent Tregs.
Apart from FoxP3 and other Treg signature molecules, we found that the canine CD4+CD25high T cells expressed transcripts encoding transcription factors specific to pro-inflammatory T helper (Th) cells in greater abundance than CD4+CD25− T cells. For instance, CD25high T cells preferentially expressed gata3 and irf4 of Th2 cells51,52,53 and, batf, ikzf3, ikzf4 and rorα of Th17 cells54,55,56,57. A trivial explanation of this phenomenon was enrichment of effector Th cells within the CD25high T cells, which were not exclusively FoxP3+ and likely to be contaminated by Th cells. Healthy dogs are exposed to environmental antigens at mucosal surfaces on a continuous basis, with subsequent polarisation of a proportion of the local T cells and escape of these cells into the peripheral blood. An alternative explanation was that some of the peripheral Tregs themselves expressed Th-specific transcription factors, as has been previously documented25,58,59,60,61,62. The CD4+CD25high T cells also expressed a number of homing receptor transcripts at greater abundance than the CD4+CD25− T cells. For instance, CD25high T cells preferentially expressed Th2-associated chemokine receptor transcripts ccr3, ccr4 and ccr863,64,65, in line with the greater expression of Th2 transcription factor transcripts gata3 and irf4. Other chemokine receptors enriched in canine CD25high T cells are expressed by human and murine Tregs resident in various tissues and organs, i.e. CXCR6 and CCR3 in adipose tissue66, CCR2, CCR5 and CXCR3 in pancreas67, CCR4 in skin68,69 and, CCR2, CCR5 and CCR8 in muscle70. In contrast, CD25high T cells expressed three transcripts encoding naïve T cell homing molecules CD62L (L-selectin), CCR7 and IL7R71,72,73,74 in lower abundance. Trafficking of Tregs to peripheral lymphoid and non-lymphoid niches is critical to their functions in homeostasis, autoimmune disease and cancer in humans and mice, and expression of homing receptors may vary with developmental stage and target locations of Tregs68,70,75,76,77,78,79,80. Single-cell RNA-seq would be required to distinguish whether these differential expression patterns were attributable to contaminant Th cells or to bona fide Tregs. Nevertheless, these data raise the intriguing possibility of ectopic expression of Th-specific transcripts by Tregs in dogs, as in other species: for instance, human Tregs isolated ex vivo from healthy donors express gata3 and ccr4 of Th2 cells25, and murine Tregs incorporate irf4 to suppress Th2 response58.
Pathways associated with the development and function of canine Tregs were identified in our dataset. A cascade of signal transduction pathways is engaged upstream and downstream of FoxP3, dedicating Tregs to lineage-specific commitment81,82,83,84,85,86,87,88. Phospholipase C signalling is a critical transduction pathway downstream of TCR activation in Tregs, and its defect causes profound autoimmune lesions in mice89. The dominant mediator phospholipase C produces secondary messenger molecules 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG)90,91,92. IP3 activates calcium flux, which then triggers the transcription factor nuclear factor of activated T cells (NFAT) to interact with FoxP389,91. DAG functions in a cascade upstream of p38-MAPK signalling, which regulates the cell cycle and is indispensable in the induction of anergy and maintenance of Treg suppressive function93. The upregulation of phospholipase C, p38-MAPK and cell cycle regulation pathways in canine Tregs accords with these observations.
We interrogated expression signatures of Tregs across species, reasoning that similarity of transcripts would speak to their core function in Tregs. Canine Tregs resembled both human and murine Tregs, yielding 31 common differentially expressed transcripts. More than half of the 31 consensus transcripts encode Treg-specific molecules, indicative of interspecies conservation of Treg signature. Of the 12 transcripts not hitherto related to Tregs, hip1 has potential immunoregulatory relevance. Hip1 is a serine hydrolase protein embedded in cell envelopes of Mycobacterium tuberculosis, which reside intracellularly in macrophages and dendritic cells (DCs) of the host, evading immune responses by impeding functions of these primary APCs using Hip194,95,96,97. First, M. tuberculosis deactivates Toll-like receptor 2 and MyD88-dependent pathways via Hip1, reducing activation and cytokine production of macrophages and DCs94,96. Second, M. tuberculosis disrupts interactions between CD4+ T cells and APCs through GroEL2, a product of Hip1 hydrolysis95,97. Therefore, Hip1 may be another mechanism by which Tregs negatively modulate APCs. Fam129a and Alpha actinin-4 encoded by fam129a and actn4 inhibit cell apoptosis98,99, and Cathepsin Z, encoded by ctsz, promotes angiogenesis and metastasis100,101. These three proteins could potentially be blocked by specific mAb to attenuate the number and function of Tregs in the cancer microenvironment. The remaining eight transcripts are involved in T cell activation: protein products of cadm1, frmd4b, lmna, anxa2, galm, pou2f2, csf1 and ptprj may directly or indirectly enhance TCR signalling or interaction of T cells with APCs102,103,104,105,106,107,108,109. Single cell RNA-seq would be required to further explore the significance of these transcripts to Tregs, along with confirmation of differential expression of their protein products and their role in suppressive function, if any.
In conclusion, we have characterised the phenotype, function, and transcriptomic signature of canine Tregs. We have delineated a core set of 31 transcripts that show differential expression by the Tregs of three mammalian species, including humans. More than half of these transcripts have been previously associated with Tregs in mice and humans. However, 12 transcripts have hitherto not been associated with Tregs in any species, prompting further questions about their role in this cellular context. This comparative approach is a powerful tool in generating hypotheses that may yield fresh mechanistic insights or novel immunotherapeutic targets in this important, yet elusive, area of immunology.
Data Availability
Raw and processed canine RNA-seq data of this study have been deposited to Gene Expression Omnibus (GEO), accession number GSE132068.
References
Ohkura, N., Kitagawa, Y. & Sakaguchi, S. Development and maintenance of regulatory T cells. Immunity 38, 414–423, https://doi.org/10.1016/j.immuni.2013.03.002 (2013).
Sasada, T., Kimura, M., Yoshida, Y., Kanai, M. & Takabayashi, A. CD4+CD25+ regulatory T cells in patients with gastrointestinal malignancies: possible involvement of regulatory T cells in disease progression. Cancer 98, 1089–1099, https://doi.org/10.1002/cncr.11618 (2003).
Curiel, T. J. et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 10, 942–949, https://doi.org/10.1038/nm1093 (2004).
Bennett, C. L. et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nature genetics 27, 20–21, https://doi.org/10.1038/83713 (2001).
Shitara, K. & Nishikawa, H. Regulatory T cells: a potential target in cancer immunotherapy. Ann N Y Acad Sci 1417, 104–115, https://doi.org/10.1111/nyas.13625 (2018).
Sharabi, A. et al. Regulatory T cells in the treatment of disease. Nature reviews. Drug discovery. https://doi.org/10.1038/nrd.2018.148 (2018).
Romano, M., Tung, S. L., Smyth, L. A. & Lombardi, G. Treg therapy in transplantation: a general overview. Transplant international: official journal of the European Society for Organ Transplantation 30, 745–753, https://doi.org/10.1111/tri.12909 (2017).
Benoist, C. & Mathis, D. Treg cells, life history, and diversity. Cold Spring Harb Perspect Biol 4, a007021, https://doi.org/10.1101/cshperspect.a007021 (2012).
Sakaguchi, S., Vignali, D. A., Rudensky, A. Y., Niec, R. E. & Waldmann, H. The plasticity and stability of regulatory T cells. Nat Rev Immunol 13, 461–467, https://doi.org/10.1038/nri3464 (2013).
Brockmann, L. et al. Molecular and functional heterogeneity of IL-10-producing CD4(+) T cells. Nat Commun 9, 5457, https://doi.org/10.1038/s41467-018-07581-4 (2018).
Gagliani, N. et al. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat Med 19, 739–746, https://doi.org/10.1038/nm.3179 (2013).
Zhang, S., Wu, M. & Wang, F. Immune regulation by CD8(+) Treg cells: novel possibilities for anticancer immunotherapy. Cell Mol Immunol 15, 805–807, https://doi.org/10.1038/cmi.2018.170 (2018).
Wang, R. F. CD8+ regulatory T cells, their suppressive mechanisms, and regulation in cancer. Hum Immunol 69, 811–814, https://doi.org/10.1016/j.humimm.2008.08.276 (2008).
Okamura, T. et al. CD4+CD25−LAG3+ regulatory T cells controlled by the transcription factor Egr-2. Proc Natl Acad Sci USA 106, 13974–13979, https://doi.org/10.1073/pnas.0906872106 (2009).
Okamura, T., Yamamoto, K. & Fujio, K. Early growth response gene 2-expressing CD4(+)LAG3(+) regulatory T cells: the therapeutic potential for treating autoimmune diseases. Front Immunol 9, 340, https://doi.org/10.3389/fimmu.2018.00340 (2018).
Li, X. et al. Generation of human regulatory gammadelta T cells by TCRgammadelta stimulation in the presence of TGF-beta and their involvement in the pathogenesis of systemic lupus erythematosus. J Immunol 186, 6693–6700, https://doi.org/10.4049/jimmunol.1002776 (2011).
Wesch, D., Peters, C. & Siegers, G. M. Human gamma delta T regulatory cells in cancer: fact or fiction? Front Immunol 5, 598, https://doi.org/10.3389/fimmu.2014.00598 (2014).
Krovi, S. H. & Gapin, L. Invariant natural killer T cell subsets-more than just developmental intermediates. Front Immunol 9, 1393, https://doi.org/10.3389/fimmu.2018.01393 (2018).
Lam, P. Y., Nissen, M. D. & Mattarollo, S. R. Invariant natural killer T cells in immune regulation of blood cancers: harnessing their potential in immunotherapies. Front Immunol 8, 1355, https://doi.org/10.3389/fimmu.2017.01355 (2017).
Itoh, M. et al. Thymus and autoimmunity: production of CD25+CD4+ naturally anergic and suppressive T cells as a key function of the thymus in maintaining immunologic self-tolerance. J Immunol 162, 5317–5326 (1999).
Sakaguchi, S., Miyara, M., Costantino, C. M. & Hafler, D. A. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol 10, 490–500, https://doi.org/10.1038/nri2785 (2010).
Kitagawa, Y. et al. Guidance of regulatory T cell development by Satb1-dependent super-enhancer establishment. Nature immunology 18, 173–183, https://doi.org/10.1038/ni.3646 (2017).
Stephens, G. L., Andersson, J. & Shevach, E. M. Distinct subsets of FoxP3+ regulatory T cells participate in the control of immune responses. The Journal of Immunology 178, 6901–6911, https://doi.org/10.4049/jimmunol.178.11.6901 (2007).
Hansmann, L. et al. Dominant Th2 differentiation of human regulatory T cells upon loss of FOXP3 expression. J Immunol 188, 1275–1282, https://doi.org/10.4049/jimmunol.1102288 (2012).
Duhen, T., Duhen, R., Lanzavecchia, A., Sallusto, F. & Campbell, D. J. Functionally distinct subsets of human FOXP3+ Treg cells that phenotypically mirror effector Th cells. Blood 119, 4430–4440, https://doi.org/10.1182/blood-2011-11-392324 (2012).
Miyara, M. et al. Sialyl Lewis x (CD15s) identifies highly differentiated and most suppressive FOXP3high regulatory T cells in humans. Proc Natl Acad Sci USA 112, 7225–7230, https://doi.org/10.1073/pnas.1508224112 (2015).
Garden, O. A., Pinheiro, D. & Cunningham, F. All creatures great and small: regulatory T cells in mice, humans, dogs and other domestic animal species. Int Immunopharmacol 11, 576–588, https://doi.org/10.1016/j.intimp.2010.11.003 (2011).
Pinheiro, D. et al. Dissecting the regulatory microenvironment of a large animal model of non-Hodgkin lymphoma: evidence of a negative prognostic impact of FOXP3+ T cells in canine B cell lymphoma. PloS one 9, e105027, https://doi.org/10.1371/journal.pone.0105027 (2014).
Richards, K. L. & Suter, S. E. Man’s best friend: what can pet dogs teach us about non-Hodgkin’s lymphoma? Immunological reviews 263, 173–191, https://doi.org/10.1111/imr.12238 (2015).
Biller, B. J., Elmslie, R. E., Burnett, R. C., Avery, A. C. & Dow, S. W. Use of FoxP3 expression to identify regulatory T cells in healthy dogs and dogs with cancer. Vet Immunol Immunopathol 116, 69–78, https://doi.org/10.1016/j.vetimm.2006.12.002 (2007).
Pinheiro, D. et al. Phenotypic and functional characterization of a CD4(+) CD25(high) FOXP3(high) regulatory T-cell population in the dog. Immunology 132, 111–122, https://doi.org/10.1111/j.1365-2567.2010.03346.x (2011).
Abrams, V. K. et al. A novel monoclonal antibody specific for canine CD25 (P4A10): selection and evaluation of canine Tregs. Vet Immunol Immunopathol 135, 257–265, https://doi.org/10.1016/j.vetimm.2009.12.006 (2010).
Viguier, M. et al. Foxp3 expressing CD4+CD25high regulatory T cells are overrepresented in human metastatic melanoma lymph nodes and inhibit the function of infiltrating T cells. The Journal of Immunology 173, 1444–1453, https://doi.org/10.4049/jimmunol.173.2.1444 (2004).
Rodriguez-Perea, A. L., Arcia, E. D., Rueda, C. M. & Velilla, P. A. Phenotypical characterization of regulatory T cells in humans and rodents. Clin Exp Immunol 185, 281–291, https://doi.org/10.1111/cei.12804 (2016).
Nishioka, T., Shimizu, J., Iida, R., Yamazaki, S. & Sakaguchi, S. CD4+CD25+Foxp3+ T cells and CD4+CD25−Foxp3+ T cells in aged mice. The Journal of Immunology 176, 6586–6593, https://doi.org/10.4049/jimmunol.176.11.6586 (2006).
Knueppel, A. et al. Phenotypic and functional characterization of freshly isolated and expanded canine regulatory T cells. Exp Anim 60, 471–479 (2011).
Archer, T. M. et al. In vivo effects of aspirin and cyclosporine on regulatory T cells and T-cell cytokine production in healthy dogs. Vet Immunol Immunopathol 197, 63–68, https://doi.org/10.1016/j.vetimm.2018.01.003 (2018).
Palatucci, A. T. et al. Circulating regulatory T cells (Treg), leptin and induction of proinflammatory activity in obese Labrador Retriever dogs. Vet Immunol Immunopathol 202, 122–129, https://doi.org/10.1016/j.vetimm.2018.07.004 (2018).
Pfaffl, M. W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic acids research 29, e45 (2001).
Albert, M. H. et al. MiRNome and transcriptome aided pathway analysis in human regulatory T cells. Genes and immunity 15, 303–312, https://doi.org/10.1038/gene.2014.20 (2014).
Afgan, E. et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic acids research 46, W537–W544, https://doi.org/10.1093/nar/gky379 (2018).
Auray, G. et al. Characterization and transcriptomic analysis of porcine blood conventional and plasmacytoid dendritic cells reveals striking species-specific differences. J Immunol 197, 4791–4806, https://doi.org/10.4049/jimmunol.1600672 (2016).
Bhairavabhotla, R. et al. Transcriptome profiling of human FoxP3+ regulatory T cells. Hum Immunol 77, 201–213, https://doi.org/10.1016/j.humimm.2015.12.004 (2016).
Birzele, F. et al. Next-generation insights into regulatory T cells: expression profiling and FoxP3 occupancy in Human. Nucleic acids research 39, 7946–7960, https://doi.org/10.1093/nar/gkr444 (2011).
Bonnal, R. J. et al. De novo transcriptome profiling of highly purified human lymphocytes primary cells. Sci Data 2, 150051, https://doi.org/10.1038/sdata.2015.51 (2015).
Zemmour, D. et al. Single-cell gene expression reveals a landscape of regulatory T cell phenotypes shaped by the TCR. Nature immunology 19, 291–301, https://doi.org/10.1038/s41590-018-0051-0 (2018).
Zheng, C. et al. Landscape of infiltrating T Cells in liver cancer revealed by single-cell sequencing. Cell 169, 1342–1356 e1316, https://doi.org/10.1016/j.cell.2017.05.035 (2017).
Dong, S. et al. Multiparameter single-cell profiling of human CD4+FOXP3+ regulatory T-cell populations in homeostatic conditions and during graft-versus-host disease. Blood 122, 1802–1812, https://doi.org/10.1182/blood-2013-02-482539 (2013).
Schmidl, C. et al. The enhancer and promoter landscape of human regulatory and conventional T-cell subpopulations. Blood 123, e68–78, https://doi.org/10.1182/blood-2013-02-486944 (2014).
Ubaid, U. et al. Transcriptional repressor HIC1 contributes to suppressive function of human induced regulatory T cells. Cell Rep 22, 2094–2106, https://doi.org/10.1016/j.celrep.2018.01.070 (2018).
O’Garra, A. & Gabrysova, L. Transcription factors directing Th2 differentiation: Gata-3 plays a dominant role. J Immunol 196, 4423–4425, https://doi.org/10.4049/jimmunol.1600646 (2016).
Lohoff, M. et al. Dysregulated T helper cell differentiation in the absence of interferon regulatory factor 4. Proc Natl Acad Sci USA 99, 11808–11812, https://doi.org/10.1073/pnas.182425099 (2002).
Rengarajan, J. et al. Interferon regulatory factor 4 (IRF4) interacts with NFATc2 to modulate interleukin 4 gene expression. The Journal of Experimental Medicine 195, 1003–1012, https://doi.org/10.1084/jem.20011128 (2002).
Martinez, G. J. & Dong, C. BATF: bringing (in) another Th17-regulating factor. J Mol Cell Biol 1, 66–68, https://doi.org/10.1093/jmcb/mjp016 (2009).
Yang, X. O. et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity 28, 29–39, https://doi.org/10.1016/j.immuni.2007.11.016 (2008).
van Hamburg, J. P. & Tas, S. W. Molecular mechanisms underpinning T helper 17 cell heterogeneity and functions in rheumatoid arthritis. J Autoimmun 87, 69–81, https://doi.org/10.1016/j.jaut.2017.12.006 (2018).
Liu, S. Q., Jiang, S., Li, C., Zhang, B. & Li, Q. J. miR-17-92 cluster targets phosphatase and tensin homology and Ikaros Family Zinc Finger 4 to promote Th17-mediated inflammation. J Biol Chem 289, 12446–12456, https://doi.org/10.1074/jbc.M114.550723 (2014).
Zheng, Y. et al. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature 458, 351–356, https://doi.org/10.1038/nature07674 (2009).
Ayyoub, M. et al. Human memory FOXP3+ Tregs secrete IL-17 ex vivo and constitutively express the T(H)17 lineage-specific transcription factor RORgamma t. Proc Natl Acad Sci USA 106, 8635–8640, https://doi.org/10.1073/pnas.0900621106 (2009).
Schmidl, C. et al. Epigenetic reprogramming of the RORC locus during in vitro expansion is a distinctive feature of human memory but not naive Treg. European journal of immunology 41, 1491–1498, https://doi.org/10.1002/eji.201041067 (2011).
Chaudhry, A. et al. CD4+ regulatory T cells control Th17 responses in a Stat3-dependent manner. Science 326, 986–991, https://doi.org/10.1126/science.1172702 (2009).
Koch, M. A. et al. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nature immunology 10, 595–602, https://doi.org/10.1038/ni.1731 (2009).
Sallusto, F., Mackay, C. R. & Lanzavecchia, A. Selective expression of the eotaxin receptor CCR3 by human T helper 2 cells. Science 277 (1997).
Zhou, S. F. et al. Characterization of Th1- and Th2-associated chemokine receptor expression in spleens of patients with immune thrombocytopenia. J Clin Immunol 33, 938–946, https://doi.org/10.1007/s10875-013-9883-4 (2013).
D’Ambrosio, D. et al. Selective up-regulation of chemokine receptors CCR4 and CCR8 upon activation of polarized human type 2 Th cells. J Immunol 161, 5111–5115 (1998).
Cipolletta, D. et al. PPAR-gamma is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature 486, 549–553, https://doi.org/10.1038/nature11132 (2012).
Chen, Z., Herman, A. E., Matos, M., Mathis, D. & Benoist, C. Where CD4+CD25+ T reg cells impinge on autoimmune diabetes. J Exp Med 202, 1387–1397, https://doi.org/10.1084/jem.20051409 (2005).
Sather, B. D. et al. Altering the distribution of Foxp3(+) regulatory T cells results in tissue-specific inflammatory disease. J Exp Med 204, 1335–1347, https://doi.org/10.1084/jem.20070081 (2007).
Iellem, A., Colantonio, L. & D’Ambrosio, D. Skin-versus gut-skewed homing receptor expression and intrinsic CCR4 expression on human peripheral blood CD4+CD25+ suppressor T cells. European journal of immunology 33, 1488–1496, https://doi.org/10.1002/eji.200323658 (2003).
Burzyn, D. et al. A special population of regulatory T cells potentiates muscle repair. Cell 155, 1282–1295, https://doi.org/10.1016/j.cell.2013.10.054 (2013).
Ley, K. & Kansas, G. S. Selectins in T-cell recruitment to non-lymphoid tissues and sites of inflammation. Nat Rev Immunol 4, 325–335, https://doi.org/10.1038/nri1351 (2004).
Masopust, D. & Schenkel, J. M. The integration of T cell migration, differentiation and function. Nat Rev Immunol 13, 309–320, https://doi.org/10.1038/nri3442 (2013).
Carrette, F. & Surh, C. D. IL-7 signaling and CD127 receptor regulation in the control of T cell homeostasis. Semin Immunol 24, 209–217, https://doi.org/10.1016/j.smim.2012.04.010 (2012).
Surh, C. D. & Sprent, J. Homeostasis of naive and memory T cells. Immunity 29, 848–862, https://doi.org/10.1016/j.immuni.2008.11.002 (2008).
Abadier, M. et al. Effector and regulatory T cells roll at high shear stress by inducible tether and sling formation. Cell Rep 21, 3885–3899, https://doi.org/10.1016/j.celrep.2017.11.099 (2017).
Campbell, D. J. Control of regulatory T cell migration, function, and homeostasis. J Immunol 195, 2507–2513, https://doi.org/10.4049/jimmunol.1500801 (2015).
Chow, Z., Banerjee, A. & Hickey, M. J. Controlling the fire–tissue-specific mechanisms of effector regulatory T-cell homing. Immunol Cell Biol 93, 355–363, https://doi.org/10.1038/icb.2014.117 (2015).
Ding, Y., Xu, J. & Bromberg, J. S. Regulatory T cell migration during an immune response. Trends Immunol 33, 174–180, https://doi.org/10.1016/j.it.2012.01.002 (2012).
Lim, H. W., Broxmeyer, H. E. & Kim, C. H. Regulation of trafficking receptor expression in human Forkhead Box P3+ regulatory T cells. The Journal of Immunology 177, 840–851, https://doi.org/10.4049/jimmunol.177.2.840 (2006).
Siewert, C. et al. Induction of organ-selective CD4+ regulatory T cell homing. European journal of immunology 37, 978–989, https://doi.org/10.1002/eji.200636575 (2007).
Hori, S., Nomura, T. & Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 299, 1057–1061, https://doi.org/10.1126/science.1079490 (2003).
Josefowicz, S. Z. & Rudensky, A. Control of regulatory T cell lineage commitment and maintenance. Immunity 30, 616–625, https://doi.org/10.1016/j.immuni.2009.04.009 (2009).
Zheng, Y. et al. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature 445, 936–940, https://doi.org/10.1038/nature05563 (2007).
Hill, J. A. et al. Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity 27, 786–800, https://doi.org/10.1016/j.immuni.2007.09.010 (2007).
Crellin, N. K., Garcia, R. V. & Levings, M. K. Altered activation of AKT is required for the suppressive function of human CD4+CD25+ T regulatory cells. Blood 109, 2014–2022, https://doi.org/10.1182/blood-2006-07-035279 (2007).
Sauer, S. et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci USA 105, 7797–7802, https://doi.org/10.1073/pnas.0800928105 (2008).
Marson, A. et al. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature 445, 931–935, https://doi.org/10.1038/nature05478 (2007).
Lee, W. & Lee, G. R. Transcriptional regulation and development of regulatory T cells. Exp Mol Med 50, e456, https://doi.org/10.1038/emm.2017.313 (2018).
Fu, G. et al. Phospholipase C{gamma}1 is essential for T cell development, activation, and tolerance. J Exp Med 207, 309–318, https://doi.org/10.1084/jem.20090880 (2010).
Wells, A. D. et al. Regulation of T cell activation and tolerance by Phospholipase C-1-dependent integrin avidity modulation. The Journal of Immunology 170, 4127–4133, https://doi.org/10.4049/jimmunol.170.8.4127 (2003).
Putney, J. W. & Tomita, T. Phospholipase C signaling and calcium influx. Adv Biol Regul 52, 152–164, https://doi.org/10.1016/j.advenzreg.2011.09.005 (2012).
Kawakami, T. & Xiao, W. Phospholipase C-beta in immune cells. Adv Biol Regul 53, 249–257, https://doi.org/10.1016/j.jbior.2013.08.001 (2013).
Adler, H. S. & Steinbrink, K. MAP kinase p38 and its relation to T cell anergy and suppressor function of regulatory T cells. Cell Cycle 7, 169–170, https://doi.org/10.4161/cc.7.2.5312 (2008).
Madan-Lala, R., Peixoto, K. V., Re, F. & Rengarajan, J. Mycobacterium tuberculosis Hip1 dampens macrophage proinflammatory responses by limiting toll-like receptor 2 activation. Infect Immun 79, 4828–4838, https://doi.org/10.1128/IAI.05574-11 (2011).
Georgieva, M., Sia, J. K., Bizzell, E., Madan-Lala, R. & Rengarajan, J. Mycobacterium tuberculosis GroEL2 modulates dendritic cell responses. Infect Immun 86, https://doi.org/10.1128/IAI.00387-17 (2018).
Madan-Lala, R. et al. Mycobacterium tuberculosis impairs dendritic cell functions through the serine hydrolase Hip1. J Immunol 192, 4263–4272, https://doi.org/10.4049/jimmunol.1303185 (2014).
Naffin-Olivos, J. L. et al. Mycobacterium tuberculosis Hip1 modulates macrophage responses through proteolysis of GroEL2. PLoS Pathog 10, e1004132, https://doi.org/10.1371/journal.ppat.1004132 (2014).
Chakraborty, S. et al. Alpha-actinin 4 potentiates myocyte enhancer factor-2 transcription activity by antagonizing histone deacetylase 7. J Biol Chem 281, 35070–35080, https://doi.org/10.1074/jbc.M602474200 (2006).
Ji, H. et al. AKT-dependent phosphorylation of Niban regulates nucleophosmin- and MDM2-mediated p53 stability and cell apoptosis. EMBO reports 13, 554–560, https://doi.org/10.1038/embor.2012.53 (2012).
Bernhardt, A., Kuester, D., Roessner, A., Reinheckel, T. & Krueger, S. Cathepsin X-deficient gastric epithelial cells in co-culture with macrophages: characterization of cytokine response and migration capability after Helicobacter pylori infection. J Biol Chem 285, 33691–33700, https://doi.org/10.1074/jbc.M110.146183 (2010).
Sevenich, L. et al. Synergistic antitumor effects of combined cathepsin B and cathepsin Z deficiencies on breast cancer progression and metastasis in mice. Proc Natl Acad Sci USA 107, 2497–2502, https://doi.org/10.1073/pnas.0907240107 (2010).
Nakahata, S. & Morishita, K. CADM1/TSLC1 is a novel cell surface marker for adult T-cell leukemia/lymphoma. Journal of clinical and experimental hematopathology: JCEH 52, 17–22 (2012).
Gonzalez-Granado, J. M. et al. Nuclear envelope lamin-A couples actin dynamics with immunological synapse architecture and T cell activation. Science signaling 7, ra37, https://doi.org/10.1126/scisignal.2004872 (2014).
Marlin, R. et al. Sensing of cell stress by human gammadelta TCR-dependent recognition of annexin A2. Proc Natl Acad Sci USA 114, 3163–3168, https://doi.org/10.1073/pnas.1621052114 (2017).
Klarlund, J. K. et al. Signaling complexes of the FERM domain-containing protein GRSP1 bound to ARF exchange factor GRP1. J Biol Chem 276, 40065–40070, https://doi.org/10.1074/jbc.M105260200 (2001).
Pai, T., Chen, Q., Zhang, Y., Zolfaghari, R. & Ross, A. C. Galactomutarotase and other galactose-related genes are rapidly induced by retinoic acid in human myeloid cells. Biochemistry 46, 15198–15207, https://doi.org/10.1021/bi701891t (2007).
Corcoran, L. et al. Oct2 and Obf1 as facilitators of B:T cell collaboration during a humoral immune response. Front Immunol 5, 108, https://doi.org/10.3389/fimmu.2014.00108 (2014).
Zhu, Y. et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res 74, 5057–5069, https://doi.org/10.1158/0008-5472.CAN-13-3723 (2014).
Omerovic, J., Clague, M. J. & Prior, I. A. Phosphatome profiling reveals PTPN2, PTPRJ and PTEN as potent negative regulators of PKB/Akt activation in Ras-mutated cancer cells. The Biochemical journal 426, 65–72, https://doi.org/10.1042/bj20091413 (2010).
Acknowledgements
We are grateful to all the owners for kindly volunteering their dogs, and to the Clinical Investigation Centre of the Royal Veterinary College for collecting blood samples. We wish to thank the Oxford Genomics Centre at the Wellcome Centre for Human Genetics for performing RNA sequencing and initial data analysis. We also thank the Petplan Charitable Trust for funding this study. Additional funding resources were derived from intramural grants at the RVC and Barts Cancer Institute.
Author information
Authors and Affiliations
Contributions
Y.W. conducted the entire study and wrote the manuscript. Y.C. performed the volcano plot analysis and assisted with all the statistical analysis. A.J.S. and S.L.P. co-supervised Y.W. on project conduction and data interpretation. E.S. performed initial RNA-seq analysis and provided scripts for basic transcriptomic analysis. M.R.G. helped with suppression assay experiments. J.G. co-supervised Y.W. and co-funded the study. D.X. contributed expertise and intellectual input on all the transcriptomic data interpretation, and co-supervised Y.W. in the last year of the study. O.A.G. conceived and funded the study, recruited Y.W., served as the principal supervisor, and edited the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, Y., Chang, YM., Stell, A.J. et al. Phenotypic characterisation of regulatory T cells in dogs reveals signature transcripts conserved in humans and mice. Sci Rep 9, 13478 (2019). https://doi.org/10.1038/s41598-019-50065-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-50065-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
