
Abstract
The rice root-knot nematode, Meloidogyne graminicola, is a serious pest in most rice-growing countries. Usually, nematodes employ antioxidants to counteract the harm of reactive oxygen species (ROS) and facilitate their infection. Here the gene encoding M. graminicola protein disulphide isomerase (MgPDI) was identified. The deduced protein is highly conserved in the putative active-site Cys-Gly-His-Cys. In situ hybridization showed that MgPDI was specifically localized within esophageal glands of pre-parasitic second stage juveniles (J2s). MgPDI was significantly up-regulated in the late parasitic J2s. Characterization of the recombinant protein showed that the purified MgPDI exhibited similar activities to other oxidases/isomerases such as the refolding of the scrambled RNase and insulin disulfide reductase and the protection of plasmid DNA and living cells from ROS damage. In addition, silencing of MgPDI by RNA interference in the pre-parasitic J2s lowered their multiplication factor. MgPDI expression was up-regulated in the presence of exogenous H2O2, whereas MgPDI silencing resulted in an increase in mortality under H2O2 stress. MgPDI is localized in the apoplast when transient expression in Nicotiana benthamiana leaves. The results indicated that MgPDI plays important roles in the reproduction and pathogenicity of M. graminicola and it also contributes to protecting nematodes from exogenous H2O2 stress.
Similar content being viewed by others


Introduction
Rice (Oryza sativa) is an important cereal crop; more than half of the world population relies on rice. Among rice pathogens, rice root-knot nematode, Meloidogyne graminicola (Mg), is considered to be a quarantine pest in most of the rice-growing countries and cause significant yield reductions1,2,3. Meloidogyne graminicola is an obligate sedentary endoparasite of rice4. The second stage juvenile (J2) penetrates the epidermal root tip and migrates intercellularly along the vascular zone and becomes sedentary to induce and establish the feeding site. Then the J2 develops into a third (J3) and then fourth (J4) stage juvenile and eventually into an adult where the male moves out from the root while the female remains embedded within the root5. Individuals of Mg can produce many generations within a single rice growing season under favorable conditions6.
Reactive oxygen species (ROS) can be involved in host defense reactions through the oxidation of phenolic compounds into more toxic quinones and lignin-like compounds. Plants generate ROS as signaling molecules to control various processes including pathogen defense and programmed cell death under biotic stresses7. However, in order to survive, aerobic cells may reduce ROS-related oxidative stress by expressing a set of antioxidant enzymes, including thioredoxin (Trx)8.
The protein disulphide isomerase (PDI) family, a member of the thioredoxin superfamily, can catalyze the formation, reduction, and isomerization of disulfide bonds of proteins9. Protein disulphide isomerase includes two catalytically active thioredoxin domains containing characteristic Cys-Gly-His-Cys (CGHC) active-site motifs, namely domains a and a’ which are separated by two homologous but inactive internal domains b and b’10,11. Protein disulfide isomerase is a multifunctional enzyme because it can catalyze thiol oxidation reactions, disulfide reduction, and isomerisation in vitro under different redox conditions. Previous research has demonstrated that PDI participates in many biological processes by regulating disulfide bonds in different substrates. For example, PDI can release the cholera toxin active chain A from the endoplasmic reticulum to the cytosol of an infected cell and permit HIV to enter into host cells with infectious diseases12. The PDI from Caenorhabditis elegans was reported to play a role in the formation of the cuticle13. While HsPDI plays a role probably by inducing local changes in the redox status of infected host tissue14. Other studies have reported that PDIs of pathogens also play important roles in virulence when infecting a host15. Over-expression of Leishmania major PDI (LmPDI) can promote the virulence of parasitic strains of Leishmania species to cause leishmaniasis16. However, PDI inhibitors can hinder parasite growth in culture9. Expression of PDI from the oomycete plant parasite Phytophthora parasitica induces strong cell death in Nicotiana benthamiana leaves while the effect is reduced in a gene mutant17. Researchers have studied PDI in a number of organisms and report that PDI is evolutionarily conserved from prokaryotes to eukaryotes9. Characterizing PDI from these organisms can contribute to our understanding of redox control and provide information on novel targets for pathogen control. However, little is known about the PDI family from M. graminicola.
In the present study, a 56-kDa protein designated MgPDI that contains a CGHC active motif in the rice root-knot nematode, M. graminicola, was identified and characterized. It is localized in the apoplast when transient expression in N. benthamiana leaves. The enzymatic activities of MgPDI include oxidation, isomerisation, and reduction of substrates. MgPDI likely plays an important role in protecting M. graminicola against rice-released ROS and facilitating M. graminicola infestation of rice hosts and its role was demonstrated in this study.
Results
Cloning and sequence analysis of MgPDI
The full-length MgPDI was identified through RACE-PCR and was deposited in GenBank under accession number MH392200. The ORF of the MgPDI was from nucleotide 170–1666, encoding a putative protein of 498 amino acids with a deduced signal peptide cleavage site between residues 20 and 21. The predicted isoelectric point and molecular weights were 7.02 and 56.96 kDa, respectively. Further domain analysis revealed that the putative protein contained four conserved thioredoxin domains (a, b, b’, a’) with two catalytic domains (CGHC) (Fig. 1a,b). Protein disulphide isomerase has been found in humans and other animals and plant-parasitic and free-living nematodes. Results of a BLASTP search showed that MgPDI shared 79%, 71%, 69%, 32% and 33% identity to PDI from Heterodera schachtii, Caenorhabditis elegans [NP_491995], Ascaris suum [ERG84937], Leishmania major [AAN75008], and Brugia malayi [XP_001899304], respectively.

Sequence and structural analysis of MgPDI. (a) Alignment of MgPDI with orthologues in other nematode species. (b) Schematic drawing of MgPDI primary and tertiary structures.
In their tertiary structure, the four domains are found to be spatially organized in the shape of a twisted “U”, in which the a and a’ domains are located at the ends while the b and b’ domains formed the base of the “U”; Four conserved thioredoxin domains (a, b, b′, a′) are highlighted in purple, blue, yellow and red, respectively; The C-terminal extension is in green; Two catalytic domains (CGHC) are highlighted in the MgPDI tertiary structure as colored spheres. One sphere represents one of the atoms in the corresponding amino acids (Fig. 1b).
In situ hybridization and expression analysis
In situ hybridization was performed to localize the expression of MgPDI in the pre-parasitic juveniles of M. graminicola. The DIG-labelled antisense probe of MgPDI specifically hybridized with transcripts in the subventral gland cells of the pre-parasitic J2s (pre- J2s), while no hybridization was observed in the negative control, the labeled sense probe (Fig. 2a). To further investigate the expression pattern of MgPDI during different developmental stages of M. graminicola, we used cDNA generated from nematode RNA isolated at different pre-parasitic (eggs and freshly hatched J2s) and parasitic developmental stages (par-J2s, par-J3s/par-J4s and young females) in qRT-PCR analyses. The expression level of MgPDI increased during the sedentary stages of nematode development, reaching its maximum in the late parasitic J2s (5 days post-infection, 5dpi) with a 6-fold increase compared with pre-parasitic J2s. In J3/J4s and young females, expression decreased but was still higher when compared with unhatched J2s in eggs and hatched pre-parasitic J2s (Fig. 2b).

Expression patterns of MgPDI in M. graminicola. (a) Localization of MgPDI by in situ hybridization. Right: antisense cDNA probes; left: sense: cDNA probes. (b) Expression pattern of MgPDI in five different life stages of M. graminicola. Vertical bars represent the means ± SD.
Recombinant MgPDI: optimization and validation
We expressed MgPDI in E. coli BL21 (DE3) cells (Supplementary Fig. S1). Isopropyl-B-D-thiogalactoside induced the E. coli expression of the recombinant protein (Fig. 3, lane 2). To exclude the possible effects of the tags on the activity of MgPDI, thrombin was used to cleave N-terminal fusion tags and the recombinant protein yielded an enriched nontagged protein band after purification (Fig. 3, lane 3). The final concentration of MgPDI was 2 mg/ml after removing the thrombin. Mass spectrometry analysis was used to validate the identity of the expressed protein. The purified recombinant protein exhibited high sequence homology with the peptide fragments deduced from MgPDI where the score was 11512.77% and coverage was 72.09% (Supplementary Table S1).

The 12% SDS-PAGE analysis of MgPDI expressed in E. coli BL21 (DE3). The total proteins (lane 1), the expressed product of pET-32a-MgPDI induced by IPTG (lane 2) and enriched nontagged MgPDI (lane 3) were run on one gel.
Oxidoreductase activity of MgPDI
Oxidase/isomerase activity was analyzed using an RNase A renaturation assay, and the results indicated that MgPDI exhibited the properties of oxidative folding on RNase A, similar to the properties of human PDI. The activity of MgPDI induced a time-dependent increase in denatured RNase A refolding; the velocity of cCMP hydrolysis catalyzed by renatured RNase A increased with the time allowed for the refolding reaction (Fig. 4a). The human PDI used as a positive control showed marginally greater oxidase/isomerase activity than the MgPDI in the present study. For the MgPDI reductive assay, the decline in bovine insulin disulfide bonds with the increase in MgPDI concentrations was detected by the increasing turbidity in the reactions. The negative control reaction without MgPDI, however, showed no insulin disulphide bond reduction (Fig. 4b).

Biological activity of recombinant MgPDI. (a) Thiol isomerase activity of the purified recombinant MgPDI. (b) Dithiothreitol-mediated insulin reduction by recombinant MgPDI. (c) MgPDI protects H2O2-stressed HEK293 cells. (d) Potential of recombinant MgPDI to protect super-coiled DNA from cleavage in an MFO system. Lanes: a, pGEM-T without any treatment; b, pGEM-T incubated with 1.65 mM DTT; c, pGEM-T incubated with 16.5 mM FeCl3; d, pGEM-T incubated with MFO system; e–h, pGEM-T incubated with MFO system and different concentrations of purified MgPDI. NF, nicked form; SF, super-coiled form of pGEM-T. Vertical bars represent the means ± SD.
MgPDI function as antioxidants
The influence of MgPDI on viability of HEK293 cells under H2O2 stress was determined using a cell proliferation assay kit. Overall, viability of cells in the MgPDI-treated groups was higher than that of the BSA-treated control when exposed to the H2O2 stress. Moreover, viability increased with the increase of MgPDI concentration (Fig. 4c). The degree of DNA damage was indicated by distinct mobility-shift patterns produced by the two DNA bands after electrophoretic resolution on a gel; MgPDI appears to rescue the plasmid DNA-nicking reaction in a dose-dependent manner (Fig. 4d, lanes e to h). However, the DNA underwent maximum damage in the MFO system when MgPDI was absent (Fig. 4d, lane d).
Hydrogen peroxide content determination
The hydrogen peroxide (H2O2) level was determined in rice roots, and increased level of H2O2 was found at 3 dpi, however, H2O2 levels in knots were lower at 7 dpi and 14 dpi compared with the root tips without Mg infection (Supplementary Fig. S2, Fig. 5a).

MgPDI expression is triggered by H2O2 and increases H2O2 tolerance. (a) The H2O2 content per gram of fresh root tissue. (b) Mortality rate of freshly hatched J2s exposed to H2O2. (c) The relative mRNA expression levels of MgPDI in response to the H2O2 stress. Vertical bars represent the means ± SD.
MgPDI expression is induced by H2O2 and increases H2O2 tolerance
In this study we observed that J2s can survive up to 90 min in 10 mM H2O2 without a significant increase in mortality rate; however, overall mortality of J2s noticeably increased with increasing H2O2 concentrations (Fig. 5b). Next, qRT-PCR was used to analyse the expression of MgPDI in response to the H2O2 stress. A significant increase in transcript abundance of MgPDI was observed when comparing juveniles that were exposed to H2O2 (20 mM–80 mM) for 30 min with J2s that were treated with water (Fig. 5c).
Influence of RNAi silencing of MgPDI on nematode parasitism
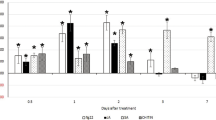
In vitro RNAi targeting of MgPDI was performed to analyze whether MgPDI plays an important role in parasitism. The results of qRT-PCR experiments showed a decrease in the transcript abundance of MgPDI when the nematodes were soaked with dsRNA against MgPDI, indicating the effective silencing of MgPDI (Fig. 6a). We examined the mortality rates of MgPDI or GFP dsRNA-treated J2s after soaking them in various concentrations of H2O2 like those used in the transcript expression experiment. A significantly lower percentage in survival of MgPDI dsRNA-treated J2s was observed than of GFP dsRNA-treated J2s where both groups were exposed to H2O2 (Fig. 6b).

Effect of MgPDI expression on rice susceptibility to M. graminicola infection. (a) MgPDI expression in nematode after treatment with dsRNA. Asterisks indicate significant differences based on Tukey’s test (*P < 0.05; **P < 0.01). (b) Effect of MgPDI silencing on H2O2 stress tolerance. (c) Average numbers of knots and the MF values at 15 dpi. Vertical bars represent the means ± SD.
The infection studies showed that the treatment of J2 with MgPDI dsRNA significantly reduced the reproductive ability of nematodes (MF = 6.22) compared to that of the treatment of J2 with GFP dsRNA (MF = 11.46) (Fig. 6c).
MgPDI is localised in the plant apoplastic space
To investigate the subcellular localization of MgPDI in plant cells, the reporter protein GFP was fused to the C-terminus of the MgPDI protein. The GFP signal from the MgPDI:GFP fusion expressed in N. benthamiana was observed at the cell periphery. However, the GFP signal was observed in the apoplastic space when the plant cells were plasmolysed by treating with 1 M NaCl2 which indicated that the localization of MgPDI was in the apoplast (Fig. 7).

Subcellular localization of transiently expressed MgPDI:GFP fusions in N. benthamiana leaves. The first and third panel were MgPDI:GFP/GFP in normal cell, and the second and forth panel was MgPDI:GFP/GFP in plasmolysed cell.
Discussion
The present study identifies MgPDI encoding a typical PDI in the endoparasitic nematode M. graminicola, and MgPDI contains a 20-amino acid signal peptide at its N-terminal and two catalytic thioredoxin-like domains (active sites), each containing the canonical CGHC motif, and two non-catalytic domains (Fig. 1a,b). PDI is specifically responsible for folding proteins into the endoplasmic reticulum18. In addition, due to the active motifs in PpPDI1, its enzymatic function purportedly causes cell death in P. parasitica17. Our results also showed that the active catalytic motifs are highly conserved in the sequences under investigation (Fig. 1a). The described sequence was annotated as MgPDI for its arrangement of PDI classical domains.
In situ hybridization showed that MgPDI was expressed in oesophageal gland cells of M. graminicola pre-parasitic J2s, which is said to be nematode secretory effector proteins origin and presumed to be involved in the early parasitic stages of Mg. In addition, the expression analysis in different life stages of MgPDI found that transcription of MgPDI was up-regulated during early parasitic stages and peaked at 5 dpi, which is similar with the expression pattern of HsPdi in H. schachtii14.
To explore other functional characteristics of MgPDI, a large quantity of active recombinant MgPDI was obtained by induction at optimal conditions (20 °C overnight with 0.5 mM IPTG). Previous studies demonstrated that the enzymatic activity of PDI lies in its roles in oxidization, isomerization, and reduction9. In addition, the oxidase/isomerase activity of MgPDI utilizes the refolding of scrambled RNase19. A classical assay of insulin disulfide bridge-reduction in the presence of DTT was performed to analyze the enzymatic activity of PDI reduction, and the results indicated that MgPDI catalysed the reduction of insulin disulfide bonds, as seen in previous studies9. Protein disulphide isomerase is a member of the thioredoxin superfamily, which is composed of several redox proteins that play key roles in many essential antioxidant and redox-regulatory processes20,21. As for the enzymatic activity of antioxidants, first, our results indicated that MgPDI functioned in HEK293 cells to reduce oxidative stress by H2O2, and in particular, the recombinant protein exerted its protective influence in a dose-dependent manner (Fig. 4c). To further confirm the ability of MgPDI to function as an antioxidant, the plasmid DNA nicking assay was performed to demonstrate the ability of MgPDI to protect super-coiled DNA from nicking. Results indicated that MgPDI (at 0.1, 1, 10 and 100 μg/ml) effectively protects the super-coiled DNA from damage (Fig. 4d). DNA protection against oxidative damage by thioredoxin has also been demonstrated in Haemonchus contortus22. This is the first report that a functional recombinant PDI protein was obtained from the plant parasitic nematode M. graminicola and confirmed that MgPDI encodes a functional protein disulfide isomerase.
To determine the importance of MgPDI in the parasitism of M. graminicola, MgPDI expression was knocked down using RNAi, which has been effectively used to research both sedentary and migratory endoparasites, such as plant-parasitic nematodes and the function of their pathogenicity factors23. Knocking down the expression of MgPDI in preparastitic nematodes reduced the MF by more than 45% compared with the MF when in the non-specific presence of dsRNA (GFP). Our results are in accordance with Habash et al., who reported that RNAi silencing of the HsPDI gene of H. schachtii resulted in a significant reduction in the number of eggs produced14. These findings indicated that protein disulfide isomerase is essential in the reproduction of M. graminicola.
Studies have shown that ROS causes damage to cellular organelles and inhibits cell functions by oxidizing DNA, proteins, and lipids24. However, plant-parasitic nematodes can encode various antioxidant enzymes including superoxide dismutase (SOD), catalase, peroxiredoxins, glutathione peroxidases and protein disulfide isomerase to protect against ROS14,25,26,27,28. Results of the present study support that the primary role of MgPDI is to protect Mg from damage by ROS. Previous research reported that the production of ROS during the oxidative burst reflected the defense response in plants29. In order to test whether the reactive oxygen species of rice infected with Mg were affected. The H2O2 level was determined in rice roots, and we found that H2O2 levels in knots were lower at 7 dpi and 14 dpi compared with the root tips without Mg infection. What’s more, we observed a significant increase in MgPDI expression in response to the exogenous stress of H2O2. In addition, the tolerance of MgPDI-silenced nematodes to different concentrations of H2O2 decreased. This result is consistent with Habash et al., who reported that HsPDI plays a role in protecting nematodes against ROS14, and Dubreuil et al., who reported that silencing peroxiredoxin expression in M. incognita impaired the nematodes infectivity on tomato and their tolerance to exogenous H2O228.
Previous research of immunolocalization indicated that effector proteins were translocated to the plant apoplast30. The HaEXPB2 from Heterodera avenae containing the signal peptide could also target the protein to the apoplast when overexpressed in N. benthamiana31. Our studies of subcellular localization showed that MgPDI is localized in apoplast, but how it functioned in the apoplast need to be researched in the further studies.
In conclusion, the combined results from the gene discovery, expression, localization, silencing, and recombinant protein characterization suggest that MgPDI potentially plays an important role in combating the burst of ROS in host plants during the infection process. In brief, MgPDI is a putative virulence factor facilitating M. graminicola infestation of rice hosts.
Materials and Methods
M. graminicola culture and maintenance
The M. graminicola isolates ZJJH were maintained on rice (O. sativa cv. ‘Nipponbare’) grown in potting soil in a growth chamber under a 16 h/8 h light/dark photoperiod at 28 °C/26 °C32. Pre-parasitic J2 and parasitic stage nematodes were collected as described by Huang et al. and Haegeman et al.32,33.
MgPDI gene discovery and sequence analysis
Total RNA was extracted from about 10000 freshly hatched second-stage juveniles using the TRIzol reagent (Invitrogen, California, USA). Based on M. graminicola transcriptome data34, the full-length cDNA sequence of MgPDI was obtained by rapid amplification of cDNA ends using the SMART RACE cDNA Amplification kit (Clontech, USA) according to the manufacturer’s instructions. PCR-amplified DNA fragments were cloned into pGEM-T Easy Vector (Promega, USA) and then sequenced by Sangon Biotech (Shanghai, China). All primers used in this study were synthesized by Invitrogen Biotechnology Co. Ltd. and are listed in Table 1.
The deduced protein sequence, pI (isoelectric point) and MW (molecular weight) of MgPDI were predicted by DNAMAN V.6 (Lynnon Biosoft, Que-bec, Canada). An online protein structure homology-modeling server SWISS-MODEL was used to predict the protein tertiary structure. The signal peptide of MgPDI was determined using SignalP 4.1 Server at http://www.cbs.dtu.dk/services/SignalP/ 35. Homologous PDI protein sequences from various species were retrieved from the NCBI database and aligned using the program of DNAMAN 6.0.
In situ hybridization
The specific primers PDI-T7-P/PDI-AP and PDI-P/PDI-T7-AP (Table 1) were designed to synthesize DIG-labelled sense and antisense RNA probes (307 bp) using DIG RNA labelling mix (Roche, Basel, Switzerland). In situ hybridization was performed according to a modified method of De Boer et al. on about 8000 freshly hatched J2s36. After hybridization, the probe was detected with an anti-digoxin-FITC monoclonal antibody (diluted 1:1000), and nematodes were imaged using a Laser Scanning Confocal Microscope (Zeiss LSM800, Oberkochen, Germany).
Developmental expression analysis
Total RNA was extracted from M. graminicola nematodes at different life stages as described previously37 using the TRIzol method (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. The cDNA was synthesized using the ReverTra Ace qRT-PCR RT kit (Toyobo, Osaka, Japan). Quantitative RT-PCR was performed with the primer pair MgqRT-PCR-F/MgqRT-PCR-R (Table 1) and actin genes of M. graminicola were amplified as a reference with the primers Mg-ACT-Q-F/Mg-ACT-Q-R34. The qRT-PCR reactions were performed on a CFX Connect Real-Time System (BIO-RAD, USA) using THUNDERBIRD qRT-PCR Mix (Toyobo, Osaka, Japan). Three technical replicates for each reaction were performed and three independent experiments were performed under the following thermal cycler conditions: 95 °C for 60 sec, 40 cycles at 95 °C for 15 sec, and 60 °C for 30 sec. The relative changes in gene expression were determined using the 2−ΔΔCT method38.
Expression, purification, and validation of recombinant MgPDI
The cDNA fragments encoding MgPDI without signal peptide were amplified by PCR primers with restriction sites (Table 1). Products of the PCR assembly were ligated into the pET-32a vector (Novagen, USA) using T4 DNA ligase (Promega, USA). The resulting recombinant vector was validated by PCR product sequencing and was transformed into an Escherichia coli BL21 (DE3) competent cell (TaKaRa, China) for protein expression. Transformed E. coli was cultured at 37 °C and agitated at 180 rpm in lysogeny broth (LB) containing ampicillin (50 μg/ml) until optical density 600 (OD600) reached 0.5. Isopropyl-B-D-thiogalactoside (IPTG) was added to the final concentration of 0.5 mM and then incubated at 20 °C, while shaking at 180 rpm. After incubating for 1 day, the bacterial cells were precipitated by centrifugation and expression analyzed on 12% SDS-PAGE. Thrombin (Sigma, MO, USA) was used to cleave the tags from MgPDI in HisTALON™ Gravity Columns (Clontech, USA) to purify the recombinant proteins, and p-aminobenzamidine-agarose (Sigma, MO, USA) was applied to bind thrombin after cleavage. Lastly, the supernatant was collected and the concentration was measured by absorbance at 280 nm and stored at −80 °C39. The purified MgPDI protein band was cut out from the gel after SDS-PAGE and staining by coomassie blue. In-gel proteins were digested overnight in 12.5 ng/mL trypsin in 25 mmol/L NH4HCO3. The peptide mixtures were injected into the trap column, with a flow rate of 10 μl/min, of a Thermo Scientific Easy nanoLC 1000 (LTQ-Orbitrap Elite, Thermo Fisher Scientific, Waltham, MA, USA) for 2 min to analyze the purified proteins according to the method described by Xu et al.40.
PDI activity assays
Isomerization activity of PDI was determined by a standard assay using the modified method previously reported by Hong & Soong9. A preparation of 8 μM scrambled RNase A (Sigma) was incubated with 1.4 μM purified recombinant MgPDI protein in a buffer containing 4.5 mM cytidine 2′,3-cyclic monophosphate (cCMP) (Sigma), 1 mM reduced glutathione (Sigma), 0.2 mM oxidized glutathione (Sigma), 2 mM EDTA, and 100 mM Tris–Cl (pH 8.0). The reduction of cCMP by active RNase into CMP was monitored by the absorbance at 296 nm for 60 min in a Microplate Reader (Thermo, Varioskan Flash). For the PDI reductive assay, the MgPDI enzyme activity to reduce insulin was determined by a modified method41,42. Reaction mixtures (200 μl) included 100 mM Tris-Cl (pH 6.8), 2 mM ethylenediaminetetraacetic acid (EDTA), 0.13 mM insulin from bovine pancreas (Sigma), 0.33 mM dithiothreitol (DTT) and increasing concentrations (ranging from 2.5 to 10 μM) of purified MgPDI protein. The turbidity of the reaction mixture (Dithiothreitol-mediated insulin reduction by MgPDI) was monitored by measuring the increase of absorbance at 650 nm using a Microplate Reader (Thermo, Varioskan Flash). The reduction of insulin by DTT was recorded in a solution without MgPDI as a negative control.
Human cell viability assay
The method which assessed cell viability activity of MgPDI on Human Embryonic Kidney 293 (HEK293) cells was modified from the previously described study43. The HEK293 cells were adjusted to 3 × 104 cells/well and cultured overnight at 37 °C. Afterwards, the final concentration of MgPDI added to the medium was 0.1, 10 or 1000 ng/ml. After a further 6 h of incubation, H2O2 was added to a final concentration of 100 μM and cells were incubated for another 30 min in order to induce oxidative stress. Cell viability was measured using a CCK-8 Cell Counting Kit (Vazyme, China) at a wavelength of 450 nm. Cells incubated with 5 μg/ml bovine serum albumin (BSA) were used as a control. Each group was tested in triplicate.
Protective effect of MgPDI against oxidative damage by an MFO system
A mixed function oxidase (MFO) system was used to generate thiol radicals to damage the plasmid DNA44. The MFO system consisting of 1.65 mM DTT, 16.5 mM FeCl3 and different concentrations of MgPDI (from 0.1 to 100 μg/mL) were pre-incubated at 37 °C for 2.5 h. Subsequently, pGEM-T Easy plasmid DNA (500 ng, Promega) was added and incubated for 1 h at 37 °C. Nicking of DNA was evaluated by ethidium bromide staining after electrophoresis in 0.8% agarose gels45.
Hydrogen peroxide content determination
The H2O2 level was determined according to Ji et al.46. Each sample consisted of approximately 0.1 g fresh roots, collected from knots or tips. Root samples were collected at 3, 7 and 14 dpi, The experiment was performed twice, each time with three replicate samples.
Double stranded RNA (dsRNA) and infection assay
The forward and reverse primers contained T7 promoter sequences at their 5′ ends for in vitro RNA synthesis (Table 1). Double stranded RNA (dsRNA) was synthesized and purified using the T7 RiboMAXTM Express kit (Promega, USA) according to the manufacturer’s instructions. The GFP template was used for the synthesis of a dsRNA construct as a negative control.
RNAi soaking was performed using the modified method of Rosso et al. and Huang et al.47,48. Twenty-five thousand freshly hatched J2s of M. graminicola were soaked in the dsRNA solution (2 mg mL−1 dsRNA, 3 mM spermidine, 50 mM octopamine, and 0.05% gelatin, adjusted with 0.25 × M9 buffer) for 36 h at room temperature in the dark on a rotator. For each reaction, approximately 8000 J2s were used for the plant infection assay; approximately 3000 J2s were used to determine nematode survival after the H2O2 stress, Freshly hatched J2s of M. graminicola were soaked in MgPDI dsRNA or GFP dsRNA (control), dsRNA-treated nematodes were soaked in 20, 40 and 80 mM H2O2 or in sterile water (0 mM) and live nematodes were counted after 30 min; and all the remaining J2s were used for the qRT-PCR to evaluate the level of MgPDI silencing.
For the infection assay, Pluronic F-127 (PF-127) (Sigma-Aldrich) gel was used as previously reported49,50. Eighty second-stage juveniles were soaked in dsRNA targeted against MgPDI or GFP and then inoculated each rice seedling according to Tian51. Roots of rice plants were stained with acid fuchsin52 and the number of eggs was counted after dissecting the stained galls under a microscope at 15 dpi. To determine the reproductive potential of M. graminicola, a nematode multiplication factor [MF = (number of egg masses × number of eggs per egg mass) ÷ nematode inoculum level] was calculated.
Sensitivity of M. graminicola to hydrogen peroxide (H2O2)
Approximately 200 freshly hatched J2s were incubated in each treatment of 0, 10, 20, 40 or 80 mM of H2O2. Dead nematodes were counted after 30, 60, 90 and 120 min and the percentage of survival was calculated. Treatments were replicated three times and the experiment was repeated three times. To determine MgPDI gene expression in individuals under H2O2 stress, 5000 freshly hatched J2s were incubated in each treatment of 20, 40 or 80 mM H2O2 for 30 min, and washed in sterile tap water. Individuals of J2s incubated in sterile tap water were used as the control. The relative expression levels were quantified in H2O2-incubated nematodes relative to J2s soaked in sterile distilled water (0 mM H2O2) by qRT-PCR as described above.
Subcellular localization of MgPDI
The coding sequences of MgPDI was cloned into the pGD-eGFP vector, and the MgPDI:: pGD-eGFP construct was transformed into the Agrobacterium tumefaciens strain EHA 105. The method of Agrobacterium-mediated transient expression was carried out as previously described14,31. The A. tumefaciens suspensions were adjusted with the infiltration buffer to an OD600 of 1.0 and mixed with an RNA-silencing suppressor P19 at 1:1. After incubation for 2 h at RT, the A. tumefaciens strains carrying the constructs were injected in the leaves abaxial side of 6 week-old Nicotiana benthamiana plant using 1 mL hypodermic syringe without needle. To distinguish the cell wall and the plasma membrane, the leaf cells were plasmolysed by infiltrating 1 M NaCl2 solution for 10 min before observation. Infiltrated plants were incubated in the growth chamber (16 hrs light, 8 hrs dark and at 25 °C) for 5 days31. The primers sequences are given in Table 1.
Data Availability
All data generated or analysed during this study are included in this published article (and its Supplementary Information files).
References
Bridge, J. & Page, S. L. J. The rice root-knot nematode, Meloidogyne graminicola, on deep water rice (Oryza sativa subsp. indica). Revue de Nematol. 5, 225–232 (1982).
Pokharel, R. R. et al. Variability and the recognition of two races in Meloidogyne graminicola. Australas Plant Path. 39, 326–333 (2010).
Kyndt, T., Fernandez, D. & Gheysen, G. Plant-parasitic nematode infections in rice: molecular and cellular insights. Annu. Annu. Rev. Phytopathol. 52, 135–153 (2014).
Abad, P., Favery, B., Rosso, M. N. & Castagnone-Sereno, P. Root-knot nematode parasitism and host response: molecular basis of a sophisticated interaction. Mol Plant Pathol. 4, 217–224 (1982).
Dubreuil, G., Magliano, M., Deleury, E., Abad, P. & Rosso, M. N. Transcriptome analysis of root-knot nematode functions induced in the early stages of parasitism. New Phytol. 176, 426–436 (2007).
Shrestha, R., Uzzo, F., Wilson, M. J. & Price, A. H. Physiological and genetic mapping study of tolerance to root-knot nematode in rice. New Phytol. 176, 665–672 (2007).
Pandhair, V. & Sekhon, B. S. Reactive oxygen species and antioxidants in plants: An overview. J Plant Biochem Biot. 15, 71–78 (2006).
Yao, P. et al. Molecular cloning, expression and antioxidant characterisation of a typical thioredoxin gene (AccTrx2) in Apis cerana cerana. Gene. 527, 33–41 (2013).
Hong, B. X. & Soong, L. Identification and enzymatic activities of four protein disulfide isomerase (PDI) isoforms of Leishmania amazonensis. Parasitol Res. 102, 437–446 (2008).
Edman, J. C., Ellis, L., Blacher, R. W., Roth, R. A. & Rutter, W. J. Sequence of protein disulfide isomerase and implications of its relationship to thioredoxin. Nature. 317, 267–270 (1985).
Kemmink, J., Darby, N. J., Dijkstra, K., Nilges, M. & Creighton, T. E. The folding catalyst protein disulfide isomerase is constructed of active and inactive thioredoxin modules. Curr Biol. 7, 239–245 (1997).
Tsai, B., Rodighiero, C., Lencer, W. I. & Rapoport, T. A. Protein disulfide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell. 104, 937–948 (2001).
Eschenlauer, S. C. P. & Page, A. P. The Caenorhabditis elegans ERp60 homolog protein disulfide isomerase-3 has disulfide isomerase and transglutaminase-like cross-linking activity and is involved in the maintenance of body morphology. J. Biol Chem. 278, 4227–4237 (2003).
Habash, S. S. et al. Identification and characterization of a putative protein disulfide isomerase (HsPDI) as an alleged effector of Heterodera schachtii. Sci Rep-Uk. 7, 13536 (2017).
Stolf, B. S. et al. Protein Disulfide Isomerase and Host-Pathogen. Interaction. The scientific world journal. 11, 1749–1761 (2011).
Ben Achour, Y., Chenik, M., Louzir, H. & Dellagi, K. Identification of a disulfide isomerase protein of Leishmania major as a putative virulence factor. Infect Immun. 70, 3576–3585 (2002).
Meng, Y. et al. The protein disulfide isomerase 1 of Phytophthora parasitica (PpPDI1) is associated with the haustoria-like structures and contributes to plant infection. Front Plant Sci. 6, 632 (2015).
Soares Moretti, A. I. & Martins Laurindo, F. R. Protein disulfide isomerases: Redox connections in and out of the endoplasmic reticulum. Arch Biochem Biophys. 617, 106–119 (2017).
Lyles, M. M. & Gilbert, H. F. Catalysis of the oxidative folding of ribonuclease-a by protein disulfide isomerase - pre-steady-state kinetics and the utilization of the oxidizing equivalents of the isomerase. Biochemistry. 30, 619–625 (1991).
Ferrari, D. M. & Soling, H. D. The protein disulphide-isomerase family: unravelling a string of folds. Biochem J. 339, 1–10 (1999).
Nakamura, T. et al. Redox regulation of lung inflammation by thioredoxin. Antioxid Redox Sign. 7, 60–71 (2005).
Sotirchos, I. M., Hudson, A. L., Ellis, J. & Davey, M. W. A unique thioredoxin of the parasitic nematode Haemonchus contortus with glutaredoxin activity. Free Radical Bio Med. 46, 579–585 (2009).
Maule, A. G. et al. An eye on RNAi in nematode parasites. Trends Parasitol 27, 505–513 (2011).
Baker, C. J. & Orlandi, E. W. Active oxygen in plant pathogenesis. Annu Rev Phytopathol. 33, 299–321 (1995).
Molinari, S. & Miacola, C. Antioxidant enzymes in phytoparasitic nematodes. J Nematol. 29, 153–159 (1997).
Robertson, L. et al. Cloning, expression and functional characterisation of a peroxiredoxin from the potato cyst nematode Globedera rostochiensis. Mol Biochem Parasit. 111, 41–49 (2000).
Jones, J. T., Reavy, B., Smant, G. & Prior, A. E. Glutathione peroxidases of the potato cyst nematode Globodera rostochiensis. Gene. 324, 47–54 (2004).
Dubreuil, G. et al. Peroxiredoxins from the plant parasitic root-knot nematode, Meloidogyne incognita, are required for successful development within the host. Int J Parasitol. 41, 385–396 (2011).
Levine, A. et al. H2O2 from the oxidative burst orchestrates the plant hypersensitive disease resistance response. Cell. 79, 583–593 (1994).
Vieira, P. et al. The plant apoplasm is an important recipient compartment for nematode secreted proteins. J Exp Bot. 62, 1241–1253 (2011).
Liu, J. et al. Molecular Characterization of A Novel Effector Expansin-like Protein from Heterodera avenae that Induces Cell Death in Nicotiana benthamiana. Sci Rep. 6, 35677 (2016).
Huang, W. K., Ji, H. L., Gheysen, G. & Kyndt, T. Thiamine-induced priming against root-knot nematode infection in rice involves lignification and hydrogen peroxide generation. Mol Plant Pathol. 17, 614–624 (2016).
Haegeman, A., Bauters, L., Kyndt, T., Rahman, M. M. & Gheysen, G. Identification of candidate effector genes in the transcriptome of the rice root knot nematode Meloidogyne graminicola. Mol Plant Pathol. 14, 379–390 (2013).
Petitot, A. S. et al. Dual RNA-seq reveals Meloidogyne graminicola transcriptome and candidate effectors during the interaction with rice plants. Mol Plant Pathol. 17, 860–874 (2016).
Tamura, K., Dudley, J., Nei, M. & Kumar, S. MEGA4: Molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 24, 1596–1599 (2007).
Boer, D., Yan, Y., Smant, G., Davis, E. L. & Baum, T. J. In-situ hybridization to messenger RNA in Heterodera glycines. J Nematol. 30, 309–312 (1998).
Chen, J. et al. A novel Meloidogyne graminicola effector, MgGPP, is secreted into host cells and undergoes glycosylation in concert with proteolysis to suppress plant defenses and promote parasitism. Plos Pathog. 13, e1006301 (2017).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods. 25, 402–408 (2001).
Wang, Z. Z., Zhan, L. Q. & Chen, X. X. Two types of lysozymes from the whitefly Bemisia tabaci: Molecular characterization and functional diversification. Dev Comp Immunol. 81, 252–261 (2018).
Xu, W. et al. Melatonin enhances thermotolerance by promoting cellular protein protection in tomato plants. J Pineal Res. 61, 457–469 (2016).
Holmgren, A. Thioredoxin catalyzes the reduction of insulin disulfides by dithiothreitol and dihydrolipoamide. J Biol Chem. 254, 9627–9632 (1979).
Liu, Q. et al. Molecular characterization and immunolocalization of a protein disulfide isomerase from Angiostrongylus cantonensis. Parasitol Res. 110, 2501–2507 (2012).
Kim, D. H., Kim, J. W., Jeong, J. M., Park, H. J. & Park, C. I. Molecular cloning and expression analysis of a thioredoxin from rock bream, Oplegnathus fasciatus, and biological activity of the recombinant protein. Fish Shellfish Immun. 31, 22–28 (2011).
Wang, X. W., Liou, Y. C., Ho, B. & Ding, J. L. An evolutionarily conserved 16-kDa thioredoxin-related protein is an antioxidant which regulates the NF-kappa B signaling pathway. Free Radical Bio Med. 42, 247–259 (2007).
Umasuthan, N., Revathy, K. S., Lee, Y., Whang, I. & Lee, J. Mitochondrial thioredoxin-2 from Manila clam (Ruditapes philippinarum) is a potent antioxidant enzyme involved in antibacterial response. Fish Shellfish Immun. 32, 513–523 (2012).
Ji, H., Kyndt, T., He, W., Vanholme, B. & Gheysen, G. β-aminobutyric acid-induced resistance against root-knot nematodes in rice is based on increased basal defence. Mol Plant Microbe In. 28, 519 (2015).
Rosso, M. N., Dubrana, M. P., Cimbolini, N., Jaubert, S. & Abad, P. Application of RNA interference to root-knot nematode genes encoding esophageal gland proteins. Mol Plant Microbe In. 18, 615–620 (2005).
Huang, G., Allen, R., Davis, E. L., Baum, T. J. & Hussey, R. S. Engineering broad root-knot resistance in transgenic plants by RNAi silencing of a conserved and essential root-knot nematode parasitism gene. P Natl Acad Sci USA 103, 14302–14306 (2006).
Wang, C., Lower, S. & Williamson, V. M. Application of Pluronic gel to the study of root-knot nematode behaviour. Nematology. 11, 453–464 (2009).
Dutta, T. K., Powers, S. J., Kerry, B. R., Gaur, H. S. & Curtis, R. H. C. Comparison of host recognition, invasion, development and reproduction of Meloidogyne graminicola and M. incognita on rice and tomato. Nematology. 13, 509–520 (2011).
Tian, Z. L., Barsalote, E. M., Li, X. L., Cai, R. H. & Zheng, J. W. First Report of Root-knot Nematode, Meloidogyne graminicola, on Rice in Zhejiang, Eastern China. Plant Dis. 101, 2152–2153 (2017).
Bybd, D. W., Kirkpatrick, T. & Barker, K. R. An improved technique for clearing and staining plant-tissues for detection of nematodes. J Nematol. 15, 142–143 (1983).
Acknowledgements
This research was supported by the Special Fund for Agro-scientific Research in the Public Interest in China (No: 201503114).
Author information
Authors and Affiliations
Contributions
Conception and design of study: Z.L.T. and J.W.Z. Acquisition of data: Z.L.T. and Z.H.W. Analysis and/or interpretation of data: Z.L.T., Z.H.W., Q.N. and M.M. Drafting the manuscript: Z.L.T. and Z.H.W. Revising the manuscript critically for important intellectual content: M.M. and J.W.Z. Approval of the version of the manuscript to be published: Z.L.T., Z.H.W., M.M., Q.N. and J.W.Z.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tian, Zl., Wang, Zh., Maria, M. et al. Meloidogyne graminicola protein disulfide isomerase may be a nematode effector and is involved in protection against oxidative damage. Sci Rep 9, 11949 (2019). https://doi.org/10.1038/s41598-019-48474-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-48474-w
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
