
Abstract
Using all-atom explicit solvent replica exchange molecular dynamics simulations with solute tempering, we study the effect of methionine oxidation on Aβ10–40 peptide binding to the zwitterionic DMPC bilayer. By comparing oxidized and reduced peptides, we identified changes in the binding mechanism caused by this modification. First, Met35 oxidation unravels C-terminal helix in the bound peptides. Second, oxidation destabilizes intrapeptide interactions and expands bound peptides. We explain these outcomes by the loss of amphiphilic character of the C-terminal helix due to oxidation. Third, oxidation “polarizes” Aβ binding to the DMPC bilayer by strengthening the interactions of the C-terminus with lipids while largely releasing the rest of the peptide from bilayer. Fourth, in contrast to the wild-type peptide, oxidized Aβ induces significantly smaller bilayer thinning and drop in lipid density within the binding footprint. These observations are the consequence of mixing oxidized peptide amino acids with lipids promoted by enhanced Aβ conformational fluctuations. Fifth, methionine oxidation reduces the affinity of Aβ binding to the DMPC bilayer by disrupting favorable intrapeptide interactions upon binding, which offset the gains from better hydration. Reduced binding affinity of the oxidized Aβ may represent the molecular basis for its reduced cytotoxicity.
Similar content being viewed by others

Introduction
Progression of Alzheimer’s disease is related to the accumulation of cytotoxic Aβ peptides1, which are naturally cleaved from membrane-spanning β-amyloid precursor proteins by β and γ secretases2. This process results in the extracellular release of several Aβ alloforms, of which the 40-residue monomer Aβ1–40 is most prevalent3. Over time, Aβ peptides aggregate to form higher-ordered oligomers capable of inducing neuronal death2. The cytotoxicity of Aβ is purportedly related to its interactions with cellular membranes, which may increase calcium permeation eventually initiating neuron apoptosis4. Depending on the concentration, Aβ can reside on membrane surface in monomeric5 or oligomeric forms6.
The Aβ amino acid sequence contains a single methionine residue at position 35. Being sensitive to reactive oxygen species, methionine can readily oxidize into methionine sulfoxide (MetO) by incorporating an oxygen atom into its side chain7,8. Because Aβ is normally localized to brain tissues, which consume 20% of the body’s oxygen, it has a high propensity to become oxidized, most likely mediated by Cu2+ ions9. Consequently, it has been estimated that oxidized Aβ species represent from 10 to 50% of all Aβ peptides found in the brain10,11. Although oxidized methionine side chains may adopt two diastereomeric forms, R and S, with slightly different hydrogen bonding properties, there is no evidence for natural preference of either of the two7,12. Most experimental studies indicate that, compared to wild-type (WT) Aβ, methionine oxidation impairs formation of aggregated Aβ species13,14,15. This outcome is predominately associated with a decreased rate of assembly14,16 delaying cytotoxic effects17,18. However given enough time to aggregate, oxidized Aβ peptides have been reported to approximately match the cytotoxicity of WT Aβ9,19.
Although previous experimental and computational studies have mostly focused on WT Aβ20,21, few have also explored structural consequences of methionine oxidation for Aβ monomers in aqueous environment. Specifically, solution NMR spectroscopy has been used to investigate the effect of methionine oxidation on Aβ peptides in water22. In general, oxidation produced a more flexible peptide C-terminus by increasing its backbone mobility and preventing the G37–G38 turn. Qualitatively similar conclusions were drawn from replica exchange molecular dynamics simulations23, where WT peptides were found to be more structured than those oxidized. Constant temperature molecular dynamics simulations at physiological conditions have further suggested that oxidation increases the solvent accessible area of the C-terminus24. CD and NMR spectroscopy have been used to determine the structure of WT and methionine-oxidized Aβ in a membrane-mimicking water-SDS environment12,14. These studies have revealed a reduced helical propensity in oxidized Aβ1–40 compared to its WT counterpart. Collectively, most of previous investigations argue that the overall effect of methionine oxidation, regardless of local environment, is a disordering in the peptide C-terminus and increase in its structural fluctuations. It is worth noting that methionine oxidation also determines the polymorphic form of Aβ1–42 fibrils25,26,27.
To date, a detailed study elucidating the molecular mechanisms of binding of the methionine-oxidized Aβ peptide to cellular membranes has not been performed. Nonetheless, given the natural prevalence of oxidized Aβ species, the corresponding investigation would be highly relevant to Alzheimer’s molecular pathogenesis. To bridge this gap, we performed isobaric-isothermal all-atom replica exchange molecular dynamics simulations with solute tempering (REST)28 to probe the binding of oxidized Aβ peptides to the DMPC bilayer. As a control, we utilized our previous studies, which probed binding of WT Aβ peptides to the same bilayer29,30. Below we present our analysis, which delineates the MetO binding mechanism and distinguishes it from that governing WT binding. We have also determined the affinities of MetO and WT peptides binding to the DMPC bilayer and compared our simulation results against experimental findings.
Methods
All-atom model and simulation details
We have studied the binding of methionine oxidized (MetO) Aβ10–40 monomers to the DMPC bilayer (Fig. 1). To understand the impact of methionine oxidation on the binding mechanism, we compared our results against previous simulations of wild-type (WT) Aβ10–40 monomers binding to the same bilayer29. The amino-truncated Aβ10–40 monomer was chosen as a proxy to full-length Aβ1–40. Our previous simulations29,30 have indicated that this peptide reproduces several Aβ1–40 experimental observations, including the distribution of helical structure along the sequence and partial insertion of Aβ in the bilayer12,31,32. Additionally, Aβ10–40 peptides are naturally occurring cytotoxic and amyloidogenic species33,34,35,36. The DMPC bilayer was chosen because it is well characterized experimentally affording direct comparisons with simulation data30 and the binding of WT Aβ10–40 to DMPC bilayer approximately reproduces experimental observations for SDS micelles12,31.

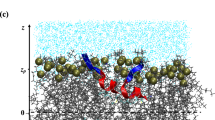
(a) Sequence of Aβ10–40 peptide with highlighted regions R1–R4. (b) Oxidation of hydrophobic methionine side chain results in polar methionine sulfoxide. (c) A DMPC lipid is divided into five structural groups: choline (G1), phosphate (G2), glycerol (G3), and two fatty acid tails (G4 and G5). The lipid headgroup is composed of three polar groups G1–G3. The bilayer hydrophobic core is formed by the tails G4 and G5. Phosphorus, oxygen, and nitrogen atoms are in purple, red, and blue, respectively. (d) A simulation snapshot illustrating binding of oxidized MetO Aβ peptides to the opposite leaflets of DMPC bilayer. The centers of mass of lipid phosphorus P atoms in each leaflet are approximately restrained at ±zP. Water is shown in blue. For clarity, we omitted ions.
Our simulation system consisted of 98 DMPC lipids forming a bilayer solvated by 4356 water molecules. Two sodium ions were introduced to compensate Aβ charge. Aβ N- and C-termini were acetylated and amidated, respectively. Aβ peptides were placed on either side of the lipid bilayer to take advantage of its symmetry and ensure that both leaflets approximately featured the same mass and pressure profiles37 (Fig. 1d). The initial size of the simulation box was 65 Å × 65 Å × 77 Å. Peptides were modeled using the CHARMM22 force field with CMAP corrections38, whereas lipids were modeled with the CHARMM36 force field. Water was represented with the modified TIP3P model39,40. The force field parameters for oxidized methionine were derived from DMSO parameters41 following previously published reports42. The force field topology and diastereomeric form were adopted from the study of Jas and Kuczera43. The resulting methionine sulfoxide was the R diastereomer, which apparently exhibits higher propensity to engage in hydrogen bonding12.
Molecular dynamics simulations were performed using NAMD44, which employed a 1 fs integration time step and periodic boundary conditions. Electrostatic interactions were computed using particle-mesh Ewald summation, and van der Waals interactions were smoothly switched off in the interval from 8 to 12 Å using the force switch option. Temperature was held constant using Langevin dynamics with a damping coefficient of 5 ps−1. Pressure was controlled via a Langevin piston method applying a semi-isotropic scheme, which results in independent fluctuations in the bilayer plane and normal. Because our simulation system included the DMPC bilayer, which could potentially disintegrate at high REST temperatures (see below), we implemented two sets of restraints to ensure bilayer integrity. The first harmonically restrained the center of mass of lipid phosphorus (P) atoms in a leaflet to the position zP = 17.35 Å with a force constant k = 6.5 kcal/mol/Å2. This force constant value was derived from restraint-free control simulations of the DMPC bilayer without Aβ peptides requiring that the restraints do not distort fluctuations of P atoms29. The second set of restraints affects Aβ or ion atoms when they come within 4 Å of the z periodic boundary (the axis normal to the bilayer plane). These were implemented as harmonic restraints with force constant k = 10 kcal/mol/Å2 and sought to inhibit interpeptide interactions. The contribution of all restraints to the potential energy at 330 K is minor (≈0.6 kcal/mol \(\lesssim \) RT). Moreover, our previous studies have shown that the effect of restraints on lipid structural properties is generally within the sampling error30,45. Because we detected no instances of interpeptide interactions via amino acid side chain contacts or cross-bridges by a third molecule, we assume that the peptides bind to the bilayer independently as monomers.
Conformational sampling was performed using isobaric-isothermal replica exchange molecular dynamics simulations with solute tempering (REST)28. Because REST formalism is described in detail elsewhere28,46, here we present its brief overview. We considered \(R=8\) replicas distributed geometrically from temperature \({T}_{0}=330\,{\rm{K}}\) to \({T}_{R-1}=430\,{\rm{K}}\). An exchange between replicas r and \(r+1\) occurs with the probability \(\omega =\,{\min }\,[1,{e}^{-{\rm{\Delta }}}]\), where \({\rm{\Delta }}={\beta }_{r}({H}_{r}({X}_{r+1})-{H}_{r}({X}_{r}))\) + \({\beta }_{r+1}({H}_{r+1}({X}_{r})-{H}_{r+1}({X}_{r+1}))\), \(\beta ={(\bar{R}T)}^{-1/2}\), H is the enthalpy, and X defines a system configuration. The solvent-solvent and solute-solvent interactions in replica r with the temperature Tr were scaled by the factors \({T}_{r}\)/\({T}_{0}\) and \({({T}_{r}/{T}_{0})}^{1/2}\), respectively. Because these scaling factors eliminate from \(\omega \) the solvent-solvent energy contribution, the number of replicas can be reduced without affecting the temperature range or exchange rates. Following our previous studies46,47 Aβ peptides and two ions were treated as solute, whereas lipids and water were considered as solvent. In our simulations, exchanges were attempted every 2 ps with the probability of success of 0.21. In total, we generated five REST trajectories collecting in all 0.8 μs of sampling (100 ns per replica). Initial 3.8 ns of sampling per replica in each trajectory were discarded as unequilibrated reducing the total equilibrated sampling to 648 ns. However, due to independent binding of two Aβ peptides to the DMPC bilayer, the effective sampling per peptide is doubled to almost 1.3 μs. Importantly, our previous work has shown that REST reproduces well the conformational sampling of Aβ10–40 binding to the bilayer collected via full-scale replica exchange molecular dynamics46.
In addition, we performed separate REST simulations probing the conformational ensemble of the MetO Aβ monomer in lipid-free water. In short, this system contained 4959 water molecules and a single sodium ion. The REST simulations utilized \(R=8\) replicas distributed geometrically within the temperature interval from \({T}_{0}=330\,{\rm{K}}\) to \({T}_{R-1}=440\,{\rm{K}}\). In all, five REST trajectories were produced collecting 0.8 μs of sampling (100 ns per replica). Initial 2 ns of REST sampling per replica were excluded as unequilibrated reducing the total equilibrated sampling to 720 ns.
Computation of structural probes
Molecular interactions were quantified by the contacts occurring between Aβ amino acid side chains and lipid structural groups G1–G5 (Fig. 1c). To detect contacts, the positions of side chains or groups were represented by the respective centers of mass. Two amino acids or an amino acid and a lipid group were in contact if the distance between them was less than 6.5 Å. To facilitate analysis, the Aβ10–40 sequence was broken up into the N-terminal region R1 (residues 10–16), central hydrophobic cluster R2 (residues 17–21), turn region R3 (residues 22–28), and hydrophobic C-terminal R4 (residues 29–40) (Fig. 1a). Aβ secondary structure was computed using STRIDE48. An amino acid was considered in a helical state if it sampled α-, 310-, or π-helix. A hydrogen bond (HB) between donor (D) and acceptor (A) was formed if the distance \({r}_{DA}\le 3.5\,\AA \) and the angle ∠\(DHA\ge {120}^{\circ }\).
To probe the localization of Aβ peptide and lipid structure, we defined several functions. The first is the probability P(z; i) for an amino acid i to be found at a distance z from the bilayer midplane. Using P(z; i) we computed the equilibrium positions of amino acids i along the z axis, \(\langle z(i)\rangle \). The insertion probability Pi(i) was derived from P(z; i) as the probability for amino acid i to reside below the average position of the center of mass of phosphorus atoms zP. The surface binding probability Ps(i) is defined as the probability for i to be localized in the lipid headgroup region (\({z}_{p} < z < {z}_{P}+6.5\,\AA \)). The unbinding probability Pu(i) represents unbound states with \(z > {z}_{P}+6.5\,\AA \). The second function is the two-dimensional number density of the bilayer lipid heavy atoms nl(r, z) computed as a function of the distance r to the Aβ center of mass and the distance z to the bilayer midplane. In these calculations, r and z were binned with the interval \({\rm{\Delta }}r={\rm{\Delta }}z=1\,\AA \). One-dimensional number densities for lipid, peptide, and water atoms were obtained in a similar way. Ordering in lipid fatty acid tails was quantified by the lipid carbon-deuterium order parameter SCD and tail tilts γ computed for sn − 2 chains30.
Thermodynamic quantities are denoted by brackets 〈…〉. Unless otherwise noted, all quantities were computed at 330 K using the weighted histogram analysis method49 derived for REST simulations46. The entropy S of a solute (DMPC bilayer with bound Aβ peptides or Aβ peptide in water) was estimated using Gibbs formula \(S=-\bar{R}\,{\sum }_{E}\,P(E){\rm{l}}{\rm{n}}\,P(E)\), where P(E) is the probability of observing the solute energy E computed from weighted histogram analysis. The bin size for E was set to 1 kcal/mol. As expected the difference in S between MetO and WT systems is approximately independent of bin size. Simulation convergence is analyzed in the Supplementary Information. Reported uncertainties are standard errors about the mean calculated from n = 5 trajectories.
Results
Effect of oxidation on Aβ structure
Our REST simulations explored the equilibrium binding of methionine oxidized (MetO) Aβ10–40 peptides to the DMPC bilayer. To gain insight into the changes in binding mechanism attributed to oxidation, we compare binding of MetO Aβ10–40 to the binding of wild-type (WT) Aβ10–40 to the same bilayer studied by us previously29. We first consider the effect of oxidation on Aβ secondary structure. The probabilities for MetO Aβ to adopt helix, turn, and random coil structure are \(\langle H\rangle =0.22\pm 0.02\), \(\langle T\rangle =0.53\pm 0.02\), and \(\langle RC\rangle =0.25\pm 0.02\), respectively. For comparison, in WT Aβ, the corresponding probabilities were 0.39 ± 0.03, 0.30 ± 0.03, and 0.31 ± 0.01. Therefore, methionine oxidation reduces helix content approximately two-fold, almost doubles turn fraction, and moderately reduces random coil structure (a 1.2-fold difference). These structural changes are further detailed in Fig. 2, which presents the distributions of secondary structure along the sequence. To aid the analysis, a secondary structure is assumed formed at amino acid i if its probability of occurrence exceeds 0.5. In MetO Aβ, there are no helical amino acids, whereas 18 residues (19–25, 27–33, 35–38) feature turn. For comparison, the WT peptide contains 11 helical residues (23–26 and 31–37), but only 5 turn residues (12–14, 21, and 22). Additionally, two residues (34, 40) in MetO Aβ adopt random coil conformations in contrast to 11 in WT Aβ (10, 16–20, 27–29, 39, and 40). These observations are supported by Table S1, which displays the fractions of helix, turn, and random coil within Aβ regions R1–R4. Indeed, in the WT a stable (namely, helical) structure is observed only in the R4 C-terminus, whereas three regions R2–R4 of MetO Aβ are dominated by turn conformations. Thus, we observe a striking differences in the WT and MetO secondary structure propensities. Collectively, our results imply that Met35 oxidation disrupts helical structure, particularly in the C-terminus, replacing it with turn conformations.

Secondary structure distribution in Aβ peptides. Helical \(\langle H(i)\rangle \) and turn \(\langle T(i)\rangle \) propensities are shown for amino acids i in panels (a and b). The data in black and red correspond to oxidized and reduced Aβ peptides bound to the DMPC bilayer. Vertical bars represent the standard error about the mean calculated from n = 5 trajectories. Regions R1–R4 are colored according to Fig. 1a. Methionine oxidation disrupts helical structure but elevates turn propensity.
We next analyze Aβ tertiary fold. The average number of amino acid contacts \(\langle C\rangle \) is 22.7 ± 0.6 in MetO Aβ and 28.4 ± 0.7 in the WT, indicating that, on average, oxidation disrupts almost 6 (20%) intrapeptide contacts. When this calculation is restricted to long-range contacts occurring between amino acids separated by at least 5 residues, the corresponding values are \(\langle {C}_{LR}\rangle =7.8\pm 0.6\) and 11.1 ± 0.7. This result suggests that 30% of long-range contacts are also destabilized by methionine oxidation, including the interactions between hydrophobic R2 and R4 regions (the number of R2–R4 contacts is reduced from 2.9 ± 0.3 to 0.6 ± 0.1). The reduction in Aβ intrapeptide interactions is visible in Fig. 3, which displays the difference contact map \(\langle {\rm{\Delta }}C(i,j)\rangle \) for WT and MetO Aβ. The list of most affected contacts is presented in Table S2. Notably, 10 out of 15 such contacts are destabilized in MetO Aβ, supporting the analysis above. All five stabilized contacts occur between proximal residues (\(|i-j|=2\) or 3) reflecting the formation of turns in MetO Aβ. The three long-range contacts in the table, all of which are destabilized, are Phe19–Ile31, the salt-bridge Asp23–Lys28, and Val24–Ile31. Their disruption implicates expansion of the Aβ structure as well as the loss of interactions between the two hydrophobic regions R2 and R4. Consistent with these observations, the radius of gyration of MetO Aβ \(\langle {R}_{g}\rangle \) is 16.8 ± 0.2 Å, but is reduced to 14.8 ± 0.7 Å for the WT. Therefore, oxidation of Met35 causes the Aβ peptide to adopt more open, expanded conformations.

The difference contact map \(\langle {\rm{\Delta }}C(i,j)\rangle \) visualizes changes in intrapeptide interactions induced by methionine oxidation. We define \(\langle {\rm{\Delta }}C(i,j)\rangle =\langle C(i,j)\rangle -{\langle C(i,j)\rangle }_{WT}\), where \(\langle C(i,j)\rangle \) and \({\langle C(i,j)\rangle }_{WT}\) are the contact maps obtained for MetO and WT29 Aβ, respectively. The values of \(\langle {\rm{\Delta }}C(i,j)\rangle \) are color-coded in the inset scale. Short-range (\(|i-j| < 5\)) and long-range (\(|i-j|\ge 5\)) contacts are placed above and below the diagonal. Regions R1–R4 are colored following Fig. 1a. Methionine oxidation generally weakens intrapeptide interactions, while strengthening some local contacts.
Aβ interactions with DMPC bilayer
To explore MetO Aβ binding and insertion into the DMPC bilayer, we computed the probability P(z; i) for amino acid i to be at the distance z from the bilayer midplane (Fig. 4). Using P(z; i) it is straightforward to obtain the average position of amino acids along the z-axis, \(\langle z(i)\rangle \). The figure shows that MetO Aβ peptide is divided into two almost equal regions - unbound N-terminus composed of amino acids 10 to 25 and bound C-terminus (26–40) (see Methods for definitions). In contrast, only a few N-terminal amino acids (10–13) in WT Aβ are classified as unbound with all others being bound to the DMPC bilayer. To supplement this conclusion, Table S3 presents the probabilities for Aβ sequence regions k = R1–R4 to be inserted Pi(k), surface-bound Ps(k), or unbound Pu(k). We found that only one MetO Aβ region, the C-terminal R4 representing 39% of the sequence, is inserted into the bilayer (\({P}_{i}(R4) > 0.5\)) compared to two in WT Aβ, R2 and R4. More dramatic differences emerge if we compare unbound regions. Indeed, three MetO Aβ regions, R1–R3 representing 61% of the Aβ sequence, are classified as unbound from the bilayer, whereas there are no unbound regions for WT Aβ.

Color coded probabilities P(z; i) for MetO Aβ amino acids i to be at the distance z from the bilayer midplane. The average positions of MetO and WT Aβ amino acids, \(\langle z(i)\rangle \), are represented by black and red lines. Vertical bars represent the standard error about the mean calculated from n = 5 trajectories. Two dashed lines separate the regions in which amino acids are considered inserted into the bilayer hydrophobic core, bound to the bilayer surface, or unbound (see Methods). An amino acid is bound to the bilayer if it is bound to its surface or inserted. Regions R1–R4 are colored according to Fig. 1a.
The analysis above relies on the location of Aβ amino acids along the bilayer normal. To directly probe Aβ binding to the DMPC bilayer, we have examined the interactions between peptides and lipids. Although half of MetO Aβ amino acids are classified above as unbound, the peptide maintains its binding to the DMPC bilayer with the probability exceeding 0.99, i.e., on average, it keeps at least one contact with lipids. In all, upon binding, MetO Aβ forms \(\langle {C}_{l}\rangle =32.3\pm 1.7\) contacts with lipid groups. Figure 5a shows that their distribution along the peptide sequence is remarkably skewed toward the C-terminus. Indeed, it follows from Fig. 5a that MetO Aβ C-terminal R4 forms 22.2 ± 3.0 contacts with lipids, whereas all other regions combined (R1–R3) establish only 10.1 ± 1.5 contacts. This result implies that 69% of MetO Aβ contacts with lipids are due to R4, which contains only 39% of all amino acids, whereas 31% of contacts are formed by R1–R3 regions representing 61% of the Aβ sequence. Although the total number of contacts between WT Aβ and the bilayer is approximately the same as for MetO Aβ (\(\langle {C}_{l}\rangle =32.9\pm 6.9\)), their distribution is notably different. In particular, the C-terminal R4 region of WT Aβ establishes only 14.6 ± 3.1 contacts with the bilayer (44% of all and 1.5-fold smaller than for MetO R4), whereas the rest of the peptide forms 18.4 ± 3.8 contacts (56%). Therefore, binding of the WT peptide implicates a more even distribution of binding interactions than in the oxidized peptide.

(a) The number of contacts \(\langle {C}_{l}(i)\rangle \) formed between Aβ amino acids i and lipid groups. The data in black and red correspond to MetO and WT Aβ. (b) Black bars show residue specific changes in the number of binding contacts \(\langle {\rm{\Delta }}{C}_{l}(i)\rangle =\langle {C}_{l}(i)\rangle -{\langle {C}_{l}(i)\rangle }_{WT}\), where \(\langle {C}_{l}(i)\rangle \) and \({\langle {C}_{l}(i)\rangle }_{WT}\) are the numbers of contacts formed by MetO or WT amino acids, respectively. Residue specific changes in the number of hydrogen bonds \(\langle {\rm{\Delta }}{N}_{HB}(i)\rangle \) (shaded bars) are defined similarly to \(\langle {\rm{\Delta }}{C}_{l}(i)\rangle \). Vertical bars represent the standard error about the mean calculated from n = 5 trajectories. Regions R1–R4 are colored according to Fig. 1a. Both panels indicate that methionine oxidation strengthens the interactions between the Aβ C-terminus and the DMPC bilayer while weakening binding of other peptide regions.
The residue specific differences in the number of binding contacts \(\langle {\rm{\Delta }}{C}_{l}(i)\rangle \) formed by MetO and WT peptides are shown in Fig. 5b. This plot reveals that the C-terminal R4 is the only region that gains binding contacts, whereas all (with very few exceptions) amino acids from other three regions R1–R3 lose interactions with lipids. To illustrate this conclusion, we determined that the average value of \(\langle {\rm{\Delta }}{C}_{l}(i)\rangle \) within R4 is 0.6, but it becomes negative (−0.4) within R1–R3. Similar implications follow from the analysis of hydrogen bonds between Aβ and the bilayer and the difference contact map \(\langle {\rm{\Delta }}{C}_{l}(i,k)\rangle \), which distinguishes interactions with lipid groups k (see Supplementary Information). Indeed, Fig. 5b implicates remarkably non-uniform changes in hydrogen bonding along the Aβ sequence with the C-terminal R4 gaining hydrogen bonds and the other regions losing them. Taken together, our data suggest that methionine oxidation “polarizes” the Aβ-bilayer binding interface by strengthening binding of the Aβ C-terminus and weakening binding elsewhere. As a result, the C-terminus of MetO Aβ is embedded into the bilayer hydrophobic core with the rest of the peptide weakly coupled with the DMPC bilayer.
Impact of Aβ on the bilayer structure
Finally, we assess the structural perturbations caused within the bilayer by Aβ binding. In our previous work30, we concluded that WT Aβ indents the bilayer, creates a lipid density void beneath its binding footprint, and disorders proximal fatty acid tails. With MetO Aβ data available, we can determine if methionine oxidation augments these results. To begin, Fig. 6 presents the number density \({n}_{l}(r,z)\) of lipid heavy atoms as a function of the distance r to the Aβ center of mass and distance z to the bilayer midplane. The lipid density depression in the bilayer, which occurs upon WT Aβ binding, is largely subdued during MetO Aβ binding. We quantified this change using the following approach45. A bilayer is divided into proximal and distant regions depending on the distance to Aβ. The boundary between the two occurs at the distance Rc = 14 Å from the Aβ center of mass, where the number of Aβ-lipid contacts reaches maximum. In the distant region (\(r > {R}_{c}\)), we define the lipid-water boundary zb from the condition \({n}_{l}(r > {R}_{c},{z}_{b})={n}_{w}({z}_{b})\equiv {n}_{b}\), where nw(z) is the water number density as a function of z. For both systems, zb ≈ 20 Å. In the proximal region (\(r < {R}_{c}\)), we determine the boundary zb(r) setting \(n(r,{z}_{b}(r))={n}_{b}\). The difference in zb(r) between the upper and lower leaflets produces the bilayer thickness D(r). The impact of Aβ can be assessed by computing the change in bilayer thickness \({\rm{\Delta }}D=D(r > 20\,\AA )-D(r < 6\,\AA )\). Then, the bilayer thinning for MetO and WT Aβ is \({\rm{\Delta }}D=4.5\pm 1.4\) and 12.7 ± 2.7 Å, respectively, indicating that the methionine-oxidized peptide produces a three-fold weaker impact on the bilayer thickness.

The number density nl(r, z) of lipid heavy atoms as a function of the distance r to the Aβ center of mass and distance z to the bilayer midplane. The left and right halves of the plot represent the indentation of the DMPC bilayer caused by WT and MetO Aβ peptides. Continuous black lines mark the bilayer boundaries. Vertical dashed lines separate proximal and distant regions. The panel demonstrates that binding of the MetO Aβ peptide causes considerably smaller thinning of the DMPC bilayer than of the WT.
To evaluate the changes in bilayer density induced by Aβ binding we used two approaches. First, we determined variations in the lipid surface number density ns. In the distant region \({n}_{s}=0.016\pm 0.000\,{\AA }^{-2}\), which is reduced to \(0.012\pm 0.000\,{\AA }^{-2}\) in the proximal region around bound MetO Aβ. Lipid density is further decreased to \(0.010\pm 0.001\,{\AA }^{-2}\) in the center of the proximal region (\(r < 6\,\AA \)), thus representing a modest 38% density drop with respect to the distant value. For comparison, WT Aβ induces larger decrease in lipid density within the proximal region to \(0.011\pm 0.002\,{\AA }^{-2}\), which becomes even more pronounced in its center, where \({n}_{s}=0.006\pm 0.003\,{\AA }^{-2}\) (a > 2.5-fold drop). Second, we considered variations in the volume number density of bilayer heavy atoms nl. In the distant bilayer region, nl averaged over a leaflet (\(0 < z < {z}_{b}\)) is \(0.035\pm 0.000\,{\AA }^{-3}\) and is reduced merely 20% to \(0.028\pm 0.001\,{\AA }^{-3}\) in the proximal region around the bound MetO Aβ peptide. In contrast, WT Aβ causes a larger (31%) corresponding decrease in nl to \(0.024\pm 0.003\,{\AA }^{-3}\). Thus, both surface lipid and volume bilayer densities point to the same conclusion that oxidized Aβ peptide creates a smaller lipid density void within its binding footprint compared to the WT peptide. This outcome is consistent with a notably smaller thinning of the DMPC bilayer induced by MetO Aβ binding. The analysis of structural disordering in fatty acid tails due to Aβ binding is presented in the Supplementary Information.
Discussion
Our isobaric-isothermal REST simulations demonstrate that adding a single oxygen atom to the Aβ monomer results in a dramatic structural reorganization in the bound peptide and DMPC bilayer. In particular, we observed almost complete disappearance of helical structure in the C-terminus of MetO Aβ. Indeed, according to Fig. 2a and Table S1, the C-terminal R4 helix fraction is reduced more than 5-fold. What is the mechanism responsible for destabilization of helical structure? In our previous study, we noted a large hydrophobic moment of the Aβ C-terminus, which matches those of other helix-forming peptides binding to cellular membranes29. Therefore, it is reasonable to suggest that, since oxidation changes the distribution of hydrophobicity along the C-terminus, its hydrophobic moment is affected. To check this conjecture, we constructed an idealized α-helix composed of all 12 C-terminal residues (Fig. S9) and computed its hydrophobic moment \(\overrightarrow{\mu }\) as described in the Supplementary Information. The absolute value of \(\overrightarrow{\mu }\) reflects the extent of partitioning of polar and apolar amino acids, whereas its direction points to the concentration of hydrophobic amino acids. Using the octanol-water hydrophobic scale from Wimley et al.50, we determined that μ for the WT Aβ C-terminus is 4.35 Å kcal/mol. Methionine oxidation adds a single polar atom (oxygen) to a hydrophobic C-terminus. Although, to our knowledge, the hydrophobicity for oxidized methionine has not been computed in the manner of the Wimley et al. scale50, the study of Black and Mould51 suggests that oxidized methionine is similar in its hydrophobicity to asparagine and glutamine. If these amino acids are substituted for Met35, the R4 hydrophobic moment decreases by 35 and 33% to 2.84 and 2.92 Å kcal/mol, respectively. A qualitatively similar observation follows if the Kyte-Doolite hydrophobicity scale52 is used. Therefore, addition of polar oxygen to Met35 decreases the C-terminus hydrophobic moment and disrupts its amphiphilic character. Figure 4 indicates that reduced Met35 in a helical state is inserted into the bilayer (\(\langle z(i)\rangle < {z}_{p}\)). Accordingly, oxidation should destabilize the WT C-terminal helix at the bilayer hydrophobic/hydrophilic interface.
The aforementioned theoretical argument rationalizing helix disruption implies that orientations of amino acids in the MetO C-terminus, particularly of Met35, with respect to the bilayer normal must differ from those in the WT. To check this expectation, we computed in Fig. S10 the angles \(\langle \theta (i)\rangle \) between the side chain vectors \({\overrightarrow{v}}_{i}\) and the reversed bilayer normal. (A vector \({\overrightarrow{v}}_{i}\) connects the Cα atom and side chain center of mass of amino acid i). The figure reveals that \(\langle \theta (i)\rangle \) for i = Met35 in the bound WT Aβ peptide is 32.6 ± 4.3°, whereas for MetO Aβ \(\langle \theta (i)\rangle =114.4\pm 9.1^\circ \). Thus, oxidation of Met35 reorients its side chain from being directed toward the hydrophobic core of the bilayer to that pointing toward solvent. Indeed, according to Fig. 4 oxidized Met35 is located closer to the bilayer polar headgroup region than its reduced form. Met35 solvation can be directly measured if we compute the average number of water molecules within 6.5 Å of the Met35 side chain center of mass. For WT and MetO peptides, there are 3.4 ± 1.9 and 14.5 ± 0.4 water molecules hydrating Met35, respectively. Collectively, these results demonstrate that adding oxygen to Met35 significantly decreases the Aβ C-terminal hydrophobic moment, reorients and displaces Met35 toward solvent, and increases its solvation by more than four-fold.
Because methionine oxidation unravels the entire C-terminal helix, its effect extends over the C-terminus. Being constrained by the helical structure, two hydrophobic residues in the WT, i = Ala30 and Leu34, are directed toward solvent (\(\langle \theta (i)\rangle > 90^\circ \)) and occur within the bilayer polar headgroup region (Fig. 4). In contrast, in MetO Aβ both amino acids are pointed toward the bilayer midplane (Fig. S10) and Leu34 is inserted into the hydrophobic core. As a result, in the MetO peptide, which is no longer constrained by the C-terminal helix, the sole R4 amino acid oriented toward solvent is Met35. Furthermore, oxidation of Met35 increases total Aβ hydration. Specifically, 228 ± 27 water molecules are bound, on average, to the WT peptide, whereas for MetO Aβ their number increases to 276 ± 8. Unraveling of C-terminal helix also disrupts intrapeptide contacts, as MetO Aβ sheds 2.3 short-range and 3.4 long-range contacts compared to WT Aβ. Loss of intrapeptide contacts further destabilizes helical structure. Taken together, these findings demonstrate that the mechanism of C-terminal helix unraveling can be ultimately traced to considerable decrease in the C-terminus hydrophobic moment in the oxidized peptide.
Based on the analysis of bilayer density, thinning, and distribution of MetO Aβ amino acids along the bilayer normal, we concluded that MetO Aβ amino acids tend to mix with lipids rather than form a peptide-only phase within the bilayer. What is then the reason for better “mixing” propensity of the oxidized peptide? To answer this, we probed the fluctuations in Aβ structure by computing the standard deviations in the peptide \(\varphi \) and \(\psi \) dihedral angles53. Figure S11 presents the difference in these dihedral angle fluctuations, \({\rm{\Delta }}\delta \varphi (i)\) and \({\rm{\Delta }}\delta \psi (i)\), for all amino acids i in MetO and WT Aβ (see Fig. S11 caption for definitions). The figure shows that 23 out of 29 amino acids (excluding the termini) have positive \({\rm{\Delta }}\delta \varphi (i)\) and/or \({\rm{\Delta }}\delta \psi (i)\) implicating enhanced flexibility of the oxidized peptide. To evaluate the bias in the distributions of \({\rm{\Delta }}\delta \varphi (i)\) and \({\rm{\Delta }}\delta \psi (i)\), we computed the ratios \({f}_{\varphi }={\sum }_{+}\,{\rm{\Delta }}\delta \varphi (i)/{\sum }_{-}\,-{\rm{\Delta }}\delta \varphi (i)\) and \({f}_{\psi }={\sum }_{+}\,{\rm{\Delta }}\delta \psi (i)/\)\({\sum }_{-}\,-{\rm{\Delta }}\delta \psi (i)\), where the subscripts + and − refer to summation over positive or negative \({\rm{\Delta }}\delta \varphi (i)\) and \({\rm{\Delta }}\delta \psi (i)\). We found that \({f}_{\varphi }=2.8 > 1\) and \({f}_{\psi }=5.2 > 1\) indicating that oxidation causes a net increase in the fluctuations in Aβ structure. Calculating \({f}_{\varphi }\) and \({f}_{\psi }\) for the C-terminus only, we find 117.4 and 4.8 suggesting that this Aβ region experiences particularly enhanced fluctuations.
In summary, these results argue that, consistent with the net loss of intrapeptide contacts, unraveling of C-terminal helix, and overall peptide expansion, MetO Aβ is more flexible than the WT. More open MetO Aβ conformations subject to large fluctuations afford better mixing with lipid atoms manifested in almost two-fold higher surface number density of lipids within the proximal region center and 1.5-fold higher number of Aβ-lipid contacts in the C-terminus compared to the WT peptide. Furthermore, though weak bilayer thinning is expected to reduce lipid structural perturbation, better mixing of lipids with amino acids evidently causes a marginal net increase in lipid structure disordering as noted in the Supplementary Information.
Finally, given considerable changes occurring in the mechanism of Aβ binding in response to methionine oxidation, it is likely that oxidized and reduced peptides bind to the DMPC bilayer with different affinities. To investigate this possibility, we computed the free energy of binding using replica exchange conformational sampling and the MM-GBSA approach54. The free energy G of a system can be decomposed as
where Emm is the molecular mechanical energy, Gsolv,p is the polar contribution to the solvation free energy computed using the Generalized Born implicit solvent model55 with the dielectric constant of 78.5, Gsolv,ap is the apolar contribution to the solvation free energy estimated from the solvent accessible surface area with a 1.4 Å probe radius and nonpolar surface tension coefficient γ = 0.005 kcal/mol/Å2, and TS is the conformational entropic term. The latter was computed using Gibbs formula for entropy (see Methods). Taking into account that our simulation system contains two non-interacting Aβ peptides, the free energy of Aβ binding to the DMPC bilayer is
where \({G}_{A\beta +DMPC}\), GAβ, and GDMPC are the free energies of the DMPC bilayer with two bound Aβ peptides, of the unbound peptide in water, and peptide-free DMPC bilayer. Then, the change in the binding free energy between MetO and WT Aβ peptides is
In Eq. (3), the free energy of the peptide-free DMPC bilayer cancels out and each remaining term is computed using Eq. (1). REST simulations of MetO Aβ bound to the DMPC bilayer were used to determine \({G}_{MetOA\beta +DMPC}\), whereas our previous REMD/REST simulations of WT Aβ in water and bound to the DMPC bilayer were utilized to compute GWTAβ and \({G}_{WTA\beta +DMPC}\)29,46,56. Additional REST simulations of oxidized Aβ10–40 monomer in water were performed to estimate GMetOAβ (see Methods). Detailed account of binding energetics is given in the Supplementary Information.
According to Eq. (3) and Table S5 \({\rm{\Delta }}{\rm{\Delta }}{G}_{b}=17\pm 9\) kcal/mol implying that MetO Aβ has lower binding affinity than the WT. It is instructive to decompose \({\rm{\Delta }}{\rm{\Delta }}{G}_{b}={\rm{\Delta }}{\rm{\Delta }}{E}_{mm}+{\rm{\Delta }}{\rm{\Delta }}{G}_{solv,p}+{\rm{\Delta }}{\rm{\Delta }}{G}_{solv,ap}-T{\rm{\Delta }}{\rm{\Delta }}S\). It follows from Table S5 that the difference in molecular mechanical energy \({\rm{\Delta }}{\rm{\Delta }}{E}_{mm}=70\pm 18\) kcal/mol is unfavorable, being partially compensated by the polar contribution to solvation \({\rm{\Delta }}{\rm{\Delta }}{G}_{solv,p}=-54\pm 23\) kcal/mol. Furthermore, the difference in apolar contribution to solvation \({\rm{\Delta }}{\rm{\Delta }}{G}_{solv,ap}=2\pm 1\) kcal/mol is minor, whereas the entropic contribution \({\textstyle \text{-}}\,T{\rm{\Delta }}{\rm{\Delta }}S\approx 0\pm 0\) kcal/mol is negligible. Thus, oxidation reduces the binding affinity of Aβ to the DMPC bilayer through the interplay of two opposing factors. The one disfavoring binding of oxidized Aβ to the DMPC bilayer is the loss of favorable intrapeptide interactions upon binding, whereas the factor favoring binding is better hydration of the bound oxidized peptide.
It is important to compare our findings with previous studies. Constant temperature molecular dynamics24,57, replica exchange molecular dynamics23, and NMR spectroscopy22 have been used to study the effects of methionine oxidation on Aβ1–40 and Aβ1–42 peptides in water. The consensus emerging from these studies is that oxidation has a destabilizing effect on the peptide structure. For example, methionine oxidation in Aβ1–42 monomers weakens the long range intrapeptide contacts23 and increases the accessible surface area of methionine by more than two-fold24. Another molecular dynamics study has reported a decrease in β structure content within the C-terminus of Aβ1–40 monomers57, whereas NMR data suggest enhanced fluctuations in the Aβ1–42 C-terminus caused by oxidation22. It is notable that, despite the difference in local environment (water vs lipid bilayer), these outcomes qualitatively agree with the observations drawn from our study. More relevant to our findings are the structures of WT and MetO Aβ1–40 in a water-SDS micelle environment determined by NMR spectroscopy (PDB IDs 1BA4 and 1BA6)12,14. To investigate their secondary structure propensities we applied STRIDE48. Because each PDB entry contains 10 conformers, we assumed that a residue is in a helical state if that state is present in more than half of all conformers. In WT Aβ, helix spans the residues Gln15–Gly37; however, for MetO Aβ, helix is contracted to the sequence interval Lys16–Ser26, i.e., the C-terminal portion of the WT helix, Asn27–Gly37, is absent. The disappearance of helix in the C-terminus of the oxidized peptide is consistent with our REST simulations, which reported seven residues forming helix in the WT Aβ C-terminus and none in the oxidized form. Several experimental studies13,14,17,18 have noted that delayed aggregation of MetO Aβ peptides is responsible for a less severe cytotoxic effect. We speculate that our central result concerning reduced affinity of binding of oxidized Aβ peptide to the lipid bilayer constitutes a molecular basis for this experimental observation. We postulate that reduced affinity of MetO Aβ to the DMPC bilayer vs the WT could play a role in slowing down the MetO Aβ aggregation offsetting its cytotoxic effects. It is also worth noting that the value of \({\rm{\Delta }}{\rm{\Delta }}{G}_{b}=17\) kcal/mol is consistent with the experimental magnitude of free energies of peptide binding to lipid bilayers58.
Conclusions
Using all-atom explicit solvent replica exchange molecular dynamics simulations with solute tempering, we showed that methionine oxidation changes the mechanism of Aβ10–40 peptide binding to the DMPC bilayer. By comparing binding of oxidized and reduced peptides, five consequences of this common post-translational modification were identified. First, Met35 oxidation changes secondary structure of the bound Aβ peptide by unraveling C-terminal helix and promoting turn conformations. Second and related to the first, oxidation destabilizes intrapeptide interactions and causes expansion of the bound peptide. These two outcomes are explained by the loss of amphiphilic character of the C-terminal helix in the oxidized peptide quantified by a reduced hydrophobic moment. Third, oxidation “polarizes” Aβ binding to the DMPC bilayer by strengthening the interactions of the C-terminus with lipids, but largely releasing the rest of the peptide from the bilayer surface. Fourth, compared to the wild-type, binding of the oxidized peptide induces significantly smaller thinning of the bilayer and causes relatively minor drop in lipid density within the binding footprint. These observations were explained by mixing of oxidized peptide amino acids with lipids promoted by enhanced Aβ conformational fluctuations. Fifth and most important, methionine oxidation reduces the affinity of Aβ binding to the DMPC bilayer mainly by disrupting favorable intrapeptide interactions upon binding, which offset the energetic gains from better hydration. We propose that reduced binding affinity of the oxidized Aβ peptide serves as a molecular basis for its reduced cytotoxicity.
Data Availability
The datasets generated and analysed during the current study are available from the corresponding author on reasonable request.
References
Hardy, J. & Selkoe, D. J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297, 353–356 (2002).
Haass, C. & Selkoe, D. J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimers amyloid β-peptide. Nature Rev. Mol. Cell. Biol. 8, 101–112 (2007).
Haass, C. et al. Amyloid β-peptide is produced by cultured cells during normal metabolism. Nature 359, 322–325 (1992).
Williams, T. L. & Serpell, L. C. Membrane and surface interactions of Alzheimer’s Aβ peptide - insights into the mechanism of cytotoxicity. FEBS J 278, 3905–3917 (2011).
Nag, S., Chen, J., Irudayaraj, J. & Maiti, S. Measurement of the attachment and assembly of small amyloid-β oligomers on live cell membranes at physiological concentrations using single-molecule tools. Biophys. J. 99, 1969–1975 (2010).
Quist, A. et al. Amyloid ion channels: A common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. USA 102, 10427–10432 (2005).
Lee, B. C. & Gladyshev, V. N. The biological significance of methionine sulfoxide stereochemistry. Free Rad. Biol. Med. 50, 221–227 (2011).
Stadtman, E. R. Oxidation of free amino acids and amino acid residues in proteins by radiolysis and by metalcatalyzed reactions. Annu. Rev. Biochem. 62, 797–821 (1993).
Smith, D. G., Cappai, R. & Barnham, K. J. The redox chemistry of the Alzheimer’s disease amyloid β peptide. Biochim. Biophys. Acta 1768, 1976–1990 (2007).
Naslund, J. et al. Relative abundance of Alzheimer Aβ peptide variants in Alzheimer disease and normal aging. Proc. Natl. Acad. Sci. USA 91, 8378–8382 (1994).
Kuo, Y.-M. et al. Comparative analysis of amyloid-β chemical structure and amyloid plaque morphology of transgenic mouse and Alzheimer’s disease brains. J. Biol. Chem. 276, 12991–12998 (2001).
Coles, M., Bicknell, W., Watson, A. A., Fairlie, D. P. & Craik, D. J. Solution structure of amyloid β-peptide (1–40) in a water-micelle environment. Is the membrane spanning domain where we think it is? Biochemistry 37, 11064–11077 (1998).
Palmblad, M., Westlind-Danielsson, A. & Bergquist, J. Oxidation of methionine 35 attenuates formation of amyloid β-peptide 1–40 oligomers. J. Biol. Chem. 277, 19506–19610 (2002).
Watson, A. A., Fairlie, D. P. & Craik, D. J. Solution structure of methionine-oxidized amyloid β-peptide (1–40). Does oxidation affect conformational switching? Biochem 37, 12700–12706 (1998).
Hou, L., K., I., Marchant, R. E. & Zagosrki, M. G. Methionine 35 oxidation reduces fibril assemply of the amyloid Aβ 1–42 peptide of Alzheimer’s disease. J. Biol. Chem. 277, 40173–40176 (2002).
Friedemann, M. et al. Effect of methionine-35 oxidation on the aggregation of amyloid-β peptide. Biochem. Biophys. Rep. 3, 94–99 (2015).
Varadarajan, S., Kanski, J., Aksenova, M., Lauderback, C. & Butterfield, D. A. Different mechanisms of oxidative stress and neurotoxicity for Alzheimer’s Aβ(1–42) and Aβ(25–35). J. Am. Chem. Soc. 123, 5625–5631 (2001).
Butterfield, D. A. & Boyd-Kimball, D. The critical role of methionine 35 in Alzheimer’s amyloid β-peptide (1–42)- induced oxidative stress and neurotoxicity. Biochim. Biophys. Acta 1703, 149–156 (2005).
Barnham, K. J. et al. Neurotoxic, redox-competent Alzheimer’s β-amyloid is released from lipid membrane by methionine oxidation. J. Biol. Chem. 278, 42969–42965 (2003).
Nasica-Labouze, J. et al. Amyloid β-protein and Alzheimer’s disease: When computer simulations complement experimental studies. Chem. Rev. 115, 3518–3563 (2015).
Press-Sandler, O. & Miller, Y. Molecular mechanisms of membrane-associated amyloid aggregation: Computational perspective and challenges. BBA Biomembranes 1860, 1889–1905 (2018).
Yan, Y., McCallum, S. A. & Wang, C. M35 oxidation induces Aβ40-like structural and dynamical changes in Aβ42. J. Amer. Chem. Soc. 130, 5394–5395 (2008).
Rosenman, D. J., Connors, C. R., Chen, W., Wang, C. & Garcia, A. E. Aβ monomers transiently sample oligomer and fibril-like configurations: Ensemble characterization using a combined MD/NMR approach. Journal of Molecular Biology 425, 3338–3359 (2013).
Triguero, L., Singh, R. & Prabhakar, R. Comparative molecular dynamics studies of wild-type and oxidized forms of full-length Alzheimer’s amyloid β-peptide Aβ(1–40) and Aβ(1–42). J. Phys. Chem. B. 112, 7123–7131 (2008).
Luhrs, T. et al. 3D structure of Alzheimers amyloid-β(1–42) fibrils. Proc. Natl. Acad. Sci. USA 102, 17342–17347 (2005).
Wälti, M. A. et al. Atomic-resolution structure of a disease-relevant Aβ(1–42) amyloid fibril. Proc. Natl. Acad. Sci. USA 113, E4976–E4984 (2016).
Colvin, M. T. et al. Atomic resolution structure of monomorphic Aβ42 amyloid fibrils. J. Amer. Chem. Soc. 138, 9663–9674 (2016).
Wang, L., Friesner, R. A. & Berne, B. J. Replica exchange with solute scaling: A more efficient version of replica exchange with solute tempering (REST2). J. Phys. Chem. B 115, 9431–9438 (2011).
Lockhart, C. & Klimov, D. K. Alzheimer’s Aβ10–40 peptide binds and penetrates DMPC bilayer: An isobaricisothermal replica exchange molecular dynamics study. J. Phys. Chem. B 118, 2638–2648 (2014).
Lockhart, C. & Klimov, D. K. Binding of Aβ peptide creates lipid density depression in DMPC bilayer. BBA. Biomembranes 1838, 2678–2688 (2014).
Jarvet, J., Danielsson, J., Damberg, P., Oleszczuk, M. & Graslund, A. Positioning of the Alzheimer Aβ(1–40) peptide in SDS micelles using NMR and paramagnetic probes. J. Biolmol. NMR 39, 63–72 (2007).
Yip, C. M. & McLaurin, J. Amyloid-β peptide assembly: A critical step in fibrillogenesis and membrane disruption. Biophys. J. 80, 1359–1371 (2001).
Sergeant, N. et al. Truncated beta-amyloid peptide species in pre-clinical Alzheimers disease as new targets for the vaccination approach. J. Neurochem 85, 1581–1591 (2003).
Ghidoni, R. et al. Novel T719P AβPP mutation unbalances the relative proportion of amyloid-β peptides. J. Alzheimer Disease 18, 295–303 (2009).
Albertini, V. et al. Optimization protocol for amyloid-β peptides detection in human cerebrospinal fluid using SELDI TOF MS. Proteomics Clin. Appl. 4, 352–357 (2010).
Paravastu, A. K., Petkova, A. T. & Tycko, R. Polymorphic fibril formation by residues 10–40 of the Alzheimer’s β-amyloid peptide. Biophys. J. 90, 4618–4629 (2006).
Tieleman, D. P. Methods and parameters for membrane simulations. RSC Biomolecular. Sciences 20, 1–25 (2010).
Brooks, B. R. et al. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comp. Chem. 4, 187–217 (1983).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
MacKerell, A. D. et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 (1998).
Strader, M. L. & Feller, S. E. A flexible all-atom model of dimethyl sulfoxide for molecular dynamics simulations. J. Phys. Chem. A 106, 1074–1080 (2002).
Oakley, A. J., Bhatia, S., Ecroyd, H. & Garner, B. Molecular dynamics analysis of apolipoprotein-D - lipid hydroperoxide interactions: Mechanism for selective oxidation of Met-93. PLoS One 7, e34057 (2012).
Jas, G. S. & Kuczera, K. Free-energy simulations of the oxidation of C-terminal methionines in calmodulin. Proteins: Struct. Funct. Gen. 48, 257–268 (2002).
Kalé, L. et al. NAMD2: Greater scalability for parallel molecular dynamics. J. Comput. Phys. 151, 283–312 (1999).
Lockhart, C. & Klimov, D. K. The Alzheimer’s disease Aβ peptide binds to the anionic DMPS lipid bilayer. BBA Biomembranes 1858, 1118–1128 (2016).
Smith, A. K., Lockhart, C. & Klimov, D. K. Does replica exchange with solute tempering efficiently sample Aβ peptide conformational ensembles? J. Chem. Theor. Comp. 12, 5201–5214 (2016).
Smith, A. K. & Klimov, D. K. Binding of cytotoxic Aβ25–35 peptide to the dmpc lipid bilayer. J. Chem. Inform. Model 58, 1053–1065 (2018).
Frishman, D. & Argos, P. Knowledge-based protein secondary structure assignment. Proteins Struct. Funct. Gen. 23, 566–579 (1995).
Ferrenberg, A. M. & Swendsen, R. H. Optimized Monte Carlo data analysis. Phys. Rev. Let 63, 1195–1198 (1989).
Wimley, W. C., Creamer, T. P. & White, S. H. Solvation energies of amino acid side chains and backbone in a family of host-guest pentapeptides. Biochem 35, 5109–5124 (1996).
Black, S. D. & Mould, D. R. Development of hydrophobicity parameters to analyze proteins which bear post- or cotranslational modifications. Anal. Biochem. 193, 72–82 (1991).
Kyte, J. & Doolittle, R. F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157, 105–132 (1982).
Lockhart, C., Kim, S. & Klimov, D. K. Explicit solvent molecular dynamics simulations of Aβ peptide interacting with ibuprofen ligands. J. Phys. Chem. B 116, 12922–12932 (2012).
Vergara-Jaque, A., Comer, J., Monsalve, L., González-Nilo, F. D. & Sandoval, C. Computationally efficient methodology for atomic-level characterization of dendrimer-drug complexes: A comparison of amine- and acetyl-terminated PAMAM. J. Phys. Chem. B 117, 6801–6813 (2013).
Onufriev, A., Bashford, D. & Case, D. A. Exploring protein native states and large-scale conformational changes with a modified Generalized Born model. Prot. Struct. Funct. Bioinf 55, 383–394 (2004).
Lockhart, C. & Klimov, D. Molecular interactions of Alzheimer’s biomarker FDDNP with Aβ peptide. Biophys. J. 103, 2341–2351 (2012).
Brown, A. M., Lemkul, J. A., Schaum, N. & Bevan, D. R. Simulations of monomeric amyloid β-peptide (1–40) with varying solution conditions and oxidation state of Met35: Implications for aggregation. Arch. Biophys. Biochem 545, 44–52 (2014).
Seelig, J. Thermodynamics of lipid-peptide interactions. Biochimica et Biophysica Acta 1666, 40–50 (2004).
Author information
Authors and Affiliations
Contributions
C.L. and D.K.K. designed the simulations; C.L. conducted the simulations; C.L., A.K.S. and D.K.K. analyzed the results. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lockhart, C., Smith, A.K. & Klimov, D.K. Methionine Oxidation Changes the Mechanism of Aβ Peptide Binding to the DMPC Bilayer. Sci Rep 9, 5947 (2019). https://doi.org/10.1038/s41598-019-42304-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-42304-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
