
Abstract
Despite the exquisite potential of siRNA as a therapeutic, the mechanism(s) responsible for the robust indirect exposure-response relationships have not been fully elucidated. To understand the siRNA properties linked to potent activity, requires the disposition of siRNA to be characterized. A technical challenge in the characterization is the detection and quantitation of siRNA from biological samples. Described herein, a Locked Nucleic Acid (LNA) Hybridization-Ligation ECL ELISA was designed for ultra-sensitive quantification of both sense and antisense strands of siRNA independent of structural modifica-tions. This assay was applied to measure siRNA in serum and tissue homogenate in preclinical species. We observed rapid clearance of siRNA from the systemic circulation which contrasted the prolonged accumulation within the tissue. The assay was also able to distinguish and quantify free siRNA from RNA-induced silencing complex (RISC) and Argonaute 2 (Ago2) associated with therapeutic siRNA. We utilized an orthogonal method, LC-MS, to investigate 3′ exonuclease activity toward the antisense strand metabolism. Taken together, we have demonstrated that the LNA Hybridization-Ligation ECL ELISA is arobust analytical method with direct application to measuring the exposure of siRNA therapeutics seamlessly across biological matrices.
Similar content being viewed by others
Introduction
Over the past two decades RNA interference (RNAi) has emerged as a powerful route for silencing gene expression1. In 1998, the term RNAi was coined referring to the phenomenon of post-translational silencing of gene expression that occurs in response to the introduction of double-stranded RNA (dsRNA) into a cell2. In 2006, Andrew Z. Fire and Craig C. Mello were awarded the Nobel Prize for their discovery of RNAi gene silencing by double-stranded RNA3. The RNAi field quickly expanded with new feats in gene expression knockdown in cell culture4. RNAi technology advancements can now be exploited to allow specific functional inhibition of almost any chosen target gene, allowing much more rapid functional genetic characterization.The natural mechanism of RNA inhibition is mediated by small, double-stranded RNA molecules of 19–25 nucleotides. It is generally accepted that the RNAi function through a multi-step mechanism5. Upon entrance to the cell, longer double-stranded RNA molecules are first processed by the RNAse enzyme, Dicer6,7,8. This functional dimer contains helicase, dsRNA binding, and PAZ (named after piwi, argonaute, and zwille proteins) domains9. The former two domains are important for double-stranded RNA unwinding and facilitation of RNA interactions. The function of the PAZ domain is not fully understood10. Dicer produces 21–23 nucleotide RNA fragments usually with two nucleotide 3′ end overhangs, which are termed siRNA (silencing RNA). The silencing mechanism of siRNA is mediated through the RNA-induced silencing complex (RISC) which, guided by the 21-23 nucleotide fragments (siRNA), recognizes complementary sequence resulting in cleavage and degradation of the targeted mRNA11. With this mechanism, gene expression is specifically inactivated at a post-transcriptional level.
Recent advances with RNAi and siRNA synthetic chemistry have fueled interest in therapeutic siRNA molecules. Antisense oligonucleotides (ASOs) have made landmark achievements in the treatment of several diseases including neurological disorders12, ocular disease13, and early cardiovascular disease14. Synthetic siRNAs have been demonstrated to target genes in vivo for multiple disease areas including cancer15,16,17 hypercholesterolemia18, liver cirrhosis19, respiratory syncytial virus20, hepatitis B21 and human papillomavirus22. Numerous synthetic siRNAs are under development for various diseases. As of August 2018, the first siRNA therapeutic received approval23. With additional clinical trials, it is anticipated that more nucleic acid-based therapeutics will soon reach the market.
Improvements in delivery and chemical stability have significantly improved tissue-specific targeting, cell entry, and sustained potency of siRNA therapeutics. The combined advancements have had dramatic impact on efficacy, thereby decreasing the effective doses. To adequately address the exposure-response relationships in complex matrices, the bioanalytical method platform needs to be carefully selected based on the structure and function of the therapeutic candidate. Quantitative, highly specific and sensitive methods are a requirement to determine the pharmacokinetic (PK) parameters and pharmacodynamic (PD) relationships, both in drug discovery as well as in the drug development process of an siRNA therapeutic.
Several methods for quantifying siRNA in mammalian cell lines and in vivo applications have been reported. Various PCR-based siRNA detection methods have been developed, including primer-extension RT-PCR24, stem-loop RT-PCR25, and competitive quantitative PCR26. These methods, however, suffer from time-consuming and costly optimization processes. Singh et al. were the first to describe the Locked Nucleic Acid (LNA) analogs which display unprecedented affinity towards both complimentary DNA and RNA27. It has been demonstrated that the incorporation of a single LNA can greatly increase duplex stability with pyrimidine interactions28. Here, we developed an ultra-sensitive ELISA-based assay for quantifying double-stranded intact siRNAs for in vivo pharmacokinetic analysis. The assay makes use of two distinct locked-nucleic acid (LNA) modified DNA probes with 5′ and 3′ labeling with biotin (capture marker) and digoxygenin (detection marker), respectively. The assay provides an easy-to-use procedure while delivering high sensitivity and double-stranded siRNA specificity from biological samples. The utility of the ELISA-based assay is demonstrated through the quantitation of intracellular siRNA and in vivo serum pharmacokinetics, allowing the efficacy of chemical modifications and various delivery systems to be readily assessed.
Results and Discussion
Design of LNA Hybridization-Ligation ECL ELISA
The overall design of the hybridization oligonucleotide sandwich was modified from a previously published ELISA-based assays29,30. Summarizing previous assay designs, two DNA-based oligonucleotide probes are implemented. One acts as a capture with its 3′ biotin and 5′ 9-mer overhang and the other acts as a detection probe with its complimentary bases to the capture overhang and 3′ digoxygenin attachment. In previously reported work, the assay structure follows a two-step hybridization-ligation design. Briefly, the analyte of interest is denatured and hybridized in solution with the capture probe and is subsequently attached to a plate via avidin-biotin association. Next, the detection probe is mixed with a T4 ligation mix and after incubation is washed to remove any non-ligated detection probe. The samples are then treated with S1 nuclease in a sodium chloride solution to cleave any truncated duplex. After blocking, their assay detects signal with an anti-digoxigenin-AP substrate solution and is read on a fluorescence plate reader.
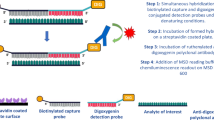
Here we have demonstrated significant assay improvements with the application of LNA chemical base modifications throughout the assay probes, re-tooling of the enzymatic reactions, and transferring the assay to an electro-chemiluminescent (ECL) platform. Figure 1 displays the assay schematic and Table 1 summarizes the improvements between previously published assays and the LNA applied ELISA reported here.

Illustration of the hybridization-ligation ECL ELISA including sequences used for the biotinylated and digoxygenin conjugated probes.
In designing optimized probes for quantification of double-stranded siRNA, LNA substitutions were interspersed into the capture and detection probes targeting a theoretical melting temperature (Tm) of 80 °C and 69 °C, respectively. It is important to note that LNA substitutions adjacent to the nucleation cut site were avoided to allow for S1 nuclease enzyme accessibility. Further, taking into consideration that the optimal hybridization temperature for LNA-containing probes is 30 degrees below the theoretical Tm, a final hybridization temperature of 52 °C was ultimately empirically determined, reflecting LNA oligonucleotide manufacturer’s recommendation of 30 degrees below theoretical Tm. As anticipated, the directed incorporation of LNA into a DNA oligomer dramatically increased probe affinity to the siRNA analyte, therefore, reducing the final assay concentrations of capture and detect probes by 4- and 100-fold, respectively, in comparison to previously published methods. In addition to increasing the affinity of the hybridization probe to the analyte of interest, an increase in thermo-stability was also gained from LNA substitution into the digoxygenin-conjugated detection probe. Moreover, a typical ligation reaction for double stranded DNA is carried out overnight at 18 °C, whereas we were able to decrease this reaction to an hour incubation at 37 °C, thereby, further minimizing the total assay duration. Moving beyond capture and detection probe optimization, further assay improvements included identification of ZnSO4 as an ideal cofactor for achieving maximal S1 nuclease activity. For analyte spikes in buffer and serum, we observed peak nuclease activity under the following reaction conditions: pH 5.5 at room temperature for 30 minutes. For more complex matrices, such as tissue homogenates, we found a dramatic improvement in the signal-to-noise ratio by increasing the incubation temperature from 25 °C to 29 °C. The impact of both metal cofactor and pH on assay performance are summarized in Supplemental Information.
Lastly, to enhance the assay’s overall dynamic range, we transferred the method from a standard fluorescent plate readout to the Meso Scale Discovery (MSD) ECL platform. An anti-digoxygenin detection antibody conjugated to a ruthenium (II) tris-bipyridine N-hydroxysuccinimide tag was incubated with samples following the nuclease reaction. In a direct comparison of DNA versus LNA probes with an anti-digoxygenin detection, we found that the addition of LNA modification dramatically increased sensitivity (See Supplemental Information). The combined assay design modifications produced an assay with a dynamic range of 0.1–10,000 pM in serum, liver and kidney homogenate with a level of detection (LOD) of 1.0 pM (Fig. 2). Most impressively, the re-tooled assay enables the measure siRNA molecules from liver homogenate in a much shorter time frame, with no change to the assay sensitivity (Table 1). Due to the duplex nature of siRNA molecules and the denaturing conditions of this assay, probes can be designed specifically to the sense or antisense strands. Although there are methods for the characterization of duplex RNA such as native Mass Spectrometry (MS)31, these methods are not nearly as sensitive enough to employ in the quantification of siRNA in vivo. For our testing of either strand, samples are homogenized and two assays for sense and antisense are run in parallel. In this manner, we conclude that overlaying values correspond to siRNA in duplex form, and a divergence in values corresponds to a single-stranded population.

LNA Hybridization-Ligation ECL ELISA assay sensitivity and specificity. (A) The assay demonstrates pM sensitivity in both rat serum and liver homogenate (n = 2). The lower limit of quantification with this assay in both serum and liver is 1.0 pM. (B) The assay is selective for both 3′ n-1 and n-2 metabolites with a response of 133% and 95% reactivity when comparing the EC50 values back to the intact siRNA molecule (n = 2). The 3′ n-3 metabolite displayed a right-shifted EC50 with a reactivity of 25% compared to full-length siRNA molecule (n = 2). Graphs fitted in SoftMax Pro (Molecular Devices, LLC. San Jose, CA) with a 4-parameter fit with fixed weighting.
Specificity of LNA Probes
Chemical modifications made to the ribose or phosphate backbone are now routinely incorporated into siRNA molecules in an effort to mitigate exo- and endo-nuclease activity within the blood and target tissue compartments. That being said, it is also well established in the literature that even fully chemically modified oligonucleotides are still subjected to nucleolytic activity in vivo32. In line with this, the specificity of our LNA capture probes were tested in the presence of synthetic metabolites created to mimic exonucleolytic degradation of the antisense strand of the siRNA duplex. These metabolites contain 2′-O-methyl chemical modifications on the ribose across all nucleotides in order to test the extent of probe cross-reactivity toward truncated oligo species. We incorporated this chemical modification for the sole purpose of gaining stability in solution. Previously published efforts using an ex vivo rat liver homogenate model demonstrated that 3′-exonucleases were predominantly responsible for mediating antisense oligonucleotide metabolism relative to either 5′-exonuclease or endonuclease activity; therefore, our work focused on LNA probe cross-reactivity with metabolites that resulted from 3′ end clipping of the antisense strand33. To ensure proper translation of rodent to NHP exonuclease activity, we assessed in vitro samples and saw clear translation of 3′-exonuclease activity, with minimal 5′-metabolites formed (See Supplemental Information for rat data). Both 3′ n-1 and n-2 metabolites (designated as one and two nucleotide subtractions, respectively) demonstrated an assay response in strong agreement with the full-length antisense strand, with EC50 values of 2.43 nM (1.3-fold of antisense strand) and 3.36 nM (0.97-fold of antisense strand), respectively, whereas 3′ n-3 exhibited a dramatic right-shifted response with an EC50 of 13.3 nM (0.25-fold of antisense strand) (Fig. 2). It is important to note that because cross-hybridization of the LNA probe was observed with 3′ n-1 and n-2 metabolites, it is possible that exclusive quantitation of the full-length antisense strand may be overestimated using this method. Regardless, LNA ELISA is able to capture a total population of full-length antisense, 3′ n-1 and n-2, of plausibly active species as it has been demonstrated that siRNA truncated species can still effectively load the RNA-induced silencing complex and mediate potent mRNA target knockdown34.
Characterization of antisense strand metabolites formed in vivo using LC-MS
Antisense oligonuceotide therapeutics are reported to undergo biotransformation in plasma35; however, unlike small molecule drugs which undergo biotransformation by phase I and phase II drug metabolizing enzymes36, siRNA biotransformation occurs by clipping of the phosphodiester bonds between nucleic acids or by innate chemical instability. Numerous reports of oligonucleotide metabolism have been described32,37, and have largely employed ion-pair reversed-phase chromatography coupled to ion trap mass spectrometers. More recently, high-resolution mass spectroscopy has been utilized for the identification of metabolites from in vitro matrices.
In this work, we applied high-resolution accurate-mass measurements to elucidate the metabolites from liver and kidney tissue of non-human primates (NHPs). The application of accurate-mass ion detection affords a high level of confidence in the metabolite identification. In an exploratory NHP study, animals were administered a subcutaneous injection of a proprietary siRNA once weekly for four weeks at a dose of 300 mg/kg. A representative extracted ion chromatogram resulting from the LC-MS data of the samples from non-human primate liver homogenate is shown in Fig. 3. The sequential degradation pattern suggests that these metabolites are the result of 3′ exonuclease activity upon the antisense strand of the duplex. Currently, little is known about the types of RNAses and/or nucleases involved in the degradation of the synthetic siRNAs, and this is an active area of research in our laboratory. Table 2 lists the percentage of intact antisense strand and metabolites in NHP liver and kidney samples. In the NHPs the major species observed in tissues is the intact antisense strand. This ensures that the assay is in line with the observed catabolites that were measured in vivo by LC/MS.

Extracted ion chromatogram of the full length antisense strand and metabolites in NHP liver. With LC-MS analysis, we observed the full siRNA antisense strand and down to six nucleotide base subtractions from the 3′ end.
All other metabolites observed were below 3% of total drug related material and thus excluded from further characterization. This data infers that the siRNA molecule evaluated herein is extremely stable in vivo, a known characteristic of fully chemically modified siRNA molecules, which contributes to the long duration of effect. The identification of the different metabolite patterns of synthetic siRNAs observed in the different tissues provides an insight into the stability and pharmacokinetic properties, in early discovery understanding the metabolite patterns can lead to the design of more stable, potent molecules. Although LC/MS can be powerful in identifying metabolite patterns in plasma and tissue, the sensitivity of LC/MS limits the applicability. Therefore, in most discovery settings, a more sensitive assay such as an ELISA, as reported here, must be utilized to measure siRNA concentrations in both plasma and in tissue.
Quantification siRNA in biological matrices from non-human primates
Serum concentration-time profiles for sense and anti-sense strands of an siRNA duplex were first determined following a single subcutaneous (SC) dose siRNA at either 0.1, 1, or 10 mg/kg in NHPs (n = 6 animals per dosing group). Peak serum concentrations (Cmax) for both sense and antisense strands were dose proportional from 0.1 to 10 mg/kg doses. The maximum concentration of drug was observed 1 hour post-dose for all three treatments (Fig. 4, Table 3 and Supplementary Information). Rapid clearance of the total dose from the serum compartment was anticipated and is in line with previously published pharmacokinetic profiles of siRNA with SC dosing38,39. This is suggestive of inherent siRNA organ distribution which is reflected in the observed volume of distributions. Furthermore, from these data, we can infer that the siRNA duplex remains intact in NHP serum up to approximately 24 to 48 hours post-dose as both the sense and antisense strand concentrations did not differ significantly. Interestingly, the antisense strand persisted in detection the entire duration of serum sampling (168 hours) and is mediated by a currently unknown mechanism, whereas the sense strand fell below the limit of assay detection (0.3 ng/mL) at all time points exceeding 48 hours- independent of dose.

Measurement of siRNA molecule in vivo. (Serum exposures of siRNA molecule in NHPs (n = 6) after a single subcutaneous dose of with 0.1, 1 or 10 mg/kg. Solid lines represent sense strand and dashed lines represent antisense strand. Liver results 28 days post-dose (B). Ago2 pulldown in liver homogenate (n = 3) to measure siRNA amount bound (C). Cropped blot of pulldown displayed within inset (Full blot image provided in Supplementary Information).
Taking this work further, we investigated tissue exposure levels of siRNA, specifically to the liver and kidneys, following four SC doses at 10 mg/kg every seven days (Fig. 4). Similar concentrations of sense and antisense strands were observed in the liver 28 days post-dose at 1,490 and 1,340 ng/mg of tissue, respectively. In contrast, the sense strand (711.3 ng/mg tissue) accumulated in the kidneys at a concentration almost 60 times that of the antisense strand (11.8 ng/mg of tissue). This tissue-specific dichotomy suggests that the sense strand may have a faster rate of clearance compared to its counterpart, which may be further elucidated with a more refined pharmacokinetic study that includes multiple tissue time points. In addition, as the siRNA mechanism of action requires Ago2 duplex loading for target mRNA degradation, we then quantitated the amount of sense and antisense strands co-immunoprecipitated with the catalytic RISC enzyme from liver homogenate. These analytes were then measured in parallel with the endogenous microRNA mir122 as a qualitative assessment for protein enrichment across experimental replicates since this RNA remains tightly associated with Ago2 under basal conditions (Fig. 4C)40. A significant difference was not observed between sense (0.028 ng/mg of tissue) and antisense (0.027 ng/mg of tissue) strand Ago2 loading and concentrations were comparable to mir122 (0.032 ng/mg of tissue). Sense strand cleavage and release from Ago2 was most likely not observed under these multiple dosing conditions due to potential saturation of the enzyme’s binding capacity as a result of high dosing paradigm implemented in this study. Further, from these data, we observed that of the total siRNA liver exposure, less than 0.01% was bound to Ago2.
Methods
Materials
Sheep polyclonal anti-digoxigenin antibody (Roche, Cat. 11222089001) was conjugated with a ruthenium label using the MSD Gold Sulfo-Tag NHS-Ester conjugation kit (Meso Scale Diagnostics, Cat. R31AA-1). Standards and sample buffer were made as follows: 10 mM Tris-HCl [pH 8.0] and 1 mM EDTA. Hybridization buffer was made as follows: 60 mM Na2PO4 [pH 7.0, dibasic], 1 M NaCl, 5 mM EDTA, and 0.02% Tween 20. Ligation buffer was made as follows: 66 mM Tris-HCL [pH 7.6], 10 mM MgCl2, 10 mM DTT, 1 mM ATP, 5 U/mL T4 DNA Ligase (Invitrogen, Cat. 15224041). Nuclease buffer was made as follows: 3 mM ZnSO4, 100 mM NaCl, and 0.8 U/μL S1 Nuclease (Thermo Fisher, Cat. 18001016). Lyophilized oligonucleotide probes were custom synthesized from Qiagen Inc. (Hilden, Germany). All chemicals and reagents were analytical grade or higher, if not specified. Metabolite-simulating 2OMe modified oligonucleotides (sequences not shown) and miR122 sequence were custom synthesized by Integrated DNA Technologies (Iowa, USA). Stock solutions of 100 μM were prepared by reconstitution in molecular grade water. The sequences of all oligonucleotide probes, standards, and metabolites are shown below.
miR122 standard: 5′-rUrGrG rArGrU rGrUrG rArCrA rArUrG rGrUrG rUrUrU rG-3′
miR122 capture probe: 5′-TCG C+TC GTG CAA ACA+CCA TTG+TCA CAC+TCC A/3Bio/-3′
antisense capture probe: 5′-TCG C+TC GTG CAG+XXX XXX XX+X XXX X+XX CGA/3Bio/-3′
sense capture probe: 5′-TCG C+TC GTG TCG+XXX XXX XX+X XXX X+XX CTG/3Bio/-3′
detection probe: 5′-CA+CGAG+CGA/3Dig_N/-3′
r(base): Ribonucleotide
+(base): LNA modification
3Bio: Biotin Conjugation with C6 Linker
3Dig_N: Digoxygenin (NHS Ester)
Hybridization-Ligation ELISA
Either duplex siRNA or single stranded controls were spiked into relative species tissue homogenate or serum matrix at a standard curve range of 2.6–166 pM (0.04–2500 ng/mL). The standard curves and samples were diluted 1:20 in sample buffer and added to a 96-well PCR plate to a final volume of 50 μL. Sequence specific capture oligonucleotide was added to hybridization buffer to a final concentration of 50 nM and 50 μL added to the samples in buffer. Sample and capture probe were hybridized on a thermal cycler under the following conditions: 90 °C for 5 minutes, 52 °C for 30 minutes, and a final hold at 12 °C. After hybridization, samples were transferred to a MSD GOLD 96-well Streptavidin SECTOR PR plate (Meso Scale Diagnostics, Cat. L13SA) for 30 minutes at room temperature. After incubation, the plate was washed with 1x wash solution (KPL, Cat. 50-63-00). Detection oligonucleotide was added to ligation buffer at 1 nM and the mix was added to the plate at a 50 μL volume with subsequent incubation at 37 °C for one hour. After washing, 50 μL of nuclease buffer was added to the wells. Specific to the sequences used, plates were incubated at 37 °C or room temperature for 30 minutes for tissue and serum samples, respectively. Plates were washed and incubated for 1 hour with 50 μL of 2 μg/mL ruthenium labeled anti-digoxygenin antibody in SuperBlock T20 TBS Blocking Buffer (Thermo Fisher, Cat. 37536). After a final wash, 150 μL of MSD Read Buffer T (Meso Scale Diagnostics, Cat. R92TD) was added and the plate was read on a MSD Sector S 600 instrument (Rockville, MD, USA).
Hybridization-Ligation Sample Preparation
Tissue samples were homogenized in lysis buffer containing 50 mM Tris HCl, 100 nM NaCl, 0.1% Triton X100, and protease inhibitor cocktail (Roche, Cat. 11836170001) to a final concentration of 200 mg/mL. Further dilution series of either tissue or serum was done in 5% tissue homogenate (200 mg/mL) or 5% serum, respectively in sample buffer.
LC-MS Sample Preparation
Oligonucleotides were extracted from rat and NHP liver homogenates using the optimized solid phase extraction protocol on a Phenomenex Clarity OTX SPE 96-well plate (Cat. 8E-S103-EGA). Prior to use, the SPE plate was conditioned with 1 mL of methanol and equilibrated with 1 mL of 50 mM NaH2PO4, 2 mM NaN3, 10 mM K2EDTA in water (pH 5.5). The liver homogenate samples were mixed with 1 volume of 10% (v/v) H3PO4 for 10 minutes and then mixed with three volumes of Phenomenex lysis-loading buffer (Cat. AL0-8579) before being loaded on to the SPE plate at low pressure (approximately 2 psi). Then the SPE plate was washed with 1 mL of 50 mM NaH2PO4, 2 mM NaN3, 10 mM K2EDTA in water (pH 5.5) once, 1 mL of 50 mM NaH2PO4 in 50:50 water:acetonitrile (pH 5.5) four times, and 1 mL of 50 mM ammonium formate in 20:80 water:acetonitrile (pH 5.5) twice before eluted with 0.5 mL of 100 mM ammonium bicarbonate, 10 mM TCEP in 50:40:10 water:acetonitrile:THF (pH 9) twice. The eluents were then evaporated under N2 to approximately 200 μL for LC-MS analysis.
LC-MS Method and Analysis
Samples were subjected to LC-MS analysis on Agilent 1290 coupled with Thermo Scientific Obritrap Fusion Tribrid Mass Spectrometer (Waltham, Ma). Chromatographic separation was performed on Waters ACQUITY UPLC Oligonucleotide BEH C18 column: 130 Ångstrm, 1.7 μm, 2.1 mm × 50 mm at 80 °C with a flow rate of 0.4 mL/min. Mobile phase A consisted of 15 mM triethylamine (TEA), 400 mM hexafluoroisopropanol (HFIP) in water, and mobile phase B consisted of the same concentrations of TEA and HFIP in methanol. Each sample (10 μL) was injected and separated on the column using the following gradient: 2% B for the initial 0.5 minutes, followed by a linear gradient to 50% B at 9.5 minutes, then another linear gradient to 95% B at 10 minutes; kept at 95% for the next 2.5 minutes; and then ramped down to 2% B at 13 minutes and kept at 2% for 1 minutes. The mass spectrometer was operated in the negative ion mode at spray voltage of −3000 V and source fragmentation energy of 60 eV. Scan range was set as 600–2400 m/z and Obritrap resolution was set at 60 K. The Oligonucleotide Mass Assembler (OMA) module in the OMA and Oligonucleotide Peak Analyzer (OPA) free software package was used to first to generate a predictive list of m/z values for all the possible metabolites that might be formed from the full-length siRNA construct41. Then the OPA module was used to analyze a representative mass spectrometry data file to confirm which metabolites were actually observed in the sample. A processing method was then generated using the top three most intense charge states and the top three isotopes of each charge state to calculate the peak area of each observed metabolite.
Nonhuman primate studies
All non-human primate studies were conducted at Charles River Laboratories (Reno, Nevada) or MPI Research Inc. (Manatwan, MI). Animals were housed in accordance with the USDA Animal Welfare Act (9 Code of Federal Regulations, parts 1, 2, and 3) and as described in the Guide for the Care and Use of Laboratory Animals. Studies were conducted under an approved IACUC protocol. For serum exposure analysis, female monkeys between 6–10 years of age were dosed subcutaneously with a dose of 0.1, 1.0, and 10 mg/kg. For tissue exposure analysis, female monkeys between 6–10 years of age were dosed at 10, 50, and 200 mg/kg once weekly for four weeks. Necropsy samples were taken 28 days post dose. Pharmacokinetic parameters were extrapolated from sense and antisense serum concentrations using WinNonLin non-compartmental analysis from time zero to 24 hours.
Argonaute-2 (Ago2) Pulldown
Homogenized tissues (25 mg/mL) were centrifuged at 12,000 × g for 10 minutes at 4 °C to pellet cellular debris. The supernatant was then added to 25 μL of either anti-mouse- or anti-human Ago2 antibody beads (Wako Chemicals, Cat. 292-67301 and 292-66701) that were pre-washed twice with 500 μL of lysis buffer (50 mM Tris HCl, 100 mM NaCl, 0.1% Triton X100). Samples were incubated with Ago2 antibody beads overnight while gently rotating at 4 °C. Following incubation, beads were washed three times with 500 μL of lysis buffer and immunoprecipitated protein was eluted with 100 μL of Quantigene Plex lysis mixture (Affymetrix, Cat.10093) while boiling at 95 °C for 5 minutes42.
References
Downward, J. Rna interference. Bmj. 328, 1245–8 (2004).
Mocellin, S. & Provenzano, M. RNA interference: learning gene knock-down from cell physiology. J. Transl. Med. 2, 39 (2004).
Fire, A. et al. Potent and specific genetic interference by double-stranded rna in caenorhabditis elegans. Nature 391, 806 (1998).
Yang, C., Qiu, L. & Xu, Z. Specific gene silencing using RNAi in cell culture. Methods Mol. Biol. 793, 457–77 (2011).
Zhou, H. J., Tsai, S. Y. & Tsai, M. J. RNAi technology and its use in studying the function of nuclear receptors and coregulators. Nucl Recept Signal 1 (2003).
Tijsterman, M. & Plasterk, R. H. A. Dicers at RISC: The mechanism of RNAi. Cell. 117, 1–3 (2004).
Hammond, S. M. Dicing and slicing. FEBS Letters 579, 5822–5829 (2005).
Jaskiewicz, L. & Filipowicz, W. Role of Dicer in Posttranscriptional RNA Silencing, 77–97 (Springer Berlin Heidelberg, Berlin, Heidelberg, 2008).
Wilson, R. Molecular mechanisms of RNA interference. Annu. Rev. Biophys. 42, 217–39 (2013).
Hall, T. & Tanaka, M. Structure and function of argonaute proteins. Structure 13, 1403–1408 (2005).
Rivas, F. V. et al. Purified argonaute2 and an siRNA form recombinant human RISC. Nature Structural & Molecular Biology. 12, 340 (2005).
Rinaldi, C. & Wood, M. J. A. Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nature Reviews Neurology 14, 9 (2017).
de Smet, M. D., Meenken, C. & van den Horn, G. J. Fomivirsen a phosphorothioate oligonucleotide for the treatment of cmv retinitis. Ocular Immunology and Inflammation 7, 189–198 (1999).
Duell, P. B. et al. Long-term mipomersen treatment is associated with a reduction in cardiovascular events in patients with familial hypercholesterolemia. Journal of Clinical Lipidology 10, 1011–1021 (2016).
Tabernero, J. et al. First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer Discovery 3, 406–417 (2013).
Schultheis, B. et al. First-in-human phase i study of the liposomal RNA interference therapeutic atu027 in patients with advanced solid tumors. Journal of Clinical Oncology 32, 4141–4148 (2014).
Bader, A., Brown, D., Stoudemire, J. & Lammers, P. Developing therapeutic micrornas for cancer. Gene. Ther. 18, 1121–6 (2011).
Fitzgerald, K. et al. A highly durable RNAi therapeutic inhibitor of PCSK9. New England Journal of Medicine 376, 41–51 (2017).
Kerr, T. A. & Davidson, N. O. Therapeutic RNA manipulation in liver disease. Hepatology 51, 1055–61 (2010).
Zhang, W. et al. Inhibition of respiratory syncytial virus infection with intranasal siRNA nanoparticles targeting the viral NS1 gene. Nature Medicine. 11, 56 (2004).
Chen, Y., Cheng, G. & Mahato, R. I. RNAi for treating hepatitis b viral infection. Pharm. Res. 25, 72–86 (2008).
Jung, H. S., Rajasekaran, N., Ju, W. & Shin, Y. K. Human papillomavirus: Current and future RNAi therapeutic strategies for cervical cancer. J. Clin. Med. 4, 1126–55 (2015).
Adams, D. et al. Patisiran, an rnai therapeutic, for hereditary transthyretin amyloidosis. New England Journal of Medicine 379, 11–21 (2018).
Raymond, C. K., Roberts, B. S., Garrett-Engele, P., Lim, L. P. & Johnson, J. M. Simple, quantitative primer-extension PCR assay for direct monitoring of microRNAs and short-interfering RNAs. Rna 11, 1737–44 (2005).
Cheng, A. et al. Stem-loop RT-PCR quantification of siRNAs in vitro and in vivo. Oligonucleotides. 19, 203–208 (2009).
Liu, W., Stevenson, M., Seymour, L., Fisher, W. & Kerry, D. Quantification of siRNA using competitive qPCR. Nucleic Acids Research 37, e4–e4 (2009).
Singh, K. S., Koshkin, A., Wengel, J. & Nielsen, P. Lna (locked nucleic acids): synthesis and high-affinity nucleic acid recognition. Chemical Communications 455–456 (1998).
Karkare, S. & Bhatnagar, D. Promising nucleic acid analogs and mimics: characteristic features and applications of PNA, LNA, and morpholino. Appl. Microbiol. Biotechnol. 71, 575–86 (2006).
Wei, X. et al. A specific picomolar hybridization-based ELISA assay for the determination of phosphorothioate oligonucleotides in plasma and cellular matrices. Pharm. Res. 23, 1251–64 (2006).
Chan, K. K. et al. A novel ultrasensitive hybridization-based elisa method for 2-methoxyphosphorothiolate micrornas and its in vitro and in vivo application. AAPS. J. 12, 556–68 (2010).
Ute Bahr, H. & Karas, M. Detection and relative quantification of siRNA double strands by MALDI mass spectrometry. Analytical Biochemistry. 80, 620–625 (2008).
Lin, Z., Li, W. & Dai, G. Application of LC-MS for quantitative analysis and metabolite identification of therapeutic oligonucleotides. J. Pharm. Biomed. Anal. 44, 330–341 (2007).
Crooke, R. M. et al. Metabolism of antisense oligonucleotides in rat liver homogenates. Journal of Pharmacology and Experimental Therapeutics 292, 140–149 (2000).
Chu, C.-Y. & Rana, T. M. Potent rnai by short rna triggers. RNA (New York, N.Y.) 14, 1714–1719 (2008).
Husser, C. et al. Identification of GalNAc-conjugated antisense oligonucleotide metabolites using an untargeted and generic approach based on high resolution mass spectrometry. Analytical Chemistry. 89, 6821–6826 (2017).
Wienkers, L. C. & Heath, T. G. Predicting in vivo drug interactions from in vitro drug discovery data. Nature Reviews Drug Discovery 4, 825 (2005).
Ewles, M., Goodwin, L., Schneider, A. & Rothhamer-Hampl, T. Quantification of oligonucleotides by LC-MS/MS: the challenges of quantifying a phosphorothioate oligonucleotide and multiple metabolites. Bioanalysis 6, 447-464 (2014).
Nair, J. K. et al. Impact of enhanced metabolic stability on pharmacokinetics and pharmacodynamics of GalNAcsiRNA conjugates. Nucleic Acids Research. 45, 10969–10977 (2017).
Willoughby, J. L. S. et al. Evaluation of galnac-sirna conjugate activity in pre-clinical animal models with reduced asialoglycoprotein receptor expression. Molecular Therapy 26, 105–114 (2018).
Valdmanis, P. N. et al. RNA interferenceinduced hepatotoxicity results from loss of the first synthesized isoform of microRNA-122 in mice. Nature Medicine. 22, 557 (2016).
Nyakas, A., Blum, L. C., Stucki, S. R., Reymond, J.-L. & Schrch, S. Oma and opasoftware-supported mass spectra analysis of native and modified nucleic acids. Journal of The American Society for Mass Spectrometry 24, 249–256 (2013).
Trubetskoy, V. S. et al. Phosphorylation-specific status of RNAi triggers in pharmacokinetic and biodistribution analyses. Nucleic Acids Res 45, 1469–78 (2017).
Author information
Authors and Affiliations
Contributions
M.T., J.L., D.D., F.X., B.B., and O.B. conceived the experiments. M.T., J.L., D.D., F.X., B.B., and N.B. conducted the experiments. M.T., J.L., D.D., F.X., B.B., and B.R. analyzed the results. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
All authors of this manuscript are employees and shareholders of Amgen, Inc.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Thayer, M.B., Lade, J.M., Doherty, D. et al. Application of Locked Nucleic Acid Oligonucleotides for siRNA Preclinical Bioanalytics. Sci Rep 9, 3566 (2019). https://doi.org/10.1038/s41598-019-40187-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-40187-4
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
