
Abstract
Longitudinal tracking of individual Plasmodium falciparum strains in multi-clonal infections is essential for investigating infection dynamics of malaria. The traditional genotyping techniques did not permit tracking changes in individual clone density during persistent natural infections. Amplicon deep sequencing (Amp-Seq) offers a tool to address this knowledge gap. The sensitivity of Amp-Seq for relative quantification of clones was investigated using three molecular markers, ama1-D2, ama1-D3, and cpmp. Amp-Seq and length-polymorphism based genotyping were compared for their performance in following minority clones in longitudinal samples from Papua New Guinea. Amp-Seq markers were superior to length-polymorphic marker msp2 in detecting minority clones (sensitivity Amp-Seq: 95%, msp2: 85%). Multiplicity of infection (MOI) by Amp-Seq was 2.32 versus 1.73 for msp2. The higher sensitivity had no effect on estimates of force of infection because missed minority clones were detected in preceding or succeeding bleeds. Individual clone densities were tracked longitudinally by Amp-Seq despite MOI > 1, thus providing an additional parameter for investigating malaria infection dynamics. Amp-Seq based genotyping of longitudinal samples improves detection of minority clones and estimates of MOI. Amp-Seq permits tracking of clone density over time to study clone competition or the dynamics of specific, i.e. resistance-associated genotypes.
Similar content being viewed by others
Introduction
Molecular-epidemiological parameters used to describe the infection dynamics of Plasmodium falciparum include the number of co-infecting parasite clones (multiplicity of infection, MOI), the rate at which different genotypes are acquired over time (molecular force of infection, molFOI) and duration of infection1. These measures are based on monitoring the presence or absence of clones in cross-sectional or longitudinal samples collected in regular intervals. In earlier studies, individual parasite clones in multi-clonal field samples were distinguished and tracked over time by genotyping the length-polymorphic marker merozoite surface protein 2 (msp2) by capillary electrophoresis-based fragment sizing (CE)2,3,4. Yet, msp2-CE genotyping has limited sensitivity for minority clone detection3,5,6. Alternative typing methods instead could perform better in detecting minority clones, but might impact measures of MOI and molFOI7,8. So far quantification of individual clones within multi-clonal infections was not feasible, as this would have required highly complex allele-specific quantitative PCR (qPCR).
SNP-based genotyping by deep amplicon sequencing (Amp-Seq) can detect low-abundant P. falciparum clones at ratios of 1:1000 in mixed infections7,8,9. Most importantly, genotyping by Amp-Seq also quantifies precisely the relative abundance of clones, as shown with artificial mixtures of clones9,10,11. From these ratios the absolute density of each clone (i.e. a certain haplotype) within a multi-clone infection can be deduced if the total parasitaemia of the sample was established by qPCR11. When analysing consecutive samples from a given study participant, presence and fluctuations in density of clones can be tracked. We explore how longitudinal information can be used to improve identification of minority clones with low densities around the detection limit.
A previous study has estimated clonal density with Amp-Seq in multi-clone infections to estimate clearance rates after antimalarial treatment11. We apply the same approach to track parasite clones longitudinally in untreated natural infections. In addition, we increase the resolution of genotyping by combining sequence information from several markers into multi-locus haplotypes.
Methods
Study design
A subset of 153 archived P. falciparum genomic DNA samples from 33 children (mean 4.3 samples [min: 2, max: 11]) aged 1–5 years were available from a cohort study described earlier12 with blood sampling over 40 weeks (first 12 weeks every fortnight, then monthly) in Papua New Guinea (PNG). The two conditions for selection of children were: ≥2/14 bleeds PCR positive, and MOI > 1 in at least one of the samples of each child. Ethical clearance was obtained from PNG Institute of Medical Research Institutional Review Board (IRB 07.20) and PNG Medical Advisory Committee (07.34). Informed written consent was obtained from all parents or guardians prior to recruitment of each child. All experiments were performed in accordance with relevant guidelines and regulations.
Genotyping using length polymorphic marker msp2
Samples were genotyped using the classical P. falciparum marker msp2 according to published protocols13. Fluorescently labelled nested PCR products were sized by CE on an automated sequencer and analysed using GeneMarker software. Fragments were accepted if the following cut-off criteria were met: peak height >500 intensity units and >10% of the height of the majority peak. Electropherograms were inspected visually to exclude obvious stutter peaks. All DNA samples were genotyped in 2 independent laboratories to assess reproducibility of clone detection and measures of MOI.
Marker selection for Amplicon deep sequencing
Amp-Seq was performed on three amplicons located in two different P. falciparum marker genes, namely PF3D7_0104100, “conserved Plasmodium membrane protein” (cpmp), and PF3D7_1133400, “apical membrane antigen 1” (ama1) whose genetic diversity has been studied in great detail7,14,15,16. Previously published primers were used for marker cpmp9. For ama1 two amplicons of 479 and 516 bp were selected that span regions of maximum diversity, i.e. subdomains 2 and 3 of the ectodomain17. Primer sequences and exact amplicon positions are listed in Supplementary Tables S1 and S2.
Sequencing library preparation
Sequencing libraries were generated by three rounds of PCR, according to previously published protocols9. After primary PCR, a 5′ linker sequence was added during nested PCR. Nested PCR products were subject to another PCR round with primers binding to the linker sequences and carrying Illumina sequence adapters plus an eight nucleotide long sample-specific molecular index to permit pooling of amplicons for sequencing and later de-multiplexing. The final sequence library was purified with NucleoMag beads. Sequencing was performed on an Illumina MiSeq platform in paired-end mode (2 × 250 bp) using Illumina MiSeq reagent kit v2 (500-cycles) together with Enterobacteria phage PhiX control (Illumina, PhiXControl v3).
Sequence read analysis and haplotype calling
Samples yielding a sequence coverage of <25 reads were excluded from the analysis. An overview of sequence read coverage for all Amp-Seq markers is given in Supplementary Table S3. Several pipelines to process Amp-Seq data have recently been published, including HaplotypR (https://github.com/lerch-a/HaplotypR.git) that was used for this study9,18,19,20,21. Haplotype calling is explained in full detail in an earlier publication9. In short: Low quality sequences were removed by trimming reads to a final size of 240 bp forward and 170 bp reverse for all amplicons. Index, linker and primer sequence (corresponding to ~50 bp) were trimmed off from forward and reverse reads. As the reference sequence, P. falciparum strain 3D7 was used (PlasmoDB release 3422). The term genotype refers to a single nucleotide polymorphism (SNP), whereas a haplotype was defined as a sequence variant of an entire amplicon. Calling a SNP required a >50% frequency of the total reads in at least two independent samples. Haplotypes containing insertions or deletions (indels) were filtered out, as well as haplotypes resulting from chimeric reads or singleton reads. The number of reads of a given haplotype over all remaining reads of the same marker within a sample is denoted by the term “within-host haplotype frequency”. Cut-off criteria for haplotype calling were as follows: a minimum of 3 reads coverage per haplotype, a within-host haplotype frequency ≥0.1% and an occurrence of this haplotype in ≥2 samples over the entire data set including technical replicates. The chosen cut-off criteria where studied in great detail and discussed in an earlier publication9.
Multi-locus haplotype inference in longitudinal samples
Amp-Seq quantifies the frequency of each haplotype within a sample. This permits the inference of multi-locus haplotypes, an approach also used earlier by software DEploid23. In this study a semi-automated procedure was applied for multi-locus haplotype inference that utilized longitudinal sample information to solve complex mixtures. A multi-locus haplotype was deduced iteratively and separately for each sample. In the first round, the multi-locus haplotype of the dominant clone of a sample was inferred by selecting each marker’s dominant haplotype (>54% within-host haplotype frequency, i.e. 50% + 3.8% standard deviation in within-host haplotype frequency between replicates). After each round, the identified dominant haplotype was ignored and in the following round the dominant haplotype was identified among the remaining reads. If several haplotypes occurred in a sample at similar frequencies, it may be impossible to identify the dominant haplotype. Nevertheless, in many cases this could be resolved by analysing the change in within-host haplotype frequency between the observed and preceding or succeeding sample of the same host. An example of our approach to multi-locus haplotype inference is shown in detail in Supplementary Text.
The final step of multi-locus haplotype inference addressed the problem of clones from a multiple infection that share by chance the same allele of one of the markers. As a consequence, the within-host frequency of a shared haplotype amounts to the sum of two or more independent clones carrying the same allele. In such cases multi-locus haplotypes were inferred by assigning the shared alleles to those haplotypes that summed up to the same proportion in the other two markers. Samples for which the multi-locus haplotype could not be established by this approach were considered unresolvable (Supplementary Table S4).
Reproducibility, sensitivity and false discovery rate
Samples were analysed in duplicate with Amp-Seq markers and msp2-CE. Performing duplicates permitted to identify and exclude false-positive haplotypes and thus prevented erroneous over-estimation of MOI. Each haplotype was classified into one of four groups (example see Supplementary Fig. S1): (1) True-positive (TP) haplotype, i.e. it passed the haplotype calling cut-off in both replicates or in one replicate plus in the preceding or succeeding bleed; (2) False-positive (FP) haplotype, i.e. it passed the haplotype calling cut-off in only one replicate and was not detected in any of the preceding or succeeding samples of that individual; (3) False-negative (FNi) haplotype, i.e. it was detected in one or both replicates but did not pass the cut-off criteria at that occasion, whereas it was detected in the preceding or succeeding bleed as TP (at least once) or FN haplotype; (4) Background noise (all other cases).
Additionally, false-negative (FNii) haplotypes were imputed for samples in which no sequence read was detected. These false-negative haplotypes were imputed only when (a) the haplotype was detected in the preceding as well as the succeeding bleed as a true-positive. Presence in only one of preceding or succeeding sample was not considered sufficient evidence for assuming a case of missed detection. For the Amp-Seq markers but not msp2-CE, false-negative haplotypes were also imputed when (b) data for the other two markers was present and the corresponding multi-locus haplotype was established in the preceding or succeeding sample.
The sensitivity to detect parasite clones was estimated based on selected individuals who had not received antimalarial treatment during the timespan analysed and harboured at least one haplotype that was detected at 3 consecutive bleeds. Sensitivity was defined as the true positive rate of a genotyping method and was calculated as TP/(TP + FN). The risk to falsely assign a haplotype not present in the sample was measured as the “false discovery rate” (FDR), calculated as FP/(TP + FP). This rate represents the extent of false haplotype calls of a genotyping method.
The reproducibility of clone detection in technical replicates (comprising all experiential procedures from PCR to sequence run) was calculated as \(\frac{2{n}_{2}}{{n}_{1}+2{n}_{2}}\), where n1 is the number of haplotypes detected in a single replicate and n2 the number of haplotypes detected in both replicates24. Only TP haplotypes were used to estimate reproducibility.
Epidemiological parameters: clone density, diversity, MOI and FOI
The density of a parasite clone was calculated by multiplying within-host haplotype frequency by parasitaemia (measured by qPCR). As late P. falciparum stages are absent from peripheral blood owing to sequestration, it was assumed that all detected clones were ring or early trophozoite stages, which each possess a single haploid genome. Thus, genome density correlates with clone density. Clone density is expressed as copies of target gene per microliter, quantified by qPCR targeting the 18S rRNA gene of P. falciparum25. The technical detection limit of qPCR was 0.4 copies/μl whole blood.
Based on true positive haplotypes, the expected heterozygosity (He) and mean MOI were determined from baseline (or first bleed available) samples for each marker as described earlier9. He was also estimated for combined markers in samples that had a resolvable multi-locus haplotype and that were separated by a treatment plus ≥2 consecutive P. falciparum negative samples from the same child.
molFOI was estimated on longitudinal sets of sample that had a complete set of replicates for all markers. Haplotypes were counted as new infection if a haplotype was (i) not present in the baseline sample but in a subsequent sample, (ii) not detected at ≥2 consecutive preceding bleeds or (iii) not detected after antimalarial treatment plus after at least one negative sample. Time at risk was calculated as the timespan between baseline and last sampling, minus 14 days for each antimalarial treatment (to account for the prophylactic effect of treatment).
An overview of sample selection criteria applied for different types of analyses is listed in Supplementary Table S5.
Results
Genetic diversity of markers
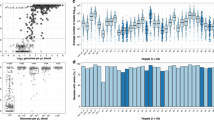
The discriminatory power of Amp-Seq markers cpmp, ama1-D2 and ama1-D3, as well as length-polymorphic marker msp2-CE was estimated in 33 baseline samples (Supplementary Table S5). The resolution was highest for amplicon marker cpmp (He = 0.961) that distinguished 30 haplotypes and yielded a mean MOI = 2.45 (Table 1, MOI distribution by marker in Supplementary Fig. S2). The second-best resolution was obtained by marker msp2-CE (He = 0.940) that distinguished 20 haplotypes and measured a mean MOI = 1.73. Haplotype and SNP frequencies of Amp-Seq markers are shown in Fig. 1 and Supplementary Fig. S2.

Frequency of individual SNPs and haplotypes of three markers in 33 baseline samples from PNG. Non-reference allelic frequency (Non-Ref AF) of each SNP (left) and frequency of haplotypes in these baseline samples (right). n, number of observations per haplotype shown for 2 most prevalent haplotypes. Total number of different haplotypes: 30 for cpmp, 15 for ama1-D2 and 22 for ama1-D3. (Frequency of haplotypes for markers msp2-CE given in Supplementary Fig. S2).
Discriminatory power can be increased by combining multiple markers. Inference of multi-locus haplotypes was not possible for all baseline samples. Instead, 47 independent samples were analysed that had fully established multi-locus haplotypes (Supplementary Table S5). These 47 samples comprised 67 fully established multi-locus haplotypes. Combining marker cpmp with either of the two ama1 fragments yielded very high diversity (54 and 56 haplotypes, He = 0.992 and 0.994 for cpmp/ama1-D2 and cpmp/ama1-D3) (Table 2 and Supplementary Fig. S3). Combining all 3 markers did not increase discriminatory power any further.
Using longitudinal genotyping data to increase detectability of clones
Imperfect detectability of parasite clones has been described previously in longitudinal genotyping studies1,26,27,28. Data from replicates and longitudinal samples can be used to make assumptions on missed clones. This permits imputing of missed haplotypes and thus improves the tracking of clonal infections within an individual over time. Two types of false-negative haplotypes were distinguished: (FNi) haplotypes that were detected below the cut-off and (FNii) haplotypes that were not detected but imputed (Supplementary Table S6). Supplementary Fig. S4 shows an example of these different types of missed haplotypes for all Amp-Seq markers.
The sensitivity to detect parasite clones was estimated for each genotyping marker by enumerating false-negative haplotypes. Sensitivity was higher for the Amp-Seq markers than for msp2-CE (in decreasing order 96.5%, 95.0%, 93.9% and 85.1% for ama1-D2, cpmp, ama1-D3 and msp2-CE) (Table 3). For ≥57% of the identified false-negative haplotypes, reads were detected but fell below cut-off criteria (category (i) above). If such haplotypes were counted as positives by relaxing the cut-off criteria, sensitivity would increase to 99.1%, 97.5% and 97.4% for Amp-Seq markers ama1-D2, cpmp and ama1-D3 (Table 3). Using the standard cut-off criteria, the false discovery rate of haplotypes for Amp-Seq markers was in the range of 0.9–4.2% (Table 3).
Reproducibility to detect parasite clones in technical replicates was greater for Amp-Seq markers than for marker msp2-CE (0.94, 0.93, 0.92 and 0.89 for ama1-D3, ama1-D2, cpmp and msp2-CE) (Supplementary Table S7). Reproducibility decreased either with decreasing clone density, decreasing within-host haplotype frequency, or decreasing sequence coverage (Supplementary Table S8 and Fig. S5)9. Differences in estimates of within-host haplotype frequency between replicates were very small: The median difference was 0.70%, 0.54% and 0.38% for cpmp, ama1-D3 and ama1-D2 (Supplementary Fig. S6).
Determination of molFOI by different molecular markers and methods
A higher sensitivity of the genotyping method does not necessary impact molFOI, i.e. new clones/year, because a missed minority clone could be detected at one of the successive bleeds. We investigated the number of new infections acquired during 40 weeks follow-up in 27 children from whom a complete data set was available (on average 4.3 samples per child [min: 2, max: 7]) (Supplementary Figs S7–S39). Mean molFOI was 2.7, 2.7, 2.3 and 2.2 new infections per year for markers ama1-D3, cpmp, msp2-CE and ama1-D2 (negative binomial regression p-value for comparison of msp2-CE to ama1-D3, cpmp and ama1-D2: 0.596, 0.649 and 0.877) (Supplementary Fig. S40). Thus, no substantial difference in mean molFOI was found for the different molecular markers and different genotyping methods. Mean molFOI of multi-locus haplotypes could not be calculated because multi-locus haplotype inference was not possible for all consecutive samples of each child (Supplementary Table S4).
Quantitative dynamics of multiple infecting P. falciparum clones
Densities of individual clones was calculated from the total parasitaemia by qPCR and the within-host haplotype frequency. Examples of individual clone density dynamics in children with multi-clone infections are shown for three Amp-Seq markers (Fig. 2). The density of some clones remained constant over time, whereas other clones showed fluctuations in density over 3 orders of magnitude (Fig. 2A,B). In some children the dominant clone remains dominant over the observation period (Fig. 2A), whereas in others switch-over between minority clone and dominant clone was observed (Fig. 2B). In highly complex field samples some clones might share the same haplotype of a given marker (Fig. 2C). Such clones can only be differentiated and quantified if multiple markers are typed and at least one of the markers is not shared between concurrent clones.

Dynamics of multi-clone infections in 4 children. Multi-marker haplotypes could be generated in panels A, B and C. Inference of multi-locus haplotypes was not possible for the child in panel D; here the dynamics of individual clones tracked by marker ama1-D2 are shown. Each colour represents a clone. Individual markers are represented by different shapes: cpmp (diamonds), ama1-D2 (circles) and ama1-D3 (squares). Solid line connecting multi-locus haplotypes represents their median frequency. Grey dotted vertical lines represent sampling dates. Red dashed lines represent day of artemisinin combination therapy. Red dash-dotted line represents end of radical cure (artesunate-primaquine) at baseline.
After artemisinin combination therapy, some of the parasite clones from multi-clone infections were cleared 14 days after antimalarial treatment, whereas others were still detectable (Fig. 2A–C). These persisting clones had decreased clone densities (<21 copies/μl) and likely represent remaining late gametocyte stages of cleared asexual infections29. Some new infections following antimalarial treatment (artesunate-primaquine) showed a rapid increase in clone density within the first 14 days after re-infection of a host, followed by a slow decrease in clone density until clearance (Fig. 2D), whereas in other infections clone density remained constant (Fig. 2C).
Discussion
While MOI and molFOI have been extensively described as epidemiological parameters, the ratio and density of individual clones within complex infections has not yet been investigated in detail. This gap in knowledge was due to shortfalls of traditional length-polymorphic markers, where the length of a fragment greatly influences the amplification efficiency in multi-clone infections with fragments competing in PCR and a strong bias favouring smaller fragments5. As a result, multi-locus haplotypes could not be inferred from traditional genotyping data in a reliable way. Such inference is required, for example, for phylogenetic or population genetic studies. In these studies, multiple-clone infections were usually excluded or only the predominant haplotype included30,31. With the possibility to establish multi-locus haplotypes from complex infections the discriminatory power will be greatly improved in future. This study explored the feasibility of multi-locus haplotype calling in complex infections and the usefulness of the Amp-Seq genotyping technique in longitudinal data.
Single Amp-Seq markers cpmp, ama1-D2, ama1-D3, and msp2-CE yielded similar resolution. Combining cpmp with either of the ama1 fragments increased further discriminatory power. The excellent performance of Amp-Seq marker cpmp had been demonstrated earlier9. Such increased resolution is of great practical value for PCR-correction in clinical drug efficacy trials, where new infections need to be reliably distinguished from those present in an individual earlier6,32,33. Discriminatory power may be increased even further by replacing one of the two ama1 fragments with another highly discriminatory marker that has no linkage to either Amp-Seq marker cpmp nor ama1.
Reproducibility of true-positive haplotype calls was measured based on two technical replicates. By definition, a true haplotype must occur in all replicates except for three cases: (1) imperfect detectability of low-density clones, where scarce template may, by chance, lead to occasional absence of the PCR template in one of the replicates, (2) template competition impeding minority clones, whereby templates of a minority clone, present at very low abundance, are outcompeted by dominant clones, and (3), insufficient sequence depth to detect the minority clone in one replicate. It is essential to differentiate between false-positive haplotype calls (caused by cross-contamination, or amplification and sequencing errors9,11) and imperfect detection. This was achieved by considering preceding or succeeding bleeds of an individual. This approach was applied for those cases only where a haplotype was missed in one of the replicates. In our data set, all missing haplotype calls of replicates could be assigned to one of the three causes: imperfect detection, template competition or insufficient sequence depth.
Genotyping longitudinal samples in duplicates enabled also an evidence-based approach to identify false-negative haplotypes. This permitted the estimation of each marker’s sensitivity to detect minority clones. The estimated sensitivities of minority clone detection should serve primarily for a comparison of different genotyping methods, as the sample’s true haplotype composition remain uncertain. Amp-Seq genotyping with markers ama1-D2, ama1-D3 and cpmp missed fewer clones compared to msp2-CE genotyping (Amp-Seq in average 5.4% versus 14.9% msp2-CE). This difference is likely due to less stringent cut-off criteria for Amp-Seq compared to msp2-CE genotyping. Minority clone detection by msp2-CE is limited by peak calling cut-off criteria, which are usually a fixed minimal signal intensity plus a minimum peak height of 10% (used in our study) or more of the dominant peak. Minority clones with an abundance of <10% of all amplified fragments will not pass these criteria. An increase of msp2-CE sensitivity would require a lower cut-off, which would lead to more false positive signals from either stutter peaks or background noise. In contrast, Amp-Seq allows the removal of PCR artefacts before haplotype calling and thus can support a much lower cut-off of <1%9.
In cohort studies where Amp-Seq genotyping is performed in successive follow up samples of the same patient, an even more relaxed definition of Amp-Seq cut-off criteria would be justifiable. In this scenario, the same evidence-based strategy of using successive samples can be used to recover minority haplotypes that were detected with read counts below the haplotype calling cut-offs. If recovery would be performed in this study, ≥57% of all false-negative haplotypes would be identified. Such recovery would increase detectability of parasite clones by Amp-Seq to >97%. In addition, multi-locus haplotypes could provide additional evidence for accurate recovery.
The higher sensitivity of Amp-Seq to detect minority clones compared to msp2-CE substantially increased MOI, but did not affect mean molFOI. Any estimation of molFOI needs to account for temporary absence of clones from the peripheral blood caused by sequestration1,26,27,28. A clone that is temporarily undetectable owing to density fluctuations is likely observed at either the preceding or succeeding bleed. Therefore, a clone is usually only counted as new infection if it was not detected in ≥2 consecutive blood samples. As a consequence, a clone missed at a single bleed will not necessarily lead to a decrease of molFOI.
A clone that was intermittently missed at one bleed by msp2-CE was always detected by Amp-Seq. This observation supports the practice in earlier publications where intermittently missed clones were imputed27,28. Counting a recurrent haplotype as new infection after a single negative bleed would lead to an overestimation of molFOI1,3,26,27,28. The statistical power of the current study was limited and a larger sample size is needed to fully explore the effect of the typing method used on estimates of MOI or molFOI.
A major advantage of Amp-Seq over msp2-CE is that the density of an individual clone in multi-clone infections can be calculated. Quantifying the density of individual parasite clones over time permits the studying of dynamics, and thus fitness, of parasite clones exposed to within-host competition34. For example, the relative densities of new infections can be compared to clones already persisting in a host, and their densities in respect to extrinsic factors or clinical symptoms can be investigated.
For infections with high multiplicity (MOI ≥ 3), inference of multi-locus haplotypes remains challenging (example in Supplementary Fig. S41). Inference is straightforward if a haplotype occurs at a distinctive abundance in any of the longitudinal samples (Supplementary Table S4). In contrast, if haplotypes are equally abundant in one sample and also remain so over several time points, the multi-locus haplotype cannot be inferred. Inference also is impossible for complex patterns with shared haplotypes, i.e. if a haplotype has a high population frequency and therefore is present in 2 or more clones of a blood sample. Shared haplotypes may even lead to inference of wrong multi-locus haplotypes, e.g. when three clones were present at an equal within-host frequency, though only two haplotypes were measured at each locus. However, the risk of erroneous multi-locus haplotype inference decreases if more than 2 unlinked markers are used, as the likelihood of shared multi-locus haplotypes decreases with increasing number of loci. In the present study, multi-locus haplotypes up to MOI = 3 were inferred. For multiplicity >3 and for resolving complex patterns of shared haplotypes, additional longitudinal samples could be analysed simultaneously, for example by incorporating the within-host haplotype frequencies of all consecutive samples of an individual into DEploid software23.
Conclusion
Amplicon sequencing improves clone detectability compared to msp2-CE owing to its greater sensitivity for detection of minority clones. Our results confirm earlier assumptions on clone persistence with intermittent missed observation. This validates the imputation of false negatives to correct for imperfect detection of clones, a strategy also used in previous studies on clone dynamics. Using multi-locus haplotypes for genotyping permitted to identify robustly individual clones and improved differentiation between new and recurring clones. Construction of multi-locus haplotypes are of great value to compensate the effects of highly abundant haplotypes in the population. The option to quantify individual clones enables new approaches to investigate effects of parasite fitness or superinfection in multi-clone infections.
Data Availability
The datasets generated and analysed during the current study are available in NCBI Sequence Read Archive repository under accession number SRX2704363 (https://www.ncbi.nlm.nih.gov/sra/SRX2704363). The source code for software HaplotypR is available at https://github.com/lerch-a/HaplotypR.
References
Felger, I. et al. The dynamics of natural Plasmodium falciparum infections. PLoS One 7, e45542 (2012).
Hofmann, N. E. et al. The complex relationship of exposure to new Plasmodium infections and incidence of clinical malaria in Papua New Guinea. Elife 6, 1–23 (2017).
Koepfli, C. et al. How much remains undetected? Probability of molecular detection of human Plasmodia in the field. PLoS One 6, e19010 (2011).
Sondén, K. et al. Asymptomatic Multiclonal Plasmodium falciparum Infections Carried Through the Dry Season Predict Protection Against Subsequent Clinical Malaria. J. Infect. Dis. 212, 608–16 (2015).
Messerli, C., Hofmann, N. E., Beck, H.-P. & Felger, I. Critical Evaluation of Molecular Monitoring in Malaria Drug Efficacy Trials and Pitfalls of Length-Polymorphic Markers. Antimicrob. Agents Chemother. 61, AAC.01500–16 (2017).
Juliano, J. J., Gadalla, N., Sutherland, C. J. & Meshnick, S. R. The perils of PCR: can we accurately ‘correct’ antimalarial trials? Trends Parasitol. 26, 119–24 (2010).
Miller, R. H. et al. A deep sequencing approach to estimate Plasmodium falciparum complexity of infection (COI) and explore apical membrane antigen 1 diversity. Malar. J. 16, 490 (2017).
Juliano, J. J. et al. Exposing malaria in-host diversity and estimating population diversity by capture-recapture using massively parallel pyrosequencing. Proc. Natl. Acad. Sci. USA 107, 20138–43 (2010).
Lerch, A. et al. Development of amplicon deep sequencing markers and data analysis pipeline for genotyping multi-clonal malaria infections. BMC Genomics 18, 864 (2017).
Levitt, B. et al. Overlap Extension Barcoding for the Next Generation Sequencing and Genotyping of Plasmodium falciparum in Individual Patients in Western Kenya. Sci. Rep. 7, 41108 (2017).
Mideo, N. et al. A deep sequencing tool for partitioning clearance rates following antimalarial treatment in polyclonal infections. Evol. Med. public Heal. 2016, 21–36 (2016).
Betuela, I. et al. Relapses contribute significantly to the risk of Plasmodium vivax infection and disease in Papua New Guinean children 1-5 years of age. J. Infect. Dis. 206, 1771–80 (2012).
Falk, N. et al. Comparison of PCR-RFLP and Genescan-based genotyping for analyzing infection dynamics of Plasmodium falciparum. Am. J. Trop. Med. Hyg. 74, 944–50 (2006).
Arnott, A. et al. Distinct patterns of diversity, population structure and evolution in the AMA1 genes of sympatric Plasmodium falciparum and Plasmodium vivax populations of Papua New Guinea from an area of similarly high transmission. Malar. J. 13, 233 (2014).
Cortés, A. et al. Allele specificity of naturally acquired antibody responses against Plasmodium falciparum apical membrane antigen 1. Infect. Immun. 73, 422–30 (2005).
Cortés, A. et al. Geographical structure of diversity and differences between symptomatic and asymptomatic infections for Plasmodium falciparum vaccine candidate AMA1. Infect. Immun. 71, 1416–26 (2003).
Hodder, A. N. et al. The disulfide bond structure of Plasmodium apical membrane antigen-1. J. Biol. Chem. 271, 29446–52 (1996).
Hathaway, N. J., Parobek, C. M., Juliano, J. J. & Bailey, J. A. SeekDeep: single-base resolution de novo clustering for amplicon deep sequencing. Nucleic Acids Res. https://doi.org/10.1093/nar/gkx1201, 1–13 (2017).
Neafsey, D. E. et al. Genetic Diversity and Protective Efficacy of the RTS,S/AS01 Malaria Vaccine. N. Engl. J. Med. 373, 2025–37 (2015).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Early, A. M. et al. Amplicon deep sequencing of low-density. bioRxiv, https://doi.org/10.1101/453472 (2018).
Bahl, A. et al. PlasmoDB: the Plasmodium genome resource. A database integrating experimental and computational data. Nucleic Acids Res. 31, 212–5 (2003).
Zhu, S. J., Almagro-Garcia, J. & McVean, G. Deconvolution of multiple infections in Plasmodium falciparum from high throughput sequencing data. Bioinformatics 34, 9–15 (2018).
Bretscher, M. T. et al. Detectability of Plasmodium falciparum clones. Malar. J. 9, 234 (2010).
Rosanas-Urgell, A. et al. Comparison of diagnostic methods for the detection and quantification of the four sympatric Plasmodium species in field samples from Papua New Guinea. Malar. J. 9, 361 (2010).
Sama, W., Owusu-Agyei, S., Felger, I., Dietz, K. & Smith, T. Age and seasonal variation in the transition rates and detectability of Plasmodium falciparum malaria. Parasitology 132, 13–21 (2006).
Sama, W., Owusu-Agyei, S., Felger, I., Vounatsou, P. & Smith, T. An immigration-death model to estimate the duration of malaria infection when detectability of the parasite is imperfect. Stat. Med. 24, 3269–88 (2005).
Smith, T., Felger, I., Fraser-Hurt, N. & Beck, H. P. Effect of insecticide-treated bed nets on the dynamics of multiple Plasmodium falciparum infections. Trans. R. Soc. Trop. Med. Hyg. 93(Suppl 1), 53–7 (1999).
Bousema, T. et al. Revisiting the circulation time of Plasmodium falciparum gametocytes: molecular detection methods to estimate the duration of gametocyte carriage and the effect of gametocytocidal drugs. Malar. J. 9, 136 (2010).
MalariaGEN Plasmodium falciparum Community Project. Genomic epidemiology of artemisinin resistant malaria. Elife 5, 1–29 (2016).
Barry, A. E., Schultz, L., Buckee, C. O. & Reeder, J. C. Contrasting population structures of the genes encoding ten leading vaccine-candidate antigens of the human malaria parasite, Plasmodium falciparum. PLoS One 4, e8497 (2009).
Porter, K. A. et al. Uncertain outcomes: adjusting for misclassification in antimalarial efficacy studies. Epidemiol. Infect. 139, 544–51 (2011).
Plucinski, M. M., Morton, L., Bushman, M., Dimbu, P. R. & Udhayakumar, V. Robust Algorithm for Systematic Classification of Malaria Late Treatment Failures as Recrudescence or Reinfection Using Microsatellite Genotyping. Antimicrob. Agents Chemother. 59, 6096–100 (2015).
de Roode, J. C., Culleton, R., Cheesman, S. J., Carter, R. & Read, A. F. Host heterogeneity is a determinant of competitive exclusion or coexistence in genetically diverse malaria infections. Proceedings. Biol. Sci. 271, 1073–80 (2004).
Acknowledgements
We are grateful to the study participants and their guardians and to the field and laboratory team of the PNG Institute of Medical Research, in particular Alice Ura. We would also like to thank Stephen Wilcox for supervising sequence library preparation and sequencing. This work was supported by the Swiss National Science Foundation [grant number 310030_159580] and the International Centers of Excellence for Malaria Research [grant number 1U19AI129392]. A.L. was partly funded by Novartis Foundation for Medical-Biological Research. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: I.F., I.M., A.L., C.K. Performed the experiments: A.L., C.K., J.H.K., N.H., A.R.U. Supervised field work: I.B., A.R.U. Analysed the data: A.L. Supervision: I.F. Writing - draft: A.L., I.F. All Co-authors have read the manuscript and agreed with the final version.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lerch, A., Koepfli, C., Hofmann, N.E. et al. Longitudinal tracking and quantification of individual Plasmodium falciparum clones in complex infections. Sci Rep 9, 3333 (2019). https://doi.org/10.1038/s41598-019-39656-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-39656-7
This article is cited by
-
Microsatellites reveal high polymorphism and high potential for use in anti-malarial efficacy studies in areas with different transmission intensities in mainland Tanzania
Malaria Journal (2024)
-
Genotyping Plasmodium falciparum gametocytes using amplicon deep sequencing
Malaria Journal (2024)
-
Persistent and multiclonal malaria parasite dynamics despite extended artemether-lumefantrine treatment in children
Nature Communications (2024)
-
Molecular tools are crucial for malaria elimination
Molecular Biology Reports (2024)
-
Amplicon deep sequencing of five highly polymorphic markers of Plasmodium falciparum reveals high parasite genetic diversity and moderate population structure in Ethiopia
Malaria Journal (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
