
Abstract
Sensitive detection of altered proteins expression in plasma membranes is of fundamental importance, for both diagnostic and prognostic purposes. Surface-Enhanced Raman Scattering (SERS) has proven to be a quite sensitive approach to detect proteins, even in very diluted samples. However, proteins detection in complex environment, such as the cellular membrane, is still a challenge. Herein, we demonstrate a SERS-based platform to reveal the overexpression of target proteins in cell membranes. As a proof of concept, we implemented ectopic expression of carbonic anhydrase IX (CA IX) and epidermal growth factor receptor (EGFR) in the plasma membrane of the SKOV3 tumor cell line. Our outcomes demonstrate that SERS signals from cells put in contact with a hyperuniform SERS substrate allow highlighting subtle differences in the biochemical composition of cell membranes, normally hidden in spontaneous Raman confocal microscopy. This opens new opportunities for a label-free membrane analysis and bio-sensing in a broader sense.
Similar content being viewed by others

Introduction
The plasma membrane of cells defines the border between the interior of the cell and the outside environment. It rules many critical cell functions, such as cell signalling and trafficking, thanks to the presence of a large variety of membrane proteins (MPs), specifically coordinating selected cell functions. Alteration of this complex biochemical machinery is often associated to serious diseases, including the malignant transformation of cells. It is thus not surprising that MPs are largely studied for therapeutic, diagnostic and drug-delivery related purposes1,2. Most of MPs studies involve extraction and solubilisation steps, and are therefore performed outside from the MP native environment3,4. Fluorescent tagging is surely a quite sensitive approach for MPs in situ detection5, but it suffers from some limitations, such as background fluorescence, poor spectral selectivity and photobleaching. Surface enhanced Raman scattering nanoparticles (SERS-NPs), constitute an attractive alternative to fluorescent probes for biological labelling due to their photo-stability and multiplexing capabilities6,7,8,9. SERS-NPs have been used to reveal the distribution of specific cell surface biomarkers in immunolabelled endothelial cells10 and breast cancer cells11. However, the use of SERS-NPs is not optimal for live-cell imaging applications, due to NP internalisation and trafficking by cells11. Recently, we demonstrated a label-free, SERS based approach for the analysis of erythrocytes membranes12, in which unlabelled cells were put in contact with a high uniform and densely packed SERS substrate. Clearly, due to the short-range response of the SERS mechanism13, only bio-molecules exposed on the membrane can be efficiently detected, conferring a high-contrast with respect to the bulk Raman contribution14,15,16. It is worth noticing that the spatial uniformity of the plasmonic substrate enhancement factor (EF)17 is a crucial parameter of such type of investigation, minimising the intrinsic signal fluctuations in SERS imaging12. Herein, we demonstrate a SERS-based approach to reveal the overexpression of a selected protein on the plasma membrane of cells. As proof of concept, we established a model of ectopic expression of two model proteins, carbonic anhydrase IX (CA IX) and epidermal growth factor receptor (EGFR), in the tumour cell line SKOV3. CA IX is a trans-membrane protein, typically expressed in normal cells after hypoxic treatments. Cancer cells constitutively express CA IX, to support the metabolic shift towards anaerobic glycolysis. In particular, CA IX overexpression represents the expedient by which cancer cells manage to neutralise the acidic pH resulting from the anaerobic metabolism. Accordingly, CA IX expression represents a negative prognostic marker in cancer18. EGFR is an integral membrane protein, able to elicit intracellular signalling mediated by a tyrosine kinase activity, after binding to cognate ligands in the extracellular environment19. Its activity is often de-regulated in cancer, so that EGFR, as well as additional members of its family of tyrosine kinase receptors, are molecular targets for innovative cancer therapeutics20.
Our outcomes demonstrate that SERS data from cancer cells placed in contact with a hyperuniform21,22 SERS substrate, combined with a robust statistical analysis of data, allows to highlight differences in the biochemical composition of cell membranes hidden by spontaneous Raman confocal microscopy. Our results support the use of a SERS-based platform for a label-free membrane analysis, a competence which is of fundamental importance for both diagnostic and prognostic purposes.
Results and Discussion
Quantitative Assessment of ectopic expression of CA IX on SKOV3 cell membrane
In order to demonstrate the suitability of SERS to detect protein expression on cell surface, we selected the tumour cell line SKOV3 for overexpression of the CA IX model protein by transfection. Gene transfection efficiency is affected by several factors23. Therefore, in order to face the statistical variability of such procedure and to distinguish successfully transfected cells from untransfected ones, SKOV3 cells were transfected with CA IX in the presence of a vector encoding a fluorescent, EGFP protein (nEGFP), specifically expressed within the nuclear compartment24. nEGFP reporter protein was preferred to other versions of the EGFP (i.e., cytosolic or membrane-associated), in order to avoid possible interference of a cytoplasmic fluorescence with SERS signal detection at the membrane level. In particular, a 1:10 molar ratio of the expression vectors EGFP:CA IX was selected. Figure 1 shows a typical fluorescence-assisted cell sorting (FACS) profile of co-tranfected cell populations. The counts of cells positive to nEGFP expression (nEGFP+), corresponding to gates Q4 and Q2 of Fig. 1, reveals that around 95% of the nEGFP+ cells are certainly positive for CA IX expression on the cell surface (in the following indicated as CAIX+). Accordingly, visual selection of nEGFP+ cells for SERS analysis resulted in a 95% probability of selecting CAIX+ cells. In the following, we will indicate as CAIX− SKOV3 cells not transfected with CAIX but successfully transfected with the nEGFP reporter protein. CAIX− cells will be used as control sample. It is worth noticing that this choice allows the factorisation of any possible interferences of nEGFP in the acquired signals, as well as any possible cellular stress due to the transfection process.

FACS analysis of transfected SKOV3 cell sub-populations. The x axes indicate fluorescence intensity for EGFP expression, while the Y axes show the fluorescence levels of the APC dye (anti-CA IX antibody). The left panel shows untransfected cells for gating purpose. Untransfected SKOV3 cells show very low basal expression of CA IX and absence of auto fluorescence in the nEGFP channel. The Panel on the right shows the FACS pattern of SKOV3 cells transfected with both nEGFP and CA IX expression vectors. The percentage of the different cell populations, gated within the Q1 to Q4 sectors, is reported in the accompanying table. The values of Q2 (36.4%) and Q4 (2.0%) gates show that just 5% of cells within the population of EGFP+ cells (Q2 + Q4) can be recognised as false positive for CA IX expression.
SERS-Assay Design and Data Processing
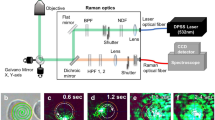
SERS measurements on transfected cells were performed with a commercial confocal micro-Raman apparatus (WITec Alpha 300 system), endowed with a Raman probe at 532 nm. This system was modified in order to include an optical branch used to check the fluorescence of SKOV3 cells due to nEGFP expression (see Materials and Methods). Hyperuniform plasmonic SERS substrates were prepared according to the procedure fully described in ref.22 and shortly summarised in the Materials and Methods paragraph. In Fig. 2a we show a 1 × 1 μm TEM image of our SERS substrate, which approximately corresponds to the area shined by the laser spot. It reveals the dense packing of Ag nanoislands, exhibiting an average interparticles gap of about 2–3 nm. Cells cultured on a glass coverslip were put in contact with the SERS substrate by simply laying down the cell coverslip onto the plasmonic substrate, allowing cells adhesion by gravity. In order to avoid water evaporation, the substrate/coverslip sandwich was sealed-off with a thin layer of low vapour vacuum grease (a schematic representation is given in Fig. 2b). Optimal adhesion of cells on the plasmonic layer, revealed by the presence of high intensity SERS signals, was obtained after a few tens of minutes from sample preparation.

SERS analysis of SKOV3 cells. (a) TEM image of a 1 μm × 1 μm Ag nanoislands pattern of our hyperuniform plasmonic substrate. The average nanoislands gap is about 2–3 nm. The dotted line denotes the laser spot area (about 0.8 μm2), within which about 400 hot-spot sites occur. This guarantees a high EF value and a very good spatial reproducibility. (b) Schematic drawing of the sample cell used for SERS analysis. The structure is comprised of a nano-patterned Ag-substrate covered by a glass coverslip on which SKOV3 cells were cultured. The two layers were glued with low vapour vacuum grease in order to avoid water evaporation. (c) Cartoon of SERS data acquisition process. Only molecules at nanometric distance from Ag nanoislands give rise to a detectable SERS signal. (d) Bright field image from SKOV3 cells analysed in this work. (e) Four background subtrated SERS spectra acquired for the cell shown in (d). They were acquired with a 10 μw laser power (on the sample) and an integration time of 2s. Spectra were selected in order to highlight the variability, in terms of bands positions, among the acquired signals owning to the same cell.
SERS spectra were acquired at different times after transfection: session 1 (48 h), session 2 (60 h), session 3 (72 h) and session 4 (96 h). SERS data acquisition protocol was applied only on CAIX+ and CAIX− cells positive to the in-line nEGFP assay. It consisted in the acquisition of SERS signals in N = 20 different points of a line connecting two opposite points of the cell border (see Fig. 2d). Typically, the obtained spectra exhibited a high variability, concerning both intensity and bands spectral positions; an example is shown in Fig. 2e. Given the spatial uniformity of the EF for the employed SERS substrate12, there are many causes that can give rise to these fluctuations. The irregular morphology of the cell membrane could be surely an issue, determining changes of the substrate-membrane distance. Moreover, even at a given point, some deleterious impact may be induced by the intermittent contact between membrane and surface at nanometric scale, due to the intrinsic fluidity of the membrane itself12. Overall, we deemed it appropriate to carry out spatial and temporal averaging of SERS signals to deal with any unwanted fluctuations. The criteria we followed are described in details in the Material and Methods paragraph.
Multivariate Analysis of SERS spectra
SERS spectra resulting from the previously mentioned averaging procedure were processed by Principal Component Analysis, PCA25. Figure 3a reports the outcomes of PCA obtained for a single measurement session (session 2). As shown, we found a quite good separation between CAIX+ cells (red symbols) and CAIX− cells (green symbols). Similar results were obtained for all the other sessions. Intriguingly, a higher clusterization of points was revealed for cells prepared at lower confluence and analysed after a longer time lapse from the transfection process. We speculate that this could be ascribed to a higher presence of daughter cells in the sample, split from parent cells during the mitosis cycles occurred between the two measurement sessions. This reasonably could contribute to homogenise the cellular sample. The capacity to highlight the presence of CA IX on the cellular membrane is still conserved by processing by PCA the spectra collected in all the investigated 4 sessions (see Fig. 3b), although at a slightly lower level with respect to the single session. This is absolutely not an obvious outcome because the complex membrane biochemistry might render intrinsically different samples acquired in different sessions. Figure 3b also shows that a certain clusterization of cells belonging to the same session can still be appreciated, especially along the PC3 score coordinate. Looking at the loading relative to the PC3 component (see Fig. 3d), we notice that all the peaks of this loading can be attributed to protein components, while no lipid-related feature can be observed (e.g. in the region around 1440 cm−1), suggesting that the difference highlighted by this loading is not amenable to modification of lipids induced by the transfection process.

(a) PCA score plot obtained in a single measurements session. CAIX+ and CAIX− cells are represented by red and green symbols, respectively. (b) Score plot obtained from spectra collected in 4 different experimental sessions (dots: session 1; triangles: session 2; diamonds: session 3; squares: session 4). Reddish/greenish colour corresponds to CAIX+/CAIX− cells. Differentiation occurs mainly along the PC3 coordinate. (c) 2D scatter plot (PC1-PC3 plane) for data shown in part b but without distinguishing the four sessions. The 95% confidence ellipses relative to CAIX+ (red) and CAIX− (green) are also shown. (d) PC3 loading plot from data shown in part b. Labels highlight the main spectral feature of PC3 loading.
In order to test the reliability of our SERS based assay of cellular membranes, we used the leave-one-out cross validation (LOOCV) procedure. Calling N the total number of analysed cells, PCA was calculated using N-1 spectra (training set), and the scores of the left-out spectrum was calculated projecting it on the PC vectors of the training data. Therefore, this spectrum was attributed to a given sample by evaluating its position with respect to the 95% confidence ellipse of the training data (whose classification was assured known) in the space of the first three scores. This procedure was iteratively repeated until all the spectra set were left out. Figure 3c reports the 95% ellipse relative to all data. It is worth noticing that these ellipses remain substantially unchanged in the iterative procedure of LOOCV, which suggests a sufficient data sampling of our analysis. The results of LOOCV analysis were summarised by the confusion matrix shown in Table 1. From the elements of this matrix, it was possible to estimate a sensitivity ~94%, a specificity ~93% and a global accuracy ~94% of our SERS-based approach for CAIX detection.
Spontaneous Raman vs SERS to reveal membrane proteins overexpression
Another interesting question is whether the differences found from SERS could also be observed by the simpler spontaneous Raman spectroscopy (RS). This issue was verified by analysing the same cellular sample, first by RS and then by SERS, by covering the sample with the plasmonic substrate. In both cases, the signal associated to each cell was obtained by averaging 20 signals from a line scan. Figure 4a reports a comparison between a typical Raman (left panel) and SERS (right panel) signal from SKOV3 cells. Notably, both signals were acquired by using the same integration time (2s), while the laser power was three orders of magnitude higher for the spontaneous Raman case. Besides, we remark that Raman signals exhibits DNA related spectral features (such as the peak at 784 cm−1) coming from the nuclear region, while all SERS features can be ascribed to lipids and proteins26. Figure 4b reports the PCA score plot obtained from both spontaneous Raman (left) and SERS (right) spectra of SKOV3 cells. In both cases, red and green symbols represent CAIX transfected/untrasfected cells, respectively. No difference between CAIX+ and CAIX− cells was detected for spontaneous RS measurements for the first three score orders, the same being true for higher orders PC (data not shown). Instead, a complete separation between CAIX+ and CAIX− cells was obtained in SERS analysis, highlighting a higher sensitivity of SERS to membrane modifications.

Comparison between Raman and SERS analysis of SKOV3 cells. (a) Typical Raman (left) and SERS (right) spectrum. In both cases, the main spectral features are highlighted by labels reporting their spectral position. (b) Score plot resulting from the analysis of Raman (left) and SERS (right) spectra from CAIX+ (red symbols) and CAIX− (green symbols).
Insight on the selectivity to proteins overexpression
To further asses the strength of our SERS based approach to reveal the modification of the chemical pattern of cytoplasmic membrane associated with overexpression of proteins, the same analysis was repeated with another protein, the EGFR. Also for this case, cells were co-transfected with EGFP, in order to solve the statistical uncertainty associated with the transfection process. Again, SERS analysis reveals a quite relevant capacity to distinguish cells overexpressing EGFR (EGFR+ cells) with respect to cells transfected in order to express only nEGFP (EGFR−), as clearly shown in Fig. 5a. Moreover, as for the CAIX case, we verified that spontaneous Raman is not able to discriminate cells overexpressing EGFP on the cytoplasmic membrane. As a further step, we checked the level of selectivity of our SERS-based approach to distinguish the overexpression of two target proteins. Hence, we applied PCA to CAIX+, EGFR+ and control cells, which lead to the scatter plot reported in Fig. 5b. Clearly, in this plot, CAIX+ and EGFR+ cells (red and blue squares, respectively) are separated from the control sample, while no difference was found between CAIX+ and EGFR+ cells, which, in fact, occupy the same score plot region. As a matter of fact, cells showing CA IX or EGFR expression may result indistinguishable due to elicitation of similar membrane perturbations, which may not be dependent on the size, the protein sequences and the glycosylation patterns of the extracellular domains exposed on the plasma membrane. However, by limiting analysis to only CAIX+ and EGFR+ cells (therefore not including control cells), a good differentiation between the two different samples is again obtained, with a global accuracy ~90% (data not shown). This outcome suggested us to check the discrimination capability (among CAIX+, EGFR+ and control) at higher PCs. Discrimination among samples was revealed, to a certain extent, by PC7, a component which describes only ~2% of variation of SERS data. Figure 5c shows this outcome. In the same plot, the 95% confidence ellipses for all the samples are also displayed. It is worth noticing that control cells exhibit a relatively small area in the plot, which suggests a more pronounced homogeneity of control cells with respect to the CAIX+ and EGFR+ samples. However, it should be noticed that the use of a high order PC (describing therefore only a little variation among data), casts doubts upon the reproducibility of the here obtained experimental outcome.

(a) PCA score plot obtained by SERS analysis of EGFR+ and EGFR− cells, represented by blue and green symbols, respectively. (b) Score plot obtained by PC analysis of SERS spectra from EGFR+ (blue squares), CAIX+ (red squares) and Control (green squares). Globally, the first 3 PCs take into account of ~92% of data variability. (c) 2D scatter plot (PC7-PC8 plane) from the same data analysed in (b). Samples are differentiated, to a good extent, along the PC7 coordinate, which describes around 2% of variability among samples. The 95% confidence ellipses relative to the three samples are also shown.
Conclusions
Herein, we have demonstrated that SERS analysis of cell membranes is able to reveal membrane alteration induced by overexpression of a model membrane protein. This intrinsically label-free approach could be quite useful in many contests. For instance, it could provide precious information on the dynamic transformation of cells induced by external stresses, including drugs administration. Moreover, SERS could be a quite powerful tool for early diagnosis of overexpression of cancer-related proteins, such as CA IX and EGFR, exposed on the cell membrane. Our SERS-based approach suffer from some limits, also highlighted in this paper, concerning in particular its selectivity. In particular, our approach fails in discriminating among CAIX+, EGFR+ and control cells, unless pushing PCA to high order, and therefore poor significant, components. Moreover, its efficiency reasonably depends upon the specific protein involved in the investigation. Nevertheless, our results demonstrate the possibility to highlight differences in the protein framework of cellular membranes.
In particular, it is reasonable to envisage a quite good sensitivity of our approach to reveal membrane transformation, since such processes typically involve a consistent change of the membrane protein pattern (e.g. malignant transformations). Globally, our results pave the way for a label-free membrane analysis, a fundamental issue for both fundamental biomedical studies and applied researches devoted to sensors development.
Materials and Methods
DNA constructs, cell cultures, transfections and FACS analysis
The vector expressing nEGFP, pEGFP NLS-SV40(TAg), was obtained by cloning the T antigen polyA signal from SV40 in the pEGFP C1 vector, as described in ref.24; the pRcCMV backbone was used to generate vectors expressing the full length CA IX27 and EGFR proteins. SKOV3 cells were cultured in RPMI Medium 1640-GlutaMAXTM-I. The medium was supplemented with 10% heat-inactivated fetal bovine serum (FBS), 50 UI ml−1 penicillin, 50 μg ml−1 streptomycin, 2mM L-glutamine. All the reagents for cell culturing were from GibcoTM, Thermo Fisher Scientific. Cell lines were purchased from the American Type Culture Collection (ATCC) and cultured in a humidified atmosphere containing 5% CO2 at 37.
For SERS analysis, 5 · 104, 7 · 104 or 1 · 105 SKOV-3 cells were seeded in 12 well plate and cultured on sterile round cover glass. 24 hours after seeding, cells were co-transfected with a 1:10 molar ratio of the expression vector pRc/CMV-CA IX encoding CA IX or EGFR (1 μg) and pEGFP-NLS-C1 (100 ng), encoding nEGFP by using 2 μL Lipofectamine Transfection Reagent (Life Technologies, Inc.). As control cell population, SKOV-3 cells were co-transfected with pEGFP-NLS-C1 and empty pRc/CMV vector.
FACS analysis was performed at the CEINGE in-house facility. 1 · 106 SKOV-3 cells were seeded in 100 mm dishes. 24 hours later, cells were co-transfected with 1:10 molar ratio pRc/CMV-CA IX or empty pRc/CMV vectors (10 μg) and pEGFP-NLS-C1 (1 μg) by using 20 μL Lipofectamine. 48 hours after transfection, cells were detached by using PBS EDTA 5 mM, and washed 2 times with PBS. Cells were stained for 30 min with
10 μL of Human Carbonic Anhydrase IX/CA9 APC-conjugated Antibody FAB2188A, R & D System. After 2 washes, APC membrane fluorescence and EGFP were analysed and monitored by cell-sorter Becton Dickinson FACSAria.
Plasmonic Substrates preparation and characterisation
Plasmonic substrate preparation is described in detail in ref.22. Briefly, BCP micelles were prepared starting from 97.2 mg PS-b-P4VP (Polymer Source Inc) dissolved in a solution of 7.0 g of tetrahydrofuran and 10.5 g of toluene taken under stirring at 700 rpm for 3 h at 25, then for 2 h at 67, before slowly cooling down the solution at room temperature. Therefore, micelles were loaded with Ag+ by adding 203.7 mg of AgNO3 and stirring (700 rpm) for 24 h. Ag+ reduction was accomplished by adding drop-wise 440 μl of a solution obtained by dissolving 440 mg of fine dry powder of NaBH4 in 4.4 ml of the same solvent THF/toluene used for micelles preparation. After filtering with a 200 nm PTFE syringe filters, this solution was centrifuged at 11 krpm for 20 min to separate the possible unloaded supernatant micelles and sonicated for 22 min. Finally, 100 μl of the resulting solution was left over the supporting glass for 30 s before starting the spin-coating at 1.0 krpm speed for 60 s. This produced an uniform film coating of nanoislands with an average size of ~26 nm and an interparticles gap ~2–3 nm. Finally, UV exposure for ~20 h at 254 nm (Hg lamp, Sankyo Denki G15T8) at an energy density of 1.7 mJ cm−2, removed the copolymer. The achieved reddish plasmonic film on glass appeared as a transparent optical coating with a green backscattering glow.
In order to estimate the EF factor, we used Crystal Violet (CV) as probe molecule. Therefore, we measured both spontaneous Raman and SERS signals from a proper CV aqueous solution. In order to detect SERS signals produced by a controlled number of molecules, we set-up an evaporation cell constituted by two parallel substrates (24 × 24 mm) distanced by 4.5 μm beads used as spacers, and sealed. Therefore, a volume of 100 μl of a 380 nM CV solution was infiltrated in this cell and allowed to dry slowly. In this way, a nearly uniform CV film is produced, in which Nsurf = 218 molecules are present within the scattering area of 0.44 μm2. SERS signal ISERS was acquired by using an impinging power (on the sample) of PSERS = 4 μW and an integration time of 2s. Then, we acquired the spontaneous Raman signal (IRaman) from a 5.57 mM CV aqueous solution, by using a power PRaman = 0.5 mW and an integration time of 2s. This signal results from Nvol = 7 × 106 molecules present in the confocal detection volume (~2 μm2). The EF was therefore estimated as:
which provided us the value EF = 1.2 × 107. To test the EF spatial reproducibility test, SERS signal was acquired over 50 mm × 50 times region, with a 500 nm step. These measurements allowed to estimate a spatial uniformity of ~5% on this scale.
Confocal Microscopy
Raman spectra were acquired by using a commercial Raman system (WITec, alpha 300). It consists in an inverted confocal microscope endowed with a Raman excitation source at 532 nm. This laser is focused on the sample through a 60X dry objective (NA = 0.8), providing an almost diffraction limited spot on the sample. In-elastically back-scattered radiation was guided toward the spectrograph by a 50 μm core optical fibre, which assures the system confocality. In order to verify fluorescence of cells induced by GFP transfection, an optical branch devoted to fluorescence excitation/detection was added, as depicted in Fig. 6. In brief, blue light (470 nm) from a fluorescence optical fibre (M470L3-C1) was firstly properly collimated and spectrally filtered, than it was injected into the same microscope objective used as condenser for bright field illumination. Cell fluorescence imaging was accomplished by using the same inverted microscope objective used for Raman excitation, coupled to a CCD camera (Hamamatsu C5985) through a switchable mirror. Samples consisted in culture coverslips, covered by cells at a confluence of ~50% and put in contact with the plasmonic substrate. In order to avoid water evaporation, this sandwich was glued by vacuum grease. Measurements were performed by acquiring 20 spectra on a line connecting two opposite cell borders. Each spectrum was acquired by using an integration time of 2s and an impinging power (on sample) of 10 μW. Spectra were acquired over the spectral range from 700–1800 cm−1 (1024 points), with a spectral resolution of ~1.5 cm−1. In order to better highlight SERS bands, acquired signals were background-subtracted. Background mainly originates from the presence of polymeric residues on the Ag-nanostructures not completely eliminated by substrate exposure to UV lamp. It presents two quite broad bands in the 1300–1600 cm−1 region, which can be easily eliminated by a polynomial fit.

Schematic representation of the Raman/fluorescence optical set-up used for this investigation.
Data processing
The ability to distinguish by SERS the effect of overexpression of a single protein at membrane level is based on the assumption that it is possible to assign to each cell a single SERS spectrum, holding information on the complex molecular structures of its membrane. This is absolutely not an easy task. As a matter of facts, several co-factors should be taken into account. A first source of spectra variability is due to the intermittent contact of the fluid cell membrane with the SERS substrate. This kind of fluctuations can be reduced by a proper choice of the integration time (2s in our case) which provides a temporal averaging. A second important source of fluctuations arises from the membrane heterogeneity which usually occurs at length scale below 200 nm (due to the presence, e.g., of lipid rafts). Part of these fluctuations are attenuated by the averaging effect produced by the diffracted laser spot area (1 μm2). This average changes, however, moving from point to point on the cell membrane. For this reason, we performed a preliminary study to face this issue. We proceeded by defining a distance \({{\rm{\Delta }}}_{{s}_{a},{s}_{b}}\) between two spectra sa and sb acquired in two different points of the cell membrane, according to the relation:
the index i running on all the Np spectral points (wavenumbers). Therefore, we acquired, for a single cell, a line scan of N = 50 spectra and repeated the same procedure over 30 cells. N = 50 was chosen as a compromise to minimise cellular stress due to the laser exposure. Each spectrum was normalised to the most prominent peak and cleaned up by spurious effects (cosmic rays). Hence, we evaluated the distance \({{\rm{\Delta }}}_{{\bar{s}}_{j}-{\bar{s}}_{N}}\), i.e. the difference between the average from all the 50 spectra of the scan (\({\bar{s}}_{N}\)) and the average spectrum calculated over a set of j spectra (j < N), extracted from all the possible \((\begin{array}{c}N\\ j\end{array})\) combinations. Finally, for j ≥ 2, we evaluated \({\bar{{\rm{\Delta }}}}_{{\bar{s}}_{j}-{\bar{s}}_{N}}\), value obtained by averaging \({{\rm{\Delta }}}_{{\bar{s}}_{j}-{\bar{s}}_{N}}\) over all the possible combinations. Figure 7 shows the typical behaviour of \({\bar{{\rm{\Delta }}}}_{{\bar{s}}_{j}-{\bar{s}}_{N}}\) as function of j. Notably, it is possible to distinguish three regions, characterised by different slopes. The first one, for 2 < j < 16, exhibiting the highest slope, reflects a strong variability among spectra, so that \({\bar{s}}_{j}\) differs significantly (in terms of relative intensity of Raman features) from \({\bar{s}}_{N}\), so that it cannot be considered representative of the whole cell. For 16 < j < 36, the slope decreases because of the assessment of the relative intensity of the different spectral features. Finally, in the third region, j ≥ 36, approaches to zero (according to its definition) in a fashion depending mainly by the noise superimposed on the spectra. A similar behaviour was obtained for all the 30 analysed cells. In particular, we verified that, for all the 30 cells, \({\bar{{\rm{\Delta }}}}_{{\bar{s}}_{j}-{\bar{s}}_{N}}\) was always in the second region for j = 20, meaning that a good representation of the single cell is provided by averaging 20 SERS spectra of the scan. This outcome provided us the criterium to handle the SERS fluctuations in all the measurements shown in this work.

Typical behaviour of \({\bar{{\rm{\Delta }}}}_{{\bar{s}}_{j}-{\bar{s}}_{N}}\) as function of j.
Handling of interference from culture medium
An important issue to be considered for SERS spectra analysis concerns the role of the protein-rich medium (PBS, eventually mixed with cells secretions) in which cells lie during measurements. This medium gives rise to a detectable SERS signal (see Fig. 8a), clearly identified in this analysis. Hence, by acquiring SERS signals along a line through the cell, starting and terminating in the medium, it is possible to obtain a SERS profile as shown in Fig. 8b. This SERS profile corresponds to the integrated intensity of Amide I band (1620–1680 cm−1). Obviously, the interference from this signal becomes important in the case of poor contact of the cell with the substrate. To face this issue, cells were allowed to adhere to SERS substrate for ~30 min before analysis. Moreover, signals were acquired in the thicker cell region (nuclear region) were cells adhesion was best achieved. In these conditions, a quite good differentiation between signal form cells and from medium was achieved (see Fig. 8).

(a) A typical SERS spectrum acquired in the medium. (b) Integrated intensity of SERS signal acquired along a line across a cell, as shown in the inset. (c) PCA performed on SERS spectra acquired in the cell medium (blue triangles) and CAIX− cells (purple squares). Notably, spectra from the medium, which exhibit a relatively high similarity, occupy a quite reduced space in the PC1-PC2 score plot.
Change history
18 June 2019
A correction to this article has been published and is linked from the HTML and PDF versions of this paper. The error has been fixed in the paper.
References
Engel, A. & Gaub, H. E. Structure and mechanics of membrane proteins. Annu. Rev. Biochem. 77, 127–48 (2008).
Almeida, J. G., Preto, A. J., Koukos, P. I., Bonvin, A. M. & Moreira, I. S. Membrane proteins structures: A review on computational modeling tools. Biochimica et Biophys. Acta (BBA) - Biomembr. 1859, 2020–2039 (2017).
Zhao, Y., Zhang, W., Kho, Y. & Zhao, Y. Proteomic analysis of integral plasma membrane proteins. Anal. Chem. 76, 1817–1823 (2004).
Cutillas, R. P. et al. Proteomic analysis of plasma membrane vesicles isolated from the rat renal cortex. Proteomics 5, 101–112 (2005).
Chattopadhyay, A. & Raghuraman, H. Application of fluorescence spectroscopy to membrane protein structure and dynamics. Curr. Sci. 87, 175–180 (2004).
Moskovits, M. Surface-enhanced spectroscopy. Rev. Mod. Phys. 57, 783–826 (1985).
Stiles, P. L., Dieringer, J. A., Shah, N. C. & Van Duyne, R. P. Surface-enhanced raman spectroscopy. Annu. Rev. Anal. Chem. 1, 601–626 (2008).
Matschulat, A., Drescher, D. & Kneipp, J. Surface-enhanced raman scattering hybrid nanoprobe multiplexing and imaging in biological systems. ACS Nano 4, 3259–3269 (2010).
Fogarty, S. W., Patel, I. I., Martin, F. L. & Fullwood, N. J. Surface-enhanced raman spectroscopy of the endothelial cell membrane. PLOS ONE 9, e106283 (2014).
Hodges, M. D. et al. Combining immunolabeling and surface-enhanced raman spectroscopy on cell membranes. ACS Nano 5, 9535–9541 (2011).
Navas-Moreno, M. et al. Nanoparticles for live cell microscopy: A surface-enhanced raman scattering perspective. Sci. reports 7, 4471 (2017).
Zito, G., Rusciano, G., Pesce, G., Dochshanov, A. & Sasso, A. Surface-enhanced raman imaging of cell membrane by a highly homogeneous and isotropic silver nanostructure. Nanoscale 7, 8593–8606 (2015).
Tong, L., Li, Z., Zhu, T., Xu, H. & Liu, Z. Single gold-nanoparticle-enhanced raman scattering of individual single-walled carbon nanotubes via atomic force microscope manipulation. The J. Phys. Chem. C 112, 7119–7123 (2008).
Managó, S. et al. Bioderived three-dimensional hierarchical nanostructures as efficient surface-enhanced raman scattering substrates for cell membrane probing. ACS Appl. Mater. & Interfaces 10, 12406–12416 (2018).
Liu, Y. et al. Label and label-free based surface-enhanced raman scattering for pathogen bacteria detection: A review. Biosens Bioelectron. 15, 131–140 (2017).
Olson, A., Spies, K., Browning, A., Soneral, P. & Lindquist, N. Chemically imaging bacteria with super-resolution sers on ultra-thin silver substrates. Sci. Reports 7, 41598 (2017).
Le Ru, E. C., Blackie, E., Meyer, M. & Etchegoin, P. G. Surface enhanced raman scattering enhancement factors: A comprehensive study. The J. Phys. Chem. C 111, 13794–13803 (2007).
Ilardi, G. et al. Histopathological determinants of tumor resistance: a special look to the immunohistochemical expression of carbonic anhydrase ix in human cancers. Curr Med Chem 21, 1569–1582 (2014).
Bakker, J., Spits, M., Neefjes, J. & Berlin, I. The egfr odyssey – from activation to destruction in space and time. J. Cell Sci. 130, 4087–4096 (2017).
Du, Z. & Lovly, C. M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol Cancer. 17, s12943 (2018).
Torquato, S. & Stillinger, F. H. Local density fluctuations, hyperuniformity, and order metrics. Phys. Rev. E 68, 041113 (2003).
Zito, G. et al. Nanoscale engineering of two-dimensional disordered hyperuniform block-copolymer assemblies. Phys. Rev. E 92, 050601 (2015).
Kim, T. K. & Eberwine, J. H. Mammalian cell transfection: the present and the future. Anal Bioanal Chem. 397, 3173–3178 (2010).
Sasso, E. et al. Binding of carbonic anhydrase ix to 45s rdna genes is prevented by exportin-1 in hypoxic cells. Biomed Res Int. 674920, 10 (2015).
Jolliffe, I. T. & Cadima, J. Principal component analysis: a review and recent developments. Phil. Trans. R. Soc. A 374, 20150–202 (2016).
Thomas, G. J. Raman spectroscopy of protein and nucleic acid assemblies. Annu. Rev. Biophys. Biomol. Struct. 28, 1–27 (1999).
Buanne, P. et al. Characterization of carbonic anhydrase ix interactome reveals proteins assisting its nuclear localization in hypoxic cells. J. Proteome Res. 12, 282–292 (2013).
Acknowledgements
This work was supported by FIRB 2012-RBFR12WAPY to G.R., MIUR-PRIN2015 and Associazione Culturale DiSciMuS RFC (Biologia dei tumori ipossici) to NZ. The Authors thank A. La Rocca for help in procedure set-up, F. Monteleone for the generation of the EGFR expression vector, M. Raia, G. Scalia, F. Visconte of the CEINGE general and clinical cytometry facility for FACS analysis, and M.G. De Biasi for critical discussion.
Author information
Authors and Affiliations
Contributions
G.R., E.S., N.Z. and A.S. designed research; G.R., E.S., and A.C. performed research; all authors analysed data; G.R, A.S. and N.Z. wrote the paper.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rusciano, G., Sasso, E., Capaccio, A. et al. Revealing membrane alteration in cells overexpressing CA IX and EGFR by Surface-Enhanced Raman Scattering. Sci Rep 9, 1832 (2019). https://doi.org/10.1038/s41598-018-37997-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-37997-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
