
Abstract
Fusarium Head Blight (FHB) caused by Fusarium graminearum pathogens constitutes a major threat to agricultural production because it frequently reduces the yield and quality of the crop. The disease severity is predicted to increase in various regions owing to climate change. Integrated management where biocontrol plays an important role has been suggested in order to fight FHB. P. polymyxa A26 is known to be an effective antagonist against F. graminearum. Deeper understanding of the mode of action of P. polymyxa A26 is needed to develop strategies for its application under natural settings in order to effectively overcome the pathogenic effects. This study aims to re-evaluate a former study and reveal whether compounds other than non-ribosomal antibiotic lipopeptides could be responsible for the antagonistic effect, despite what is often reported. Wheat seedlings were grown to maturity and the spikes infected with the pathogen under greenhouse conditions. The development of FHB infection, quantified via the disease incidence severity and 100-kernel weight, was strongly correlated (r > 0.78, p < 0.01) with the content of the polysaccharide component D-glucuronic acid in the biofilm. Furthermore, while increased inoculum density from 106 to 108 cells/ml did not affect wild type performance, a significant increase was observed with the P. polymyxa mutant deficient in nonribosomal lipopeptide synthesis. Our results show that P. polymyxa A26 biofilm extracellular polysaccharides are capable of antagonizing F. graminearum and that the uronate content of the polysaccharides is of critical importance in the antagonism.
Similar content being viewed by others

Introduction
Fusarium head blight (FHB) is a devastating disease of cereals and is caused by a group of pathogens of which F. graminearum prevails in Nordic countries. The disease severity is predicted to increase in various regions owning to climate change1. FHB is of particular concern because many of the Fusarium species produce mycotoxins that contaminate infected grain and may pose a serious threat to human and domestic animal health. Grain that has been infected with the fungus may become incorporated into our staple diets. For these reasons the antifungal activity of biocontrol agents against FHB pathogens has been extensively studied and several mechanisms suggested2,3,4,5.
Paenibacillus polymyxa is known as an FHB biocontrol agent. It is generally recognised that surface-associated bacteria colonise as biofilms, which are microniches entirely different from their surroundings. This allows the bacteria to work as a functional unit, accomplishing tasks not possible for their planktonic state. Biofilms consist of cells and matrices where complex exopolysaccharides and proteins are major components, and they can provide an important bacterial survival strategy in natural systems6,7,8. P. polymyxa biocontrol ability is often linked to the large bacterial pool of bioactive compounds such as nonribosomal peptides/polyketides (NRPs/PKs)4,9. Despite enormous diversity the compounds have common regulatory systems, as they are produced nonribosomally and require activation by phosphopantetheinyl transferases (PPTase) of which Sfp-type PPTase is required for activation of peptidyl and acyl carrier domains. The broad range of activities of NRPs and PKs includes promotion of adaptation to unfavourable environments. The P. polymyxa A26 strain was isolated from the Evolution Canyon, South Facing Slope, Israel (EC SFS)10,11 (Fig. 1 and Table 1) and has co-evolved with wild progenitors of cereals over a long period of time, helping host plants to adapt and survive under diverse harsh conditions11. The genome analysis reveals regions that reflect these adaptations and the isolate is a superior plant growth promoting bacterium (PGPB) compared to isolates from more moderate environments11. In order to study the performance of the isolate we inactivated its Sfp-type PPTase gene and created a mutant that is incapable of producing enzymatically active 4′-phosphopantetheinyl transferase. This in turn results in a strain lacking enzymatically active NRPs and PKs (A26∆sfp), and the nonribosomal peptide and polyketides, often reported as active metabolites for the biocontrol4,9, are not produced.

The Evolution Canyon (EC) model. (A) Schematic diagram. (B) Cross section view of EC at Lower Nahal Oren, Mount Carmel. (C) Air view of EC (source10: Nevo, 2012 Evolution Canyon,” a potential microscale monitor of global warming across life, PNAS 109; 8) (Photo by E. Nevo).
Formerly we established a screening method for developing FHB biocontrol agents based on combining dual plate assays with a kernel assay12. Using the method, dual plate assays confirmed that NRPS/PKs products have critical importance for the antagonistic activity of A26. Studies with the more complex system employing a wheat grain assay showed, however, that in the case of one FHB-causing pathogen, F. culmorum, biofilm matrix formation may be the biocontrol agent of major importance in antagonising the pathogen. At the same time, only limited antagonism of the other pathogen, F. graminearum, was observed in the wheat grain assay9,12. This raised the question of why, in the case of F. graminearum, biofilm antagonism did not work and only the NRPS/PKs products were of critical importance.
The present study was conducted to re-evaluate the role of the P. polymyxa A26 extracellular matrix in antagonism to F. graminearum. The mutant (A26∆sfp), which lacks the ability for NRP and PK synthesis9, was studied under two inoculum densities, low (106 cells/ml) and high (108 cells/ml). We show that the mutant has comparable antagonistic ability to that of the wild type at the high inoculum density, and that the ability is correlated with the content of uronates in the biofilm polysaccharides. While many studies have been performed aiming to reveal the mechanism of antagonism of the economically important FHB pathogens, to the best of our knowledge this is the first report on the involvement of the uronate components of the biofilm polysaccharides.
Material and Methods
Experimental set-up
Three experimental series were performed:
-
1.
The wheat grain assay was performed at two inoculum densities in order to re-evaluate the role of the A26 extracellular matrix in antagonizing F. graminearum. A26∆sfp was applied at low and high inoculum densities (106 and 108 cells/ml respectively). The results clearly showed the superior effect using the higher inoculum density.
-
2.
Developing biocontrol agents requires combining screening strategies. Hence, greenhouse experiments on wheat plants grown to maturity were performed to test the effect in more natural settings. Wheat head assays evaluating disease severity (DS), disease incidence (DI) and 100-kernel weight (100-kw) were performed. We were further interested in the involvement of exopolysaccharides (EPS), which are the largest group of biofilm components, and along with wheat head assays their biofilm EPS assessment was performed.
-
3.
In order to confirm the role of EPS in disease suppression, A26∆sfp and A26 EPS assays were performed on wheat kernels along with assays of supplied levan and alginate. Commercial samples of levan and sodium alginate were used as reference substrates owing to their contrasting uronate contents (Table S1).
Microbial growth and culture conditions
P. polymyxa A26 was isolated from the South Facing Slope at the natural laboratory called the Evolution Canyon, Israel (Table 1). The Sfp-type 4-phosphopantetheinyl transferase deletion mutant strain was previously generated as described by us earlier9. Stock cultures were stored at −80 °C and were streaked for purity on half strength tryptic soy agar (1/2 TSA, pH 6.2). 10 ml of half strength tryptic soy broth (1/2 TSB, pH 6.2) was inoculated with single cells and grown at 30 ± 2 °C for 12 h. 100 µl of the preinoculum with 108 cells per ml was used for 100 ml flask culture inoculations. Bacterial strains were grown in 1/2 TSB, pH 6.2, 180 rpm at 30 ± 2 °C, for 72 h. Cultures were centrifuged and pellets resuspended in sterile water as described earlier12. Finally, the cultures were adjusted to 106 and 108 cells/ml.
The fungal pathogen F. graminearum strain A602/1998 was obtained from the National Veterinary Institute, Uppsala, Sweden and was previously characterised for virulence12. The pathogen was grown on potato dextrose agar (PDA) plates for seven days at 22 °C. Macroconidia for inoculation were obtained by flooding the surface of colonized agar with sterile or phosphate buffered saline (PBS). The resulting inoculum density of the suspension was 105/ml.
Plant treatment
Winter wheat (Stava) seeds were surface sterilised by a 60 s wash in 99% ethanol, followed by a 6 min wash in 3% sodium hypochlorite solution, a wash in 99% ethanol, and repeated rinsing in sterile water. The seedlings were grown in greenhouse soil in 30 cm diameter pots. After a month of growth, the plants were vernalized for two months. Throughout the growing season the plants were watered daily with a standard nutritional solution. The experiment was performed in four replicates, each consisting of 20 plants. Wheat heads were sprayed with 100 µl of bacterial solutions at either low or high inoculum density (106 and 108 cells per ml respectively) at the beginning of the flowering stage (BBCH 61). The control plant heads were treated with 100 µl of sterile water. One week after the antagonist treatment (end of flowering stage) 30 µl of macroconidia suspension was used to inoculate a single central floret on each wheat head (four replicates each containing twenty plants). Controls were heads treated only with water or with pathogen suspension. Inoculated spikes were misted with water and covered with plastic bags for 10 days. Immediately before the pathogen inoculation, A26 and A26∆sfp biofilms from twenty wheat heads from each treatment were carefully flushed into 10 ml sterile water in 50 ml Falcon tubes for the biofilm studies. As controls, untreated wheat heads were flushed with sterile water. Counting of bacterial colony forming units was performed on ½ TSB plates. The biofilm bacteria were re-isolated by selective plating and confirmed by PCR as described by us earlier13. Wheat heads were collected 21 days after inoculation at the fully ripe stage (BBCH 89) and scored for disease incidence and severity on a scale from 0 to 100%. Disease severity was evaluated based on visual assessment of heads exhibiting FHB disease symptoms. F. graminearum presence was randomly confirmed using PCR12 and fluorometric assay. After the FHB evaluation the heads were allowed to dry and 100-kernel weights were determined.
Wheat head biofilm assessment, and polysaccharide and glucuronate content evaluation
Wheat heads were flushed with 10 ml deionised sterile water immediately before the pathogen treatment in order to collect the biofilms developed after the A26 and A26∆sfp inoculations. The biofilm bacteria were re-isolated by selective plating, confirmed by PCR as described9,13, quantified, the supernatant polysaccharides isolated and D-glucuronate (D-GA) contents recorded. The biofilm samples in water were used for polysaccharide isolation as described earlier14. The pellet was lyophilised, weighed and stored at −4 °C until uronic acid content was assessed. The assay was based on hydrolysis of glucoside bonds that bind the polysaccharides. This step digests the polysaccharides into their component monosaccharides. Analysis of uronic acid content was performed as described earlier by Mojica et al.15 with small modifications. Briefly, the biofilm pellets were weighed and dissolved in 200 µl of deionised water. Potassium sulfamic acid (4 M, pH 1.6) was added to the biofilm solution and mixed by vortexing. Then sodium tetraborate solution in concentrated sulfuric acid (0.0125 M) was added. The solutions were incubated for 5 min in a 100 °C water bath, cooled on ice for 3 min and centrifuged at 2,000 rpm for 10 min after which 20 µl hydroxyphenol solution (0.15% v/v) was added to the supernatant. The solution was then mixed gently and the absorbance read at 520 nm. Each data point represents the average of twenty replicate measurements.
P. polymyxa A26∆sfp cell and polysaccharide assay on wheat grains
The experiments were performed as reported earlier12. Briefly, conical flasks containing 20 g wheat grains were inoculated with 15 ml A26∆sfp at 106 or 108 cells/ml. For the polysaccharide assay, the kernels were treated with 15 ml of A26∆sfp and A 26 polysaccharide solution with the titres 10 µg/ml. Cells were grown and polysaccharides isolated as indicated below. Controls were treated with sterile water. In order to study the effect of uronates, two reference polymers contrasting in uronate content were applied: alginate (Sigma) and levan (Sigma). The polysaccharide powders were dissolved in deionized sterile water to the final concentration 15 µg/ml. 20 g wheat grains were mixed with 15 ml of the polysaccharide solution.
Flasks were incubated at room temperature for 8 h, and then inoculated with 1 cm2 agar plugs from 2 week old cultures of F. graminearum.
EPS extraction
EPS extraction was performed as described earlier with small modifications14. Briefly, bacterial cultures were diluted 1:5 with distilled water and centrifuged for 30 min at 17,600 g at 20 °C to separate cells. Then, EPS were precipitated by slowly pouring the supernatant into two volumes of isopropanol while stirring at 200 rpm. The filtered polysaccharide was suspended in a digestion solution consisting of 0.1 M MgCl2, 0.1 mg/ml DNase, and 0.1 mg/ml RNase solution, and incubated for 4 h at 37 °C. Samples were extracted twice with phenol-chloroform, lyophilised using a Virtis SP Scientific 2.0 freeze dryer, taken to the initial volume and dialysed against distilled water.
DNA extraction and quantification
The grain samples were freeze-dried and ground into a fine powder using a Precellys 24 homogenizer (Bertin Technologies, France). Samples were lysed by incubating 100 mg powder for 15 min in 350 μl of glucose buffer as described by us earlier16. DNA was extracted using a hexadecyl-trimethyl-ammonium bromide-based method17. The bacterial strains were confirmed by PCR as described by us earlier13. Fungal growth was assessed visually and randomly verified using PCR and sequencing after 10 days of growth12. For F. graminearum PCR ITS1F and ITS4 primers18 (5′-CTT GGT CAT TTA GAG GAA GTAA-3′ and 5′-TCC TCC GCT TAT TGA TAT GC-3′) were used. Initial denaturation at 95 °C was followed by 35 amplification cycles: denaturation at 95 °C for 30 sec, annealing at 58 °C for 30 sec, and extension at 72 °C for 30 sec, followed by an extension step of 72 °C for 7 min. The reaction mix contained 200 µm dNTP, 2.75 mM MgCl2, 0.025 U/µl polymerase (DreamTaq Green, Thermo Scientific, Waltham, MA, USA) and 0.2 µM of each primer. For the pathogen quantification the dsDNA of the PCR mix was purified using a Nucleo Spin kit (Macherey-Nagel, PA, USA) and quantified using the Invitrogen™ Qubit™ 4 Fluorometer according to the manufacturer’s instructions.
Qualitative determination of biofilm composition by UHPLC-MS+
The chromatographic analyses have been performed using a liquid chromatograph (Nexera X2, Shimadzu, Tokyo, Japan) equipped with a diode array detector (M30A, Shimadzu, Tokyo, Japan) and a mass spectrometer (Model 8040, Shimadzu, Tokyo, Japan). The separation of compounds was performed on Nucleodur 100-5-NH2-RP column (4.6 mm i.d. × 250 mm column length, 5 µm particle size, Macherey-Nagel GmbH, Duren, Germany). The column temperature was maintained at 35 °C and the flow rate at 1 ml min−1. The solvents used for the chromatographic elution consisted of ultra-pure water with 0.1% TFA (A) and acetonitrile (B). The chromatographic elution program used was an isocratic one, with 25% A and 75% B, for 25 minutes. The injected volume of sample and standards was 10 µl. The DAD detector spectra were recorded between 200 and 600 nm. The mass spectrometer was equipped with an electrospray ionization (ESI) source operated in positive ion mode, and quantification was carried out in the multiple reaction monitoring (MRM) mode. The mass range was between m/z 15 and 1990. The ion spray temperature was maintained at 250 °C. The drying gas flow rate was 10 L/min.
Evaluation of A26∆sfp and A26 EPS and sodium alginate water holding capacity (WHC)
Bacterial cultures were grown and harvested, and polysaccharides isolated, as described above. 600 mg of A26∆sfp and A26 EPS, sodium alginate (Sigma) and levan (Sigma) were mixed with 100 g sand and determined at different wetting cycles for 24 h. The sand and biopolymer mixture was then allowed to drain for 30 min. and the weight after saturation was recorded. Following the wet weight estimation the mixture was dried in an oven, cooled in a desiccator and weighed again. Each experiment was carried out in triplicate. WHC= gain in weight at saturation point/dry weight of soil × 100.
Data confirmation and validation
To ensure reproducibility, greenhouse experiments were performed in four replicates each containing twenty plants. Twenty biological replicates of each D-glucuronate detection experiment were performed. For the wheat grain assay three biological replicates were performed. Replicated data were studied for normal distribution and analyzed by Unscrambler X15.1 and MiniTab17. One way analysis of variance (ANOVA) and a post-hoc LSD test was used to identify treatments that were significantly different from controls at p ≤ 0.05. Linear regressions (Unscrambler X15.1) were used to determine the relationships between antagonistic parameters and D-glucuronic acid content.
Results
P. polymyxa A26∆sfp antagonism against F. graminearum by kernel assay
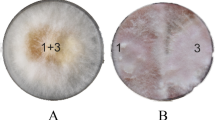
The experiment was performed following our former results on A26∆sfp F. graminearum wheat grain assay where grains inoculated with A26∆sfp showed limited pathogen antagonism12. Two initial A26∆sfp inoculum densities, 106 and 108 cells/ml, were used in the assay. Visual inspection of wheat grains over the experimental period revealed F. graminearum mycelial overgrowth in the control treatments. While limited mycelial growth was observed with A26∆sfp at 106 initial inoculum density, no fungal mycelial growth was observed at 108 A26Sfp inoculum density (Fig. 1A and Table S1). The wheat grain assay was carefully performed under axenic conditions in order to avoid contaminants from outside. F. graminearum growth randomly confirmed by PCR assays followed by QubitTM Fluorometer quantification revealed up to 0.3 ng/µl quantities of the pathogen DNA versus more than 10 ng/µl of the F. graminearum control treatment in the case of high initial inoculum (108). Similarly to the assay performed by us earlier12, F. graminearum was detected around 3 ng/µl at low inoculum density (106). The results show that the A26 mutant, deficient in nonribosomal compound synthesis, is still capable of efficiently antagonising the FHB-causing pathogen F. graminearum in the kernel assay when provided at 108 inoculum density (Fig. 2A and Table S1).

Fusarium graminearum antagonism in kernel assay. (A) Antagonistic activity of Paenibacillus polymyxa A26∆sfp at two inoculum densities, 106 and 108 cells/ml; (B) Antagonistic activity of P. polymyxa A26∆sfp and A26 extracellular polysaccharides (EPS) 15 µg/ml after 10 days incubation. See Material and Methods.
Antagonistic activity of P. polymyxa A26∆sfp and P. polymyxa A26 in the greenhouse assay
In order to confirm the results with A26∆sfp in reference to its wild-type A26 antagonistic abilities in the wheat grain assay, the experiment was performed in more natural settings, in the greenhouse. The winter wheat Stava seeds were vernalized and grown by standard procedures until the flowering stage. Then the wheat heads were sprayed with A26∆sfp and A26 at the two inoculum densities used in the wheat grain assay (106 and 108). Anthesis is the stage of the greatest susceptibility to FHB, as anthers are the common entry route into the plant. Hence, one week after the antagonist treatment (end of flowering stage) 30 µl of macroconidia suspension was used to inoculate a single central floret on each wheat head (four replicates each containing twenty plants). Wheat heads were harvested 21 days after the inoculation at the fully ripe stage and evaluated for disease incidence, disease severity and 100-kernel weight. A26 efficiently antagonises F. graminearum and the effect is not dependent on initial inoculum density (Figs 3–5 and Table S1). As shown by the disease incidence (DI), disease severity (DS) and 100- kernel weight (100-kw) (53, 10, and 3.5 respectively) the F. graminearum pathogen was equally well antagonised at both A26 inoculum densities (Figs 3–5 and Table S1). While highly similar antagonistic parameters of DI, DS and 100-kw (53, 10 and 3.6 respectively) were scored for A26∆sfp at the higher inoculum density (108), up to 25% reduced antagonistic ability was observed at 106 density (78, 30 and 2.9 respectively) (Figs 3–5 and Table S1). No disease symptoms were scored on plants after sole A26 treatment or after A26Sfp treatment, at either of the two inoculum densities, or after water treatment (Table S1). 100-kw of the efficient pathogen treatments was highly similar to the kernel weight of control treatments with sole A26 or with A26Sfp or water (3.4 ± 0.6 g).

Fusarium Head Blight disease incidence (DI) in greenhouse assay. Boxplot of DI scores of wheat plants after Paenibacillus polymyxa A26 and P. polymyxa A26∆sfp treatments at two inoculum densities, 106 (Low) and 108 (High) cells/ml. ANOVA univariate analysis was performed and post-hoc LSD tests used to identify treatments significantly different from pathogen control (p < 0.05). Different letters indicate statistically significant differences. Error bars represent standard deviations.

Fusarium Head Blight disease severity (DS) in greenhouse assay. Boxplot of DS scores after Paenibacillus polymyxa A26 and P. polymyxa A26∆sfp treatments at two inoculum densities, 106 (Low) and 108 (High) cells/ml. ANOVA univariate analysis was performed and post hoc LSD test used to identify treatments significantly different from pathogen control (p < 0.05). Different letters indicate statistically significant differences. Error bars represent standard deviations.

100 kernel weight (100-kw) in greenhouse assay. 100- kw after Paenibacillus polymyxa A26 and P. polymyxa A26∆sfp treatment at two inoculum densities, 106 (Low) and 108 (High) cells/ml. ANOVA univariate analysis was performed and post-hoc LSD tests used to control (p < 0.05). Different letters indicate statistically significant differences. Error bars represent standard deviations.
Wheat head biofilm assessment
In order to follow the performance of A26∆sfp and its wild type A26, wheat heads were flushed with water immediately before the pathogen treatment in order to collect the biofilms developed after the A26 or A26∆sfp inoculations. The biofilm bacteria were re-isolated by selective plating, confirmed by PCR as described13, quantified, supernatant polysaccharides isolated and D-glucuronate content recorded (Table 2). A26∆sfp and its wild type A26 (103 per ml) were re-isolated in the wheat head biofilms (Table 2). About 30% higher EPS content was detected in A26∆sfp biofilms in comparison to its wild type A26 strain at both initial inoculum densities (Table 2). While neither the EPS content of A26∆sfp nor that of A26 varied significantly according to the initial inoculum density, the D-GA content of the mutant was about 40% higher at 108 inoculum density. The D-GA comprised 3.5% of its EPS at 106 inoculum density while the content was 6% at 108 inoculum density (Table 2 and Fig. 6). The D-GA content in A26 was significantly lower than in A26∆sfp. i.e. 3% of A26 EPS (Table 2 and Fig. 6). The D-GA content correlated with A26∆sfp antagonistic activity (r > 0.78 p < 0.01). Additionally, the EPS titre and its D-GA content in the native biofilm layers of the control plant spikes were evaluated (Fig. 6 and Table 2). The native biofilms of all the control spikes comprised around 6% D-GA (Table 2 and Fig. 6).

D- glucuronate (D-GA) content in wheat axis biofilms. D-GA content of Paenibacillus polymyxa A26 and A26∆sfp wheat axis biofilms at two inoculum densities, 106 (Low) and 108 (High) cells/ml. ANOVA univariate analysis was performed and post-hoc LSD test used to identify treatments significantly different from water control (p < 0.05). Different letters indicate statistically significant differences. Error bars represent standard deviations.
P. polyFig.myxa A26∆sfp and P. polymyxa A26 polysaccharide fragment assessment
UHPLC-ESI-MS under positive ionization was used to determine fragment ions. Figure 7 and Table 3 depict the Q1 mass spectra of the various major chromatographic fractions (Supplementary Information). In positive ion mode, no protonated molecules [M + H]+ were detected, but we clearly identified ammonium-adducted molecules [M + NH4]+, including those at m/z ratios of 191, 388, 567, and 722. The analyses demonstrated that A26 and A26Sfp biofilm EPS at low and high inoculum densities comprised monomers (176 Da), dimers (352 Da), trimers (528 Da) and tetramers (704 Da). There were no significant quantities of oligomers larger than tetramers.
Antagonistic activity of P. polymyxa A26∆sfp and A26 polysaccharides in the kernel assay
The greenhouse A26∆sfp treatment at the high inoculum density resulted in significant increases in spike biofilm EPS D-GA. This correlated with the treatment antagonistic activity. Hence, further studies were performed to study the effect using EPS isolated from A26∆sfp and A26 flask cultures, as well as commercial sodium alginate and levan as reference substrates with contrasting uronate content (Table 2). The results show that 15 µg/ml EPS treatments of both A26∆sfp and A26 efficiently antagonise the F. graminearum pathogen (Fig. 2B and Table S1). While the levan treatment revealed fungal overgrowth comparable to that in the F. graminearum control treatment, no pathogen was visually detected in the assay with commercial sodium alginate. F. graminearum growth, randomly confirmed by PCR assays followed by fluorometric quantification, revealed up to 0.3 ng/µl quantities of the pathogen DNA in A26∆sfp and A26 EPS and alginate assay versus more than 10 ng/µl with the levan and F. graminearum control treatment (Table S1).
P. polymyxa A26∆sfp and A26 polysaccharide water holding capacity (WHC)
Addition of 0.6% (vol) biopolymers significantly increased WHC of sand. The A26 and A26Sfp enhanced the WHC about twice. The result is comparable to the enhancement by commercial sodium alginate WHC (Table 4). Levan treatment did not improve the sand WHC (Table 4).
Discussion
Here we re-evaluated the modes of antagonism of the P. polymyxa wild type A26 and its mutant A26∆sfp against F. graminearum (Figs 2–5). While A26 antagonizes effectively at both low (106 cells/ml) and high (108 cells/ml) densities of inocula used in the study, A26∆sfp F. graminearum antagonism at 108 inoculum density leads to up to 25% improved antagonistic ability, reaching the ability of the wild type to antagonize F. graminearum (Figs 2–5). At the same time, 103 bacteria of both bacterial strains at both inoculum densities were recovered from the wheat heads one week after inoculation (Table 2). It is commonly known that even though bacteria are crucial for biofilm formation, their measurement is insufficient to quantify biofilms. The EPS which form the major part of the microbial biofilm matrix19 were detected in the spike biofilm water solutions. The A26 biofilm contains 1 µg and A26 Sfp 1.5 µg of EPS per ml (Table 2). The significant increase in the mutant biofilm matrix production is in accordance with our former findings9. Owing to the huge complexity of EPS matrix components, their detailed quantification is a challenge. D-glucuronate (D-GA) has previously been suggested as a proxy for biofilm comprehensive screening and uronic acid is widely determined as representative of myco-polysaccharides in biofilms15. The assay is based on the effectiveness of chemical bond disruption and digestion of polysaccharides into component monosaccharides15. Significant differences were detected in A26∆sfp D-GA at the two inoculum densities (Fig. 6 and Table 2). The D-GA comprised 6% of EPS at the 108 inoculum density in comparison to 3.5% at the 106 inoculum density (Fig. 6 and Table 2). The result correlates well with the ability of the mutant to antagonize F. graminearum (r > 0.78, p < 0.001). The EPS titre and its D-GA content in the native biofilm layers of the control plant spikes and of the spikes prior to A26 or A26Sfp treatment were evaluated (Fig. 6 and Table 2). It is interesting to note that all the native biofilms on control spikes, similar to A26Sfp high inocula biofilms, comprised around 6% D-GA. In order to further investigate the role of uronates, two commercially available reference polymers levan and alginic acid were used. Both EPS components can be found in P. polymyxa biofilms20 The commercial sugars contrast in D-GA content (Table 2). Levan is a polymer of fructose while alginate is comprised of uronates. While the alginate treatment fully antagonized the pathogen, fungal overgrowth was observed in the case of the levan treatment. The results further confirm the role of uronates regarding the antagonistic effect against F. graminearum.
How do we reconcile the outcome that 100 times initial inoculum density differences result in significant increases in the D-GA production of A26∆sfp? The interesting phenomenon that differences in initial inoculum density can lead to variable metabolism that cannot be linked to the inocula growth stage, has been reported earlier by Jeon et al.21. The P. polymyxa GBR-1 β-amylase gene was expressed only at a high inoculum density (108 per ml). The gene was not expressed with the low density inoculum (106)21. This phenomenon is certainly connected with complex biofilm biology and requires further study using parallel sequencing and high-resolution microscopy. However, even though the D-GA composition of the EPS matrix varies between the A26 and A26∆sfp strains, their isolated EPS, applied in surplus, induce resistance to F. graminearum in the wheat kernel assay (Fig. 2B).
It is widely recognised that uronic acid moieties affect the physiological and biological properties of polysaccharides. The uronates with high content of EPS, e.g. alginate and xanthan, result in increased water holding capacity20 It is believed that uronic acid backbones lead to changes in other sugar backbones, which eventually will result in alteration of their properties and bioactivity22. It is well known that EPS biosynthesis generally involves a very sophisticated network dependent on complex factors23. EPS are combinations of monomers and polymers consisting of sugar compounds connected via glyosidic linkages14,24,25. EPS length, composition and formation vary considerably26,27,28. The main factors that influence EPS interactions include the charges, polymer functional groups, backbone and chain fitness, and the relative concentrations of the constituents14,24,25. Hence EPS are not random co-polymers but vary and occur in combinations dependent on the various C and N sources used for bacterial growth24,25. We were interested in the fragment length of the EPS in A26 and A26∆sfp wheat head biofilms. The analyses demonstrated that in biofilms from both the strains, the primary compounds were monomers (176 Da), dimers (352 Da), trimers (528 Da) and tetramers (704 Da). There were no significant quantities of oligomers larger than tetramers (Fig. 7, Table 3).

Typical examples of mass spectra recorded for diverse fractions of Paenibacillus polymyxa A26 and A26∆sfp wheat spike biofilms at low and high inoculum densities. Note the data presented also in Table 3 (A-I, B-II, C-III, D-IV, E-V). Aqueous solutions of biofilms (10 μl) were injected into the UHPLC–ESI-MS equipment. See Material and Methods.
Microorganisms in natural environments are subjected to various fluctuations in environmental conditions. It has been suggested that the ecological ‘success’ of EPS depends on their potential to favorably influence the microorganisms’ adaptation to the environment29. The EPS matrix serves as the microbial interface with the environment. In addition to environmental adaptation, EPS are also used for social skills communication, compartmentalization, competence and defense19. As the production of EPS requires copious amounts of energy, their regulatory control is important and there are many levels of EPS synthesis. Different subpopulations of eps- producing genes are activated during the different stages of biofilm development30. Several EPS molecules have high WHC which can be even as high as 15 times their weight19,20. WHC water accumulation can protect against water stress and was suggested by us as a mechanism of the drought tolerance enhancement of EC, SFS rhizobacterial strains9,10. The strains from the Hordeum spontaneum rhizosphere are likely to have machinery contributing to the wild cereal progenitors’ adaptation to the unfavorable environment9. In addition to the alleviation of plant drought stress, the strain WHC contributes to nutrient availability, maintaining water potential and physiological processes19. In our experiment the D-GA-containing EPS layer with high WHC could provide a physical barrier against F. graminearum pathogens. UHPLC-ESI-MS fragment assessments of the wheat head biofilm EPS after inoculation with A26 or A26Sfp at low and high inoculum densities showed that all biofilm EPS contain the mono-, di-, tri-, and tetramer fragment mixture (Fig. 7 and Table 3). Yet it is hard to speculate which types of D-GA-containing EPS were produced by the bacterial strains in the assay. It is clear that both A26∆sfp and A26 EPS are capable of absorbing water. The study in which sand was mixed with the biopolymers showed that A26∆sfp and A26 EPS have high WHC, which is similar to sodium alginate WHC (Table 4). It is interesting to note that the treatment with the commercial low titre (15 µg/ml1) reference substrate alginate resulted in protection against the pathogen in the model system. The result supports the idea that a biocontrol agent EPS matrix with high WHC is one of the critical factors providing a mechanism against F. graminearum pathogens. Several research groups have considered the strategy of application of pure EPS19. The work has mostly not resulted in consistent results owing to the complexity of the physical and chemical conditions of the natural environments as well as to the presence of native microbial communities which metabolize the EPS19. On the other hand, it is possible to engineer the EPS production of the native biofilm formers via the nutrient supply14.
Here we present a study where the initial observations of the antagonism of the P. polymyxa A26 biofilm to F. graminearum in the kernel assay are examined at a coarser scale in the greenhouse, and at a finer scale in experiments with commercial EPS application. We show that A26 biofilm EPS are capable of antagonizing F. graminearum and that the uronate content of the EPS is of critical importance in the antagonism.
It is clear that fundamental understanding of the genes and mechanisms of real-time biofilm formation is needed to explain the performance of the P. polymyxa native isolate A26 and its mutant A26∆sfp. Previous studies have shown that the multifaceted information about the bacterial biofilm cannot simply be adopted from studies of their domesticated strains31. It is possible that the A26 uronate-containing EPS matrix is required for efficient antagonism in the cases where the more common mechanisms of antagonism with antibiotic lipopeptides4,9 are not available (as with A26∆sfp). This would be in accordance with the fact that surplus EPS was used in the grain assay. How much the EPS produced by wild type P. polymyxa A26 contribute to its F. graminearum antagonism under different environmental conditions remains to be elucidated. The study here indicates that along with the lipopeptides4,9, the bacterial biofilm EPS compounds are capable of antagonizing F. graminearum and that the uronate content of the polysaccharides is of critical importance.
Bacterial EPS is a promising class of sustainable biopolymers to meet various industrial/agricultural requirements. Considering the presence of bacterial natural surface biofilms and that only about 1% of the bacteria can at present be cultured, many EPS are yet to be identified23,32. Even though the EPS layer is dependent upon the perception of numerous environmental signals from the host and the ecosystem, this would open the new range of application of EPS in integrated pathogen management protecting against stress factors under climate change.
Conclusions
Paenibacillus polymyxa A26 represents a promising biocontrol agent for addressing the many challenges F. graminearum poses to agricultural crop yield and quality. However, to capitalize on this potential, an improved understanding of the mechanism of action is needed. This study demonstrates that the biofilm polysaccharides containing D-GA significantly contribute to antagonism and that the initial inoculum density plays a significant role in this.
References
Chen, L., Heng, J., Qin, S. & Bian, K. A comprehensive understanding of the biocontrol potential of Bacillus velezensis LM2303 against Fusarium head blight. PLoS One 13, e0198560 (2018).
Palazzini, J., Torres y Torres Lara, C. & Chulze, T. Biological Control of Fusarium Head Blight of Wheat: From Selection to Formulation. (Springer, Dordrecht 2013).
Khan, N. et al. Antifungal activity of Bacillus species against Fusarium and analysis of the potential mechanisms used in the biocontrol. Frontiers in Microbiol (2018).
Mousa, W. K., Shearer, C. R., Limay-Rios, V., Zhou, T. & Raizada, M. N. Bacterial endophytes from wild maize suppress Fusarium graminearum in modern maize and inhibit mycotoxin accumulation. Frontiers in plant science 6, 805 (2015).
He, J., Boland, G. J. & Zhou, T. Concurrent selection for microbial suppression of Fusarium graminearum, Fusarium head blight and deoxynivalenol in wheat. J Appl Microbiol 106, 1805–1817 (2009).
Branda, S. S., Vik, S., Friedman, L. & Kolter, R. Biofilms: the matrix revisited. Trends Microbiol 13, 20–26 (2005).
Davey, M. E. & O’Toole, G. A. Microbial biofilms: from ecology to molecular genetics. Microbiol Mol Biol Rev 64, 847–867 (2000a).
Redmile-Gordon, M., Brooks, B., Evershed, R., Goulding, K. & Hirsch, P. R. Measuring the soil-microbial interface: Extraction of extracellular polymeric substances (EPS) from soil biofilms. Soil Biol Biochem 72, 163–171 (2014).
Timmusk, S. et al. Sfp- type PPTase inactivation promotes bacterial biofilm formation and ability to enhance plant drought tolerance Frontiers in Microbiol, 1–13 (2015).
Nevo, E. “Evolution Canyon,” a potential microscale monitor of global warming across life. Proc Natl Acad Sci USA 109, 2960–2965 (2012).
Timmusk, S. et al. Bacterial distribution in the rhizosphere of wild barley under contrasting microclimates. PLoS One 6, 1–8 (2011).
Abd El-Daim, I., Haggblom, P., Karlsson, M., Stenstrom, E. & Timmusk, S. Paenibacillus polymyxa A26 Sfp-type PPTase inactivation limits bacterial antagonism against Fusarium graminearum but not of F. culmorum Front. Plant Sci., 1–8 (2015).
Timmusk, S. et al. Drought-tolerance of wheat improved by rhizosphere bacteria from harsh environments: enhanced biomass production and reduced emissions of stress volatiles. PloS ONE 1–13 (2014).
Rutering, M., Schmid, J., Ruhmann, B., Schilling, M. & Sieber, V. Controlled production of polysaccharides-exploiting nutrient supply for levan and heteropolysaccharide formation in Paenibacillus sp. Carbohydr Polym 148, 326–334 (2016).
Mojica, K., Elsey, D. & Cooney, M. J. Quantitative analysis of biofilm EPS uronic acid content. J Microbiol Methods 71, 61–65 (2007).
Timmusk, S., Paalme, V., Lagercrantz, U. & Nevo, E. Detection and quantification of Paenibacillus polymyxa in the rhizosphere of wild barley (Hordeum spontaneum) with real-time PCR. J Appl Microbiol 107, 736–745 (2009).
Nygren, C. M. et al. Growth on nitrate and occurrence of nitrate reductase-encoding genes in a phylogenetically diverse range of ectomycorrhizal fungi. The New phytologist 180, 875–889 (2008).
Gardes, M. & Bruns, T. D. ITS primers with enhanced specificity for basidiomycetes–application to the identification of mycorrhizae and rusts. Mol Ecol 2, 113–118 (1993).
Costa, O. Y. A., Raaijmakers, J. M. & Kuramae, E. E. Microbial Extracellular Polymeric Substances: Ecological Function and Impact on Soil Aggregation. Frontiers in microbiology 9, 1636 (2018).
Liang, T. W. & Wang, S. L. Recent advances in exopolysaccharides from Paenibacillus spp.: production, isolation, structure, and bioactivities. Mar Drugs 13, 1847–1863 (2015).
Jeon, Y. H., Kim, S. G., Hwang, I. & Kim, Y. H. Effects of initial inoculation density of Paenibacillus polymyxa on colony formation and starch-hydrolytic activity in relation to root rot in ginseng. J Appl Microbiol 109, 461–470 (2010).
Xu, X. et al. Unsaturated guluronate oligosaccharide enhances the antibacterial activities of macrophages. FASEB J 28, 2645–2654 (2014).
Schmid, J., Sieber, V. & Rehm, B. Bacterial exopolysaccharides: biosynthesis pathways and engineering strategies. Frontiers in microbiology 6, 496 (2015).
Ruhmann, B., Schmid, J. & Sieber, V. High throughput exopolysaccharide screening platform: from strain cultivation to monosaccharide composition and carbohydrate fingerprinting in one day. Carbohydr Polym 122, 212–220 (2015).
Ruhmann, B., Schmid, J. & Sieber, V. Methods to identify the unexplored diversity of microbial exopolysaccharides. Frontiers in microbiology 6, 565 (2015).
Yegorenkova, I. V., Tregubova, K. V., Burygin, G. L., Matora, L. Y. & Ignatov, V. V. Assessing the efficacy of co-inoculation of wheat seedlings with the associative bacteria Paenibacillus polymyxa 1465 and Azospirillum brasilense Sp245. Can J Microbiol 62, 279–285 (2016).
Yegorenkova, I. V., Tregubova, K. V., Matora, L. Y., Burygin, G. L. & Ignatov, V. V. Biofilm formation by Paenibacillus polymyxa strains differing in the production and rheological properties of their exopolysaccharides. Curr Microbiol 62, 1554–1559 (2011).
Yegorenkova, I. V., Tregubova, K. V., Matora, L. Y., Burygin, G. L. & Ignatov, V. V. Use of ELISA with antiexopolysaccharide antibodies to evaluate wheat-root colonization by the rhizobacterium Paenibacillus polymyxa. Curr Microbiol 61, 376–380 (2010).
Geoghegan, M. et al. The polymer physics and chemistry of microbial cell attachment and adhesion. Faraday Discuss 139, 85–103; discussion 105–128, 419–120 (2008).
Chai, Y., Chu, F., Kolter, R. & Losick, R. Bistability and biofilm formation in Bacillus subtilis. Mol Microbiol 67, 254–263 (2008).
Aguilar, C., Vlamakis, H., Losick, R. & Kolter, R. Thinking about Bacillus subtilis as a multicellular organism. Curr Opin Microbiol 10, 638–643 (2007).
Stewart, E. J. Growing unculturable bacteria. J Bacteriol 194, 4151–4160 (2012).
Acknowledgements
The work was performed with financial support from the Carl Tryggers Stiftelse för Vetenskaplig Forskning, Swedish Research Council 2014-04035, FORMAS 222-2014-1326 and the European Commission and the Romanian Government (POSCCE 621/2014). We are indebted to Dr. D. Clapham for critically reading the manuscript.
Author information
Authors and Affiliations
Contributions
All Authors made equal contribution to the planning and development of experiments to investigate the original idea by S.T. D.C. and L.C. contributed most to the chemical aspects. S.T., T.T., E.N. and L.B. contributed most to the biological aspects.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Timmusk, S., Copolovici, D., Copolovici, L. et al. Paenibacillus polymyxa biofilm polysaccharides antagonise Fusarium graminearum. Sci Rep 9, 662 (2019). https://doi.org/10.1038/s41598-018-37718-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-37718-w
This article is cited by
-
Phenazine 1-carboxylic acid Producing Seed Harbored Endophytic Bacteria from Cultivated Rice Variety of Kerala and Its Broad Range Antagonism to Diverse Plant Pathogens
Probiotics and Antimicrobial Proteins (2023)
-
Identification and combinatorial engineering of indole-3-acetic acid synthetic pathways in Paenibacillus polymyxa
Biotechnology for Biofuels and Bioproducts (2022)
-
Effect of exopolysaccharides of Paenibacillus polymyxa rhizobacteria on physiological and morphological variables of wheat seedlings
Journal of Microbiology (2021)
-
Complete Genome Sequence of Paenibacillus sp. JZ16, a Plant Growth Promoting Root Endophytic Bacterium of the Desert Halophyte Zygophyllum Simplex
Current Microbiology (2020)
-
The impact of Paenibacillus polymyxa HY96-2 luxS on biofilm formation and control of tomato bacterial wilt
Applied Microbiology and Biotechnology (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
