
Abstract
We constructed a standardized protein folding kinetics database (PFDB) in which the logarithmic rate constants of all listed proteins are calculated at the standard temperature (25 °C). A temperature correction based on the Eyring–Kramers equation was introduced for proteins whose folding kinetics were originally measured at temperatures other than 25 °C. We verified the temperature correction by comparing the logarithmic rate constants predicted and experimentally observed at 25 °C for 14 different proteins, and the results demonstrated improvement of the quality of the database. PFDB consists of 141 (89 two-state and 52 non-two-state) single-domain globular proteins, which has the largest number among the currently available databases of protein folding kinetics. PFDB is thus intended to be used as a standard for developing and testing future predictive and theoretical studies of protein folding. PFDB can be accessed from the following link: http://lee.kias.re.kr/~bala/PFDB.
Similar content being viewed by others


Introduction
Protein folding is one of the most difficult problems in biophysics and molecular biology. Due to the accumulation of over half a century’s experimental data on reversible folding-unfolding mechanisms1,2, at least 16 protein folding kinetics datasets have been reported3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19. However, there are many problems in these datasets, including variations in temperatures (from 5 °C to 75 °C) used in kinetic folding experiments, redundant data entries, and inadequate reported data. A more complete dataset of protein folding kinetics with corrections for the above problems is thus required, and once we have such a dataset, it will be very useful for developing and testing future predictive and theoretical studies of protein folding.
Here, we thus carefully examined the existing protein folding datasets, and introduced the necessary corrections. Among the available datasets, ACPro19 and the dataset by Garbuzynskiy et al.17 (hereinafter referred to as the Garbuzynskiy dataset) were the most recent ones, which contained the most updated and largest entries. Therefore, we utilized these two datasets in the current study to construct a new database called PFDB. Furthermore, we added new protein data into the PFDB from our own collection based on extensive literature search, which resulted in the entry size of 141 globular proteins in our dataset; whose size is the biggest among the currently available protein folding datasets.
In this study, we also developed a new temperature correction method for the proteins whose kinetic folding and unfolding experiments had been carried out at a temperature different from the standard temperature (25 °C). Our temperature correction method is based on the Eyring–Kramers equation20, and the logarithmic rate constants of folding and unfolding, ln(kf) and ln(ku), respectively, at 25 °C is provided for all proteins in PFDB. Interestingly, the present study is the first to introduce the temperature corrections into the protein folding dataset, and we show that the introduction of the temperature correction has improved the quality of the database. PFDB is thus currently the most updated database of protein folding kinetics, and hence it can be used as a standard for developing future predictive and theoretical studies of protein folding.
Results and Discussions
Database construction and descriptions
We first combined the two most recent datasets of protein folding, the ACPro and Garbuzynskiy datasets, to construct the combined dataset (hereafter called “the AG dataset”) in which redundant or inappropriate entries were filtered out. We excluded the proteins containing disulfide linkages or covalently bound prosthetic groups, because the presence of these linkages or groups can significantly affect the folding kinetics. Small polypeptides with less than 34 residues were also excluded. We carefully examined each data in the AG dataset. For instance, if there is no updated protein folding kinetics data available for a protein, we included those proteins as such in PFDB, otherwise replaced with the updated data. Furthermore, we added the data of 33 new proteins into the PFDB from our own collection based on extensive literature search, resulting in the entry size of 141 globular proteins (89 two-state (2S) and 52 non-two-state (N2S) proteins) in our dataset (see Methods for details of the database construction).
Our dataset lists the following items: (i) the protein short name with a reference to the original experimental paper(s) on the folding kinetics, (ii) the PDB code, (iii) the structural class (α, β, α/β, and α + β), (iv) folds in the SCOP classification21 (http://scop.mrc-lmb.cam.ac.uk/scop/), (v) the number of residues in the PDB structure (LPDB), (vi) the actual number of residues of the protein used in the folding experiment (L), (vii) the experimental conditions (pH and temperature), (viii) the folding type (2S or N2S), (ix) the ln(kf) value reported, (x) the ln(kf) value after the temperature correction for the proteins whose folding experiments were carried out at a temperature other than 25 °C, (xi) the logarithmic rate constant of formation of a folding intermediate, ln(kI), when the value is available in the literature (only for N2S proteins), (xii) the ln(ku) value reported, (xiii) the ln(ku) value after the temperature correction, and (xiv) the Tanford β (βT) value, which is defined as βT = 1 − (mu‡/mNU), where mu‡ (kJ/mol/M) and mNU (kJ/mol/M) are the denaturant concentration dependence of the activation free energy of unfolding and the denaturant concentration dependence of the unfolding free energy from the native (N) to the fully unfolded (U) state, respectively22. The ln(kf), ln(kI) and ln(ku) values listed in PFDB are those in the absence of denaturant, usually obtained by linear extrapolation of the logarithmic rate constant along denaturant concentration.
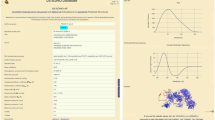
In PFDB, the folding type is thus clearly specified. The proteins that exhibited a stable folding intermediate during the kinetic folding process were classified as N2S proteins, while the proteins, exhibiting the single-exponential kinetics of folding without stable intermediates, were classified as 2S proteins even if the existence of an unstable high-energy intermediate was expected from the unfolding-limb or the folding-limb curvature of the chevron plot23. To discriminate the 2S proteins with a high-energy intermediate from the other 2S proteins, the former proteins were denoted by 2S*. Each entry of the AG dataset is also included in PFDB for comparison. A comment section is provided in the final column of the dataset and interprets discrepancies between the present and the AG datasets if any/necessary. Figure 1 depicts a snapshot of our dataset shown in the PFDB homepage.

A snapshot of our dataset in the PFDB homepage. For each protein, our dataset lists (i) protein short name, (ii) PDB code, (iii) structural class (α, β, α/β, and α + β), (iv) folds in the SCOP classification, (v) the number of residues in the PDB structure (LPDB), (vi) the actual number of residues of the protein used in the folding experiment (L), (vii) experimental conditions (pH and temperature), (viii) folding type (2S or N2S), (ix) ln(kf) reported, (x) ln(kf) after temperature correction, (xi) ln(kI) (only for N2S proteins), (xii) ln(ku) reported, (xiii) ln(ku) after temperature correction, and (xiv) Tanford β (βT). The AG dataset is also included in our database for comparison. A comment section is provided in the final column.
The protein composition in PFDB in terms of the folding type and the structural class is given in Table 1. It shows that both the 2S and N2S proteins cover all four structural classes of globular proteins. However, the 2S proteins contain only one α/β protein.
Temperature correction
Figure 2A shows a distribution of the temperature at which the ln(kf) was determined experimentally for the proteins in our dataset. Among the 141 proteins in PFDB, 99 were measured at the standard temperature of T0 (25 °C (=298.15 K)), but the other 42 (24 2S and 18 N2S proteins) were measured at different temperatures (Tx). The Tx value ranged from 5 °C to 75 °C. To maintain the consistency of folding temperature in PFDB, we developed a method for temperature correction. The predicted shape of the Eyring plot of a particular protein is determined by two parameters of the folding or unfolding reaction, the activation heat capacity (ΔCp‡) and the temperature (TH) where the activation enthalpy is zero (see Methods for more details). The predicted logarithmic rate constant at T0 (298.15 K) is thus given by the following equation:
where R is the gas constant, T0 and Tx are given by the absolute temperature, and ln[k(Tx)] is the logarithmic rate constant measured at Tx; the detailed derivation of Eq. (1) is given in Methods. We assumed that ΔCp‡ is proportional to the heat capacity change (ΔCp) of the equilibrium protein unfolding. The ΔCp is approximately proportional to the protein chain length in the PDB structure (LPDB) and empirically given by24:

(A) The temperature at which ln(kf) experimentally determined for 2S and N2S is shown. (B) Experimentally observed ln[kf(T0)] and predicted ones after temperature correction (red circles) are shown. Observed ln[kf(Tx)] values are also shown for comparison (blue crosses).
Now, it follows that:
where β is a proportionality constant. Therefore, once we have reasonable estimates of TH and β, we can evaluate ln[k(T0)] from ln[k(Tx)] and Tx by Eqs (1) and (3). It is worth mentioning that Eq. 2 is an empirical one, and theoretically, the ΔCp diminishes to zero when LPDB tends to zero. A regression equation between ΔCp and LPDB with the zero intercept has thus also been reported in the original literature as given by ΔCp = 0.058 ∙ LPDB24. Whether we used this equation or Eq. 2, the results of temperature correction were essentially identical for the proteins in our dataset, where LPDB ≥ 34.
Temperature correction for folding
We introduced the temperature corrections into the proteins whose kf values were measured at a temperature other than the standard temperature (298.15 K). First, we found that the Eyring plot or the equivalent plot of folding was well described in 14 2S proteins and 3 N2S proteins; the kf values were measured at every few degrees absolute from ~280 K to ~320 K for most of these proteins25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41. Both the TH and β values for folding kinetics, THf and βf, respectively, were more or less common among the different 2S proteins (Table 2) and also among the different N2S proteins (Table 3), except for two 2S proteins (1K9Q40 and 1PIN41), for which −ΔCp‡ for folding was larger than ΔCp. Therefore, we employed the 12 2S proteins except for these two and the 3 N2S proteins, and from their Eyring plots, we calculated the THf and ΔCpf‡. Examples of the Eyring plot for three proteins (1APS34, 1D6O35, and 1AVZ37) are shown in Figure S1. For folding kinetics, the Eyring plot is convexed, and hence, THf corresponds to the temperature of the maximum point in the Eyring plot. The ΔCpf‡ is given by the curvature of the Eyring plot, and the βf was thus evaluated by βf = ΔCpf‡/ΔCp, where ΔCp was obtained by Eq. (2); ΔCpf‡ and βf are negative because the Eyring plot is convexed. The THf and βf values thus obtained were averaged for the 12 2S proteins and for the 3 N2S proteins (Tables 2 and 3). The THf and βf values thus obtained are 315 ± 1 (standard error estimate) K and −0.62 ± 0.03 for the 2S proteins, and 305 ± 4 K and −0.75 ± 0.07 for the N2S proteins.
For the proteins whose THf and ΔCpf‡ were not available directly, we employed Eqs (1) and (3) to predict ln[kf(T0)] by assigning the THf and βf values to TH and β in the equations. However, for the proteins whose THf and ΔCpf‡ were available (1E0G28, 1HDN30, 2VH729, 1EHB27, 1HCD31, and 2CRO26), we directly calculated the ln[kf(T0)] values by Eq. (1). To distinguish ln[kf(T0)] predicted by using the averaged THf and βf and that directly calculated by Eq. (1) with the known THf and ΔCpf‡, the latter values are indicated in boldface type in our dataset. It should be also noted that the above THf and βf estimates were based on the folding data of the proteins from mesophilic organisms, and hence some care may be required when applied to the thermophilic proteins.
Next, we compared predicted ln[kf(T0)] after the temperature correction with the experimentally observed ln[kf(T0)]. For 9 2S and 5 N2S proteins (Table 4), which were not included in those used for estimating THf and βf, the experimental ln(kf) was available at both T0 and Tx. We thus applied the temperature correction to the ln[kf(Tx)] values using the above THf and βf, and compared predicted ln[kf(T0)] with the experimentally observed ln[kf(T0)]. From Fig. 2B, the predicted ln[kf(T0)] values show good agreement with the experimentally observed ones, showing the validity of our temperature correction. Although the number of data points used for this analysis is not very large (only 14 proteins), it may be enough to suggest that the temperature corrections have improved the quality of the database of protein folding.
Denaturant m values, the dependence of the free energy of unfolding on denaturant concentration, are well correlated with the ΔCp of unfolding42. Therefore, we can reasonably assume that βf is equivalent to −βT for 2S proteins. Therefore, for the 2S proteins for which the βT is available, we also calculated the ln[kf(T0)] values by assigning the THf and −βT values to TH and β in Eqs (1) and (3). The ln[kf(T0)] values thus obtained are also listed in PFDB and indicated in italic type to distinguish them from those (in roman type) predicted on the basis of THf and βf. As seen from the PFDB dataset, these two types of predicted ln[kf(T0)] are reasonably coincident with each other.
Temperature correction for unfolding
We introduced the temperature corrections into the proteins whose ku values were measured at a temperature other than the standard temperature (298.15 K), and the TH and β values for unfolding kinetics, THu and βu, respectively, were required for temperature correction. For unfolding kinetics, the Eyring plot is usually concaved with a positive βu. For 2S proteins, there is only a single transition state between U and N with a βf of −0.62 ± 0.03, and we can reasonably assume that βu = 1 + βf. Therefore, we find that βu = 0.38 ± 0.03. For N2S proteins, this simple relationship may not hold, because of a contribution from an intermediate (I) state. For the N2S proteins, however, (1 − βT) is expected to be equivalent to βu, because βT represents the relative position of the transition state between U and N in terms of the denaturant m values. The βT was reported for 38 N2S proteins in PFDB, and their average was estimated at 0.79 ± 0.02, and hence βu = 0.21 ± 0.02 for N2S proteins; 1FTG was excluded in this calculation because the I state was mostly off-pathway in this protein.
The THu corresponds to the temperature of the minimum point of the Eyring plot, but this is usually located at far below an observable temperature range of unfolding kinetics, leading to a large error in estimation of THu due to a long extrapolation along temperature. Furthermore, the Eyring plot of unfolding is not available for many of the proteins used above for estimation of THf and βf. Therefore, we had to use a different way to estimate THu. We thus chose 6 2S proteins (1IMQ13,43,1K9Q40,44, 1RFA45, 1SS146, 1U4Q47,48, and 2WXC49,50) and 3 N2S proteins (1BNI51, 1EKG52, and 1ENH53), for which the experimental ln(ku) is available at both T0 and Tx (Table 5). First, we assumed appropriate THu values (e.g., 200 K and 150 K) for 2S and N2S proteins, and assigned these THu values and the above βu values to TH and β in Eqs (1) and (3) to calculated tentative predictions of ln[ku(T0)] for 2S and N2S proteins. Then, the THu values were gradually increased or decreased until the root-mean-square deviation between the experimentally observed ln[ku(T0)] and the predicted ln[ku(T0)] values was minimized. The optimized THu values thus obtained were 224 K and 119 K for the 2S and N2S proteins, respectively. Figure 3 shows a comparison between the experimental ln[ku(T0)] values and those predicted by using the above THu and βu values, which indicates a reasonable coincidence between the experimental and predicted values.

Experimentally observed ln[ku(T0)] and predicted ones after temperature correction (red circles) are shown. Observed ln[ku(Tx)] values are also shown for comparison (blue crosses).
For the proteins whose THu and ΔCpu‡ were not available directly, we thus employed Eqs (1) and (3) to predict the ln[ku(T0)] by assigning the THu and βu values to TH and β in the equations. However, for the proteins whose THu and ΔCpu‡ were available (1EHB27 and 1HCD31), we directly calculated the ln[ku(T0)] values by Eq. (1). To distinguish the ln[ku(T0)] predicted by using the optimized THu and βu and that directly calculated by Eq. (1) with the known THu and ΔCpu‡, the latter values are indicated in boldface type in our dataset.
For the 2S proteins for which the βT is available, we also calculated the ln[ku(T0)] values by assigning the THu and (1 − βT) values to TH and β in Eqs (1) and (3). The ln[ku(T0)] values thus obtained are also listed in PFDB and indicated in italic type to distinguish them from those (in roman type) predicted on the basis of THu and βu. As seen from the PFDB dataset, these two types of predicted ln[ku(T0)] are reasonably coincident with each other.
Availability of PFDB
As a user-friendly database, PFDB is freely available at http://lee.kias.re.kr/~bala/PFDB. The database main page contains the following options: HOME, N2S, 2S, DOWNLOAD DATASET, and CONTACT. Our dataset can be downloaded by clicking the “DOWNLOAD DATASET” button.
Conclusions
In this study, we have constructed PFDB, a systematically compiled standardized database of protein folding kinetics. It is currently the most updated one with the highest number of unique entries. The quality of the dataset has been improved significantly by our temperature correction method. Therefore, our dataset can be used as a standard for developing and testing future predictive and theoretical studies of protein folding kinetics.
Methods
Construction of the AG dataset
The most recent datasets of protein folding kinetics are ACPro19 and the Garbuzynskiy dataset17. Prior to the filtering processes shown below, the ACPro dataset contained 126 proteins. Among these, we weeded out proteins with less than 34 residues (1PGB (41–56), 1L2Y and 3M48), proteins with disulfide bonds (2HQI, 1HEL, 1E65 and 1HMK), proteins with a covalently-bound prosthetic group (1YCC, 1YEA, 256B and 1HRC), proteins with irrelevant rate constants (i.e., the rate constant for formation of an intermediate instead of the actual folding rate constant (kf) for a few proteins (1AON, 1BD8 and 1JON)), and proteins whose kf was reported in the presence of denaturant (1QOP chain B). In the case of ileal lipid binding protein, the actual folding experiment was performed on the rat protein, but its PDB coordinates were not available at the time of our database creation. Instead, the reported PDB ID of 1EAL is the pig protein that is of 71.1% sequence identity with the rat protein. Since the exact PDB coordinates were not available, we excluded this protein as well as another protein without experimental references (1PSF). Furthermore, 6 proteins had duplicate entries (1NTI–2FDQ, 1SRL–1FMK, 1BF4–1BNZ, 1POH–2HPR, 1O6X–1PBA and 1EAL–2EAL) which we corrected. These filtering processes resulted in the reduction of the size of the ACPro dataset from 126 to 102 proteins. We then applied the same filtering scheme to the Garbuzynskiy dataset (107 proteins) where we weeded out proteins with less than 34 residues (1L2Y, 1T8J, 1PGB (41–56), and the 3rd entry in the Garbuzynskiy dataset), proteins with irrelevant rate constants (1AON and 1BD8), the protein 1EAL (the reason is given above), and a protein with a covalently-bound prosthetic group (256B). This change reduced the size of the Garbuzynskiy dataset from 107 to 99 proteins. When we compared the updated Garbuzynskiy (99 proteins) and ACPro (102 proteins) datasets, 6 unique proteins (1IFC, 1CBI, 1IGS, 1OPA, 2MYO and 3H08) were identified in the Garbuzynskiy dataset. Therefore, we added these 6 proteins to the ACPro dataset, and collectively named it the AG dataset (108 proteins).
Data collection and construction of PFDB
We manually collected the data of protein folding and unfolding kinetics by extensive literature search. Then we compared our collected data with those of the AG dataset. We carefully examined the data of each entry of the AG dataset, and when newer updated data did not exist, the data of that entry were included as such in our dataset of PFDB, otherwise replaced by the updated data. Finally, we added the data of 33 new proteins into the PFDB from our own collection. Of these 33 proteins, 19 are 2S proteins (1DKT, 1FGA, 1IO2, 1KDX, 1NFI,1QAU, 1RG8, 2BKF, 2GA5, 2J5A, 2JMC, 2LLH, 2L6R, 2WQG, 3O48, 3O49, 3O4D, 3ZRT (N-terminal), and 3ZRT (C-terminal)) with the remaining 14 being N2S proteins (1DWR, 1EKG, 1FA3, 1HRH, 1OKS, 1THF, 1UCH, 2BJD, 2FS6, 2KDI, 2KLL, 2X7Z, 3BLM, and 5L8I).
For 4 proteins (1RA9, 1B9C, 1FA3, and 2PQE), the presence of multiple parallel pathways of folding has been reported54,55,56, and the kf value was obtained by averaging the rate constant values along the individual pathways:
where fi and ki are the fractional amplitude and the observed rate constant, respectively, for the ith pathway of folding, and the ln(kf) values thus obtained are listed in our dataset.
The ln(kf), ln(kI) and ln(ku) values listed in PFDB are those in the absence of denaturant, usually obtained by linear extrapolation of the logarithmic rate constants along molar denaturant concentration. However, for 5 N2S proteins (1PHP (1–175)57, 1PHP (186–394)58, 1L6359, 1HNG60, and 1TTG61), the equilibria and kinetics of folding and unfolding were analyzed in terms of denaturant activity rather than the molar concentration. Whether we use the activity or the concentration in our calculation seriously affects the ln(ku) estimation, because a long extrapolation from high concentrations of denaturant back to the native condition is required. To keep consistency of our dataset, we used the linear extrapolation along the molar concentration, as far as such data were available, to estimate the ln(ku).
Derivation of Eq (1) for the temperature correction
In this study, we introduced a method for temperature correction, which gives the folding and unfolding rate constants at 25 °C (k(T0) where T0 = 298.15 K) for a protein whose rate constant at any temperature (Tx) is known. The following section will describe the derivation of Eq. (1).
According to the Eyring–Kramers equation20, we find that:
where ΔH‡(TH) and ΔS‡(TH) are the activation enthalpy and the activation entropy, respectively, at a reference temperature TH, and \({\rm{\Delta }}{C}_{{\rm{p}}}^{\ddagger }\) is the activation heat capacity; we assume that \({\rm{\Delta }}{C}_{{\rm{p}}}^{\ddagger }\) is a constant independent of temperature (T). When we set TH to the temperature where ΔH‡ is zero, i.e., the maximum or minimum point of the Eyring plot, Eq. (5) is rewritten as:
where C2 is a temperature-independent constant (C2 = C + ΔS‡(TH)/R). When \({\rm{\Delta }}{C}_{{\rm{p}}}^{\ddagger }\) and the ΔH‡(Ta) at a particular temperature (Ta) are known, TH is simply given by TH = [Ta − ΔH‡(Ta)/\({\rm{\Delta }}{C}_{{\rm{p}}}^{\ddagger }\)]. From Eq. (6), we can obtain the temperature dependence of ln(k/T), once we have TH and ΔCp‡. The difference in ln(k/T) between T0 (=298.15 K) and Tx is thus given by:
Therefore, we obtain Eq. (1).
References
Dill, K. A. & MacCallum, J. L. The protein-folding problem, 50 years on. Science 338, 1042–1046, https://doi.org/10.1126/science.1219021 (2012).
Englander, S. W. & Mayne, L. The nature of protein folding pathways. Proc Natl Acad Sci USA 111, 15873–15880, https://doi.org/10.1073/pnas.1411798111 (2014).
Bogatyreva, N. S., Osypov, A. A. & Ivankov, D. N. KineticDB: a database of protein folding kinetics. Nucleic Acids Res 37, D342–346, https://doi.org/10.1093/nar/gkn696 (2009).
De Sancho, D. & Munoz, V. Integrated prediction of protein folding and unfolding rates from only size and structural class. Phys Chem Chem Phys 13, 17030–17043, https://doi.org/10.1039/c1cp20402e (2011).
Guo, J. & Rao, N. Predicting protein folding rate from amino acid sequence. J Bioinform Comput Biol 9, 1–13 (2011).
Huang, J. T., Cheng, J. P. & Chen, H. Secondary structure length as a determinant of folding rate of proteins with two- and three-state kinetics. Proteins 67, 12–17, https://doi.org/10.1002/prot.21282 (2007).
Huang, J. T., Xing, D. J. & Huang, W. Relationship between protein folding kinetics and amino acid properties. Amino acids 43, 567–572 (2012).
Istomin, A. Y., Jacobs, D. J. & Livesay, D. R. On the role of structural class of a protein with two-state folding kinetics in determining correlations between its size, topology, and folding rate. Protein Sci 16, 2564–2569, https://doi.org/10.1110/ps.073124507 (2007).
Ivankov, D. N. & Finkelstein, A. V. Prediction of protein folding rates from the amino acid sequence-predicted secondary structure. Proc Natl Acad Sci USA 101, 8942–8944, https://doi.org/10.1073/pnas.0402659101 (2004).
Ivankov, D. N. et al. Contact order revisited: influence of protein size on the folding rate. Protein Sci 12, 2057–2062, https://doi.org/10.1110/ps.0302503 (2003).
Jung, J., Buglass, A. J. & Lee, E.-K. Topological quantities determining the folding/unfolding rate of two-state folding proteins. Journal of solution chemistry 39, 943–958 (2010).
Jung, J., Lee, J. & Moon, H. T. Topological determinants of protein unfolding rates. Proteins 58, 389–395, https://doi.org/10.1002/prot.20324 (2005).
Maxwell, K. L. et al. Protein folding: defining a “standard” set of experimental conditions and a preliminary kinetic data set of two-state proteins. Protein Sci 14, 602–616, https://doi.org/10.1110/ps.041205405 (2005).
Ouyang, Z. & Liang, J. Predicting protein folding rates from geometric contact and amino acid sequence. Protein Sci 17, 1256–1263, https://doi.org/10.1110/ps.034660.108 (2008).
Zhou, H. & Zhou, Y. Folding rate prediction using total contact distance. Biophys J 82, 458–463, https://doi.org/10.1016/S0006-3495(02)75410-6 (2002).
Zou, T. & Ozkan, S. B. Local and non-local native topologies reveal the underlying folding landscape of proteins. Phys Biol 8, 066011, https://doi.org/10.1088/1478-3975/8/6/066011 (2011).
Garbuzynskiy, S. O., Ivankov, D. N., Bogatyreva, N. S. & Finkelstein, A. V. Golden triangle for folding rates of globular proteins. Proc Natl Acad Sci USA 110, 147–150, https://doi.org/10.1073/pnas.1210180110 (2013).
Gromiha, M. M., Thangakani, A. M. & Selvaraj, S. FOLD-RATE: prediction of protein folding rates from amino acid sequence. Nucleic Acids Res 34, W70–74, https://doi.org/10.1093/nar/gkl043 (2006).
Wagaman, A. S., Coburn, A., Brand-Thomas, I., Dash, B. & Jaswal, S. S. A comprehensive database of verified experimental data on protein folding kinetics. Protein Sci 23, 1808–1812, https://doi.org/10.1002/pro.2551 (2014).
Bilsel, O. & Matthews, C. R. Barriers in protein folding reactions. Adv Protein Chem 53, 153–207 (2000).
Andreeva, A. et al. Data growth and its impact on the SCOP database: new developments. Nucleic Acids Res 36, D419–425, https://doi.org/10.1093/nar/gkm993 (2008).
Jackson, S. E. How do small single-domain proteins fold? Folding and Design 3, R81–R91 (1998).
Sanchez, I. E. & Kiefhaber, T. Evidence for sequential barriers and obligatory intermediates in apparent two-state protein folding. J Mol Biol 325, 367–376 (2003).
Robertson, A. D. & Murphy, K. P. Protein Structure and the Energetics of Protein Stability. Chem Rev 97, 1251–1268 (1997).
Candel, A. M., Cobos, E. S., Conejero-Lara, F. & Martinez, J. C. Evaluation of folding co-operativity of a chimeric protein based on the molecular recognition between polyproline ligands and SH3 domains. Protein Eng Des Sel 22, 597–606, https://doi.org/10.1093/protein/gzp041 (2009).
Laurents, D. V. et al. Folding kinetics of phage 434 Cro protein. Biochemistry 39, 13963–13973 (2000).
Manyusa, S. & Whitford, D. Defining folding and unfolding reactions of apocytochrome b 5 using equilibrium and kinetic fluorescence measurements. Biochemistry 38, 9533–9540 (1999).
Nickson, A. A., Stoll, K. E. & Clarke, J. Folding of a LysM domain: entropy-enthalpy compensation in the transition state of an ideal two-state folder. J Mol Biol 380, 557–569, https://doi.org/10.1016/j.jmb.2008.05.020 (2008).
Taddei, N. et al. Thermodynamics and kinetics of folding of common-type acylphosphatase: comparison to the highly homologous muscle isoenzyme. Biochemistry 38, 2135–2142, https://doi.org/10.1021/bi9822630 (1999).
Van Nuland, N. A. et al. Slow cooperative folding of a small globular protein HPr. Biochemistry 37, 622–637, https://doi.org/10.1021/bi9717946 (1998).
Wong, H. J., Stathopulos, P. B., Bonner, J. M., Sawyer, M. & Meiering, E. M. Non-linear effects of temperature and urea on the thermodynamics and kinetics of folding and unfolding of hisactophilin. J Mol Biol 344, 1089–1107, https://doi.org/10.1016/j.jmb.2004.09.091 (2004).
Alexander, P., Orban, J. & Bryan, P. Kinetic analysis of folding and unfolding the 56 amino acid IgG-binding domain of streptococcal protein G. Biochemistry 31, 7243–7248 (1992).
Chen, B. L., Baase, W. A. & Schellman, J. A. Low-temperature unfolding of a mutant of phage T4 lysozyme. 2. Kinetic investigations. Biochemistry 28, 691–699 (1989).
Chiti, F. et al. Structural characterization of the transition state for folding of muscle acylphosphatase. J Mol Biol 283, 893–903, https://doi.org/10.1006/jmbi.1998.2010 (1998).
Main, E. R., Fulton, K. F. & Jackson, S. E. Folding pathway of FKBP12 and characterisation of the transition state. J Mol Biol 291, 429–444, https://doi.org/10.1006/jmbi.1999.2941 (1999).
Martinez, J. C., Pisabarro, M. T. & Serrano, L. Obligatory steps in protein folding and the conformational diversity of the transition state. Nat Struct Biol 5, 721–729, https://doi.org/10.1038/1418 (1998).
Plaxco, K. W. et al. The folding kinetics and thermodynamics of the Fyn-SH3 domain. Biochemistry 37, 2529–2537, https://doi.org/10.1021/bi972075u (1998).
Schindler, T. & Schmid, F. X. Thermodynamic properties of an extremely rapid protein folding reaction. Biochemistry 35, 16833–16842, https://doi.org/10.1021/bi962090j (1996).
Tan, Y. J., Oliveberg, M. & Fersht, A. R. Titration properties and thermodynamics of the transition state for folding: comparison of two-state and multi-state folding pathways. J Mol Biol 264, 377–389, https://doi.org/10.1006/jmbi.1996.0647 (1996).
Crane, J. C., Koepf, E. K., Kelly, J. W. & Gruebele, M. Mapping the transition state of the WW domain beta-sheet. J Mol Biol 298, 283–292, https://doi.org/10.1006/jmbi.2000.3665 (2000).
Jager, M., Nguyen, H., Crane, J. C., Kelly, J. W. & Gruebele, M. The folding mechanism of a beta-sheet: the WW domain. J Mol Biol 311, 373–393, https://doi.org/10.1006/jmbi.2001.4873 (2001).
Myers, J. K., Pace, C. N. & Scholtz, J. M. Denaturant m values and heat capacity changes: relation to changes in accessible surface areas of protein unfolding. Protein Science 4, 2138–2148 (1995).
Friel, C. T., Capaldi, A. P. & Radford, S. E. Structural analysis of the rate-limiting transition states in the folding of Im7 and Im9: similarities and differences in the folding of homologous proteins. J Mol Biol 326, 293–305 (2003).
Ferguson, N., Johnson, C. M., Macias, M., Oschkinat, H. & Fersht, A. Ultrafast folding of WW domains without structured aromatic clusters in the denatured state. Proc Natl Acad Sci USA 98, 13002–13007 (2001).
Vallee-Belisle, A., Turcotte, J. F. & Michnick, S. W. raf RBD and ubiquitin proteins share similar folds, folding rates and mechanisms despite having unrelated amino acid sequences. Biochemistry 43, 8447–8458, https://doi.org/10.1021/bi0359426 (2004).
Dimitriadis, G. et al. Microsecond folding dynamics of the F13W G29A mutant of the B domain of staphylococcal protein A by laser-induced temperature jump. Proc Natl Acad Sci USA 101, 3809–3814, https://doi.org/10.1073/pnas.0306433101 (2004).
Scott, K. A., Batey, S., Hooton, K. A. & Clarke, J. The folding of spectrin domains I: wild-type domains have the same stability but very different kinetic properties. J Mol Biol 344, 195–205, https://doi.org/10.1016/j.jmb.2004.09.037 (2004).
Wensley, B. G., Gartner, M., Choo, W. X., Batey, S. & Clarke, J. Different members of a simple three-helix bundle protein family have very different folding rate constants and fold by different mechanisms. J Mol Biol 390, 1074–1085, https://doi.org/10.1016/j.jmb.2009.05.010 (2009).
Neuweiler, H. et al. Downhill versus barrier-limited folding of BBL 2: mechanistic insights from kinetics of folding monitored by independent tryptophan probes. J Mol Biol 387, 975–985, https://doi.org/10.1016/j.jmb.2008.12.056 (2009).
Neuweiler, H. et al. The folding mechanism of BBL: Plasticity of transition-state structure observed within an ultrafast folding protein family. J Mol Biol 390, 1060–1073, https://doi.org/10.1016/j.jmb.2009.05.011 (2009).
Dalby, P. A., Clarke, J., Johnson, C. M. & Fersht, A. R. Folding intermediates of wild-type and mutants of barnase. II. Correlation of changes in equilibrium amide exchange kinetics with the population of the folding intermediate. J Mol Biol 276, 647–656, https://doi.org/10.1006/jmbi.1997.1547 (1998).
Faraj, S. E., Gonzalez-Lebrero, R. M., Roman, E. A. & Santos, J. Human Frataxin Folds Via an Intermediate State. Role of the C-Terminal Region. Sci Rep 6, 20782, https://doi.org/10.1038/srep20782 (2016).
Mayor, U., Johnson, C. M., Daggett, V. & Fersht, A. R. Protein folding and unfolding in microseconds to nanoseconds by experiment and simulation. Proc Natl Acad Sci USA 97, 13518–13522, https://doi.org/10.1073/pnas.250473497 (2000).
Enoki, S., Saeki, K., Maki, K. & Kuwajima, K. Acid denaturation and refolding of green fluorescent protein. Biochemistry 43, 14238–14248, https://doi.org/10.1021/bi048733+ (2004).
Kamagata, K., Sawano, Y., Tanokura, M. & Kuwajima, K. Multiple parallel-pathway folding of proline-free Staphylococcal nuclease. J Mol Biol 332, 1143–1153 (2003).
Patra, A. K. & Udgaonkar, J. B. Characterization of the folding and unfolding reactions of single-chain monellin: evidence for multiple intermediates and competing pathways. Biochemistry 46, 11727–11743, https://doi.org/10.1021/bi701142a (2007).
Parker, M. J., Spencer, J. & Clarke, A. R. An integrated kinetic analysis of intermediates and transition states in protein folding reactions. J Mol Biol 253, 771–786, https://doi.org/10.1006/jmbi.1995.0590 (1995).
Parker, M. J. & Marqusee, S. The cooperativity of burst phase reactions explored. J Mol Biol 293, 1195–1210, https://doi.org/10.1006/jmbi.1999.3204 (1999).
Parker, M. J. et al. Domain behavior during the folding of a thermostable phosphoglycerate kinase. Biochemistry 35, 15740–15752, https://doi.org/10.1021/bi961330s (1996).
Parker, M. J., Dempsey, C. E., Lorch, M. & Clarke, A. R. Acquisition of native beta-strand topology during the rapid collapse phase of protein folding. Biochemistry 36, 13396–13405, https://doi.org/10.1021/bi971294c (1997).
Cota, E. & Clarke, J. Folding of beta-sandwich proteins: three-state transition of a fibronectin type III module. Protein Sci 9, 112–120, https://doi.org/10.1110/ps.9.1.112 (2000).
Manyusa, S. & Whitford, D. Defining folding and unfolding reactions of apocytochrome b5 using equilibrium and kinetic fluorescence measurements. Biochemistry 38, 9533–9540, https://doi.org/10.1021/bi990550d (1999).
Plaxco, K. W., Spitzfaden, C., Campbell, I. D. & Dobson, C. M. A comparison of the folding kinetics and thermodynamics of two homologous fibronectin type III modules. J Mol Biol 270, 763–770, https://doi.org/10.1006/jmbi.1997.1148 (1997).
Mizukami, T., Abe, Y. & Maki, K. Evidence for a Shared Mechanism in the Formation of Urea-Induced Kinetic and Equilibrium Intermediates of Horse Apomyoglobin from Ultrarapid Mixing Experiments. PLoS One 10, e0134238, https://doi.org/10.1371/journal.pone.0134238 (2015).
Uzawa, T. et al. Collapse and search dynamics of apomyoglobin folding revealed by submillisecond observations of alpha-helical content and compactness. Proc Natl Acad Sci USA 101, 1171–1176, https://doi.org/10.1073/pnas.0305376101 (2004).
DeVries, I., Ferreiro, D. U., Sanchez, I. E. & Komives, E. A. Folding kinetics of the cooperatively folded subdomain of the IkappaBalpha ankyrin repeat domain. J Mol Biol 408, 163–176, https://doi.org/10.1016/j.jmb.2011.02.021 (2011).
Acknowledgements
The work was supported in part by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (NRF-2017R1E1A1A01077717), and by JSPS (Japan Society for the Promotion of Science) KAKENHI Grant Numbers JP25440075 and JP16K07314. The authors thank KIAS Center for Advanced Computation for providing computing resources for this work.
Author information
Authors and Affiliations
Contributions
B.M., K.K. and J.L. designed and performed research, analyzed the data and wrote the paper.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Manavalan, B., Kuwajima, K. & Lee, J. PFDB: A standardized protein folding database with temperature correction. Sci Rep 9, 1588 (2019). https://doi.org/10.1038/s41598-018-36992-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-36992-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
