
Abstract
Injecting CO2 into oil reservoirs appears to be cost-effective and environmentally friendly due to decreasing the use of chemicals and cutting back on the greenhouse gas emission released. However, there is a pressing need for new algorithms to characterize oil/brine/rock system wettability, thus better predict and manage CO2 geological storage and enhanced oil recovery in oil reservoirs. We coupled surface complexation/CO2 and calcite dissolution model, and accurately predicted measured oil-on-calcite contact angles in NaCl and CaCl2 solutions with and without CO2. Contact angles decreased in carbonated water indicating increased hydrophilicity under carbonation. Lowered salinity increased hydrophilicity as did Ca2+. Hydrophilicity correlates with independently calculated oil-calcite electrostatic bridging. The link between the two may be used to better implement CO2 EOR in fields.
Similar content being viewed by others


Introduction
Oil will be an important energy source for the rest of the 21st century1 and carbonate reservoirs host most of the world’s oil (>60%)2. However, the recovery factor is low (<40%)3, so there is enormous motivation to improve recovery cost-effectively, and with environmentally friendly manners. CO2 EOR is attractive because it produces more oil without the expense of chemicals although CO2 injection costs energy to compress before injection, and the CO2 source availability needs to be also considered. Moreover, CO2-EOR can combat global warming by injecting CO2 into geological formations. CO2 EOR techniques include miscible4 and immiscible continuous injection5,6, carbonated water flooding7, huff and puff injection (injecting CO2 in a single well and producing from the well after CO2 equilibration with the crude oil)8,9, and water-alternating-CO2 injection10,11,12. CO2 techniques work through some combination of immiscible drive, first contact miscible drive, vaporizing-gas drive, condensing-gas drive, and vaporizing-condensing gas drive, and multiple-contact miscible drive. At the microscopic level, these processes can: promote oil-swelling, reduce oil viscosity, mitigate gravity segregation by reducing the density difference between oil and water, and, lower oil interfacial tension, all of which can increase oil recovery. The net impact of CO2 addition can be quite large, amounting to recovery of an extra 4–15% of the original oil in place in conventional reservoirs13. Moreover, CO2 huff-n-puff can achieve 14% additional oil recovery from unconventional reservoirs14.
While much is known about the effect of CO2 on oil fluid properties, oil-CO2-brine-carbonate system wettability is not well understood, which triggers intrinsic uncertainties to predict and manage the CO2 injection and reservoir performance although CO2-brine-rock system wettability has been well documented15,16. This is largely because system wettability governs subsurface multiphase flow and residual saturations17. To examine the wettability, contact angle test has been perceived as an effective means together with interpretation using Derjaguin-Landau-Verwey-Overbeek (DLVO)18,19,20 and surface complexation modelling21,22,23.
Teklu et al.10 showed that dissolving CO2 into seawater decreases oil contact angles on calcite, thus increasing hydrophilicity. Decreased salinity also decreases contact angles. Teklu et al.10 noted several potential explanations for their contact angle trends and called for a closer examination of the surface controls over wettability alteration. Venkatraman et al.24 used Gibbs free-energy function to integrate phase-behaviour computations and geochemical reactions to find equilibrium composition, but quantitative work remains to be made to understand how dissolved CO2 governs oil-brine-calcite interaction, thus wettability. Here we constrain surface chemical controls over wettability in carbonate reservoirs undergoing CO2 EOR by interpreting new oil-on-calcite contact angles in the presence of model reservoir brines containing NaCl and CaCl2 using a coupled surface complexation/CO2 and mineral dissolution model.
Results and Discussion
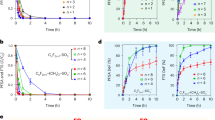
To examine the wettability of oil-brine (CO2)-carbonate system, we measured contact angle of oil on calcite substrates in the presence of carbonated brine or non-carbonated brine. Figure 1 shows oil-on-calcite contact angles measured at 25 °C and 3000 psi pressure in model brines under carbonated and non-carbonated conditions. Carbonated water lowers contact angles and produces a strongly water-wet system regardless of salinity and ion type compared to non-carbonated water. For example, non-carbonated 1 mol/L NaCl yielded a contact angle of 120°, meaning an oil-wet system. However, carbonated 1 mol/L NaCl gave a contact angle of 39°, meaning a strongly water-wet system. Similarly, Teklu et al.10 observed a contact angle shift from 116.6–133.6° (non-carbonated seawater, pH = 6.6) to 36.1–40.8° (carbonated seawater, pH = 5.5 at atmospheric condition). A secondary effect of lowered salinity decreasing contact angles and moving the system towards water wetness is also seen in Fig. 1, and was observed before by Teklu et al.10. Divalent cations (Ca2+) gave a lower contact angle compared to monovalent cations (Na+) regardless of concentration.

Oil-on-calcite contact angles in the presence of carbonated and non-carbonated brines. In total, we measured eight contact angles, four contact angles with carbonated brine and four contact angles with non-carbonated brine. The standard deviation of contact angle measurements was ±2°.
To understand how carbonation increases hydrophilicity, we develop a geochemical model that couples CO2 dissolution, mineral dissolution, and oil and calcite surface chemistry (Table 1). CO2 and calcite dissolution into brines is calculated by a standard equilibrium approach. Oil surface species are assumed to be –NH+, –COO− and –COOCa+ 23,25,26, polar surface groups expressed at, and attached to, the oil-water interface. Calcite surface species are assumed to be >CO3−, >CaCO3−, >CaOH2+, and >CO3Ca+ 21,22,23,27 (Fig. 2); where “>” denotes a calcite surface species. The primary electrostatic bridges between oppositely charged oil and calcite surface species are then the pairs, -NH+ and >CO3−, -NH+ and >CaCO3−, -COOCa+ and >CO3−, -COOCa+ and >CaCO3−, -COO− and >CaOH2+, and -COO− and> CO3Ca+. A quantitative measure of electrostatic attraction is termed the bond product sum21,23, BPS, which is equal to [-NH+] [>CO3−] + [-NH+] [>CaCO3−] + [-COOCa+] [>CO3−] + [-COOCa+] [>CaCO3−] + [-COO−] [>CaOH2+] + [-COO−] [>CO3Ca+]; where bracketed terms are calculated surface concentrations (μmol/m2). Bond product sum (electrostatic bridges) is an explicit way to reflect the electrostatic force change between the oil/brine and rock/brine interfaces. Our previous studies25,28 show that DLVO and surface complexation modelling predict similar wettability trends. This is because the physics of DLVO and surface complexation is the same as a result of diffuse double layer. We therefore decided to use BPS to reveal the interaction of oil-brine-carbonate because BPS can be practically modelled using the geochemical reactions with reservoir simulators for waterflooding and EOR studies.

Schematic of surface chemistry alteration during CO2 EOR.
Speciation of Oil/Brine Interfaces
Figures 3 and 4 show calculated oil surface speciation in non-carbonated and carbonated NaCl and CaCl2 brines. Calculation results are listed in Tables 2 and 3 in Supplementary Information. The calculated amount of -NH+ decreases with increasing pH regardless of ion type and salinity for both non-carbonated and carbonated brines as pH controls the amount of-NH+ through Reaction 1 (Table 1) shifting to the left25,29. The calculated amount of -COO− increases with increasing pH but decreases due to the formation of –COOCa+ for non-carbonated brines (Fig. 3). The same trend is observed in carbonated brines (Fig. 4), but with an increase of -COO− with increasing pH due to the formation of CO32−, which decreases Ca2+. Note: the amount of –COOCa+ depends on dissolved Ca levels and to a lesser extent ionic strength because of their effect on surface species concentrations and the Ca2+ activity coefficient25. Keeping in mind that the surface speciation responds to dissolved phase concentrations that, through calcite equilibria, are set by pH and amount of carbonation (in situ PCO2). For example, Ca2+ levels and ionic strength are higher at low pH and in carbonated brine. The PHREEQC surface complexation calculation tracks each of the competing factors while maintaining equilibrium with calcite.

pH-dependent oil surface speciation in non-carbonated brine.

pH-dependent oil surface speciation in carbonated brine.
Speciation of Calcite/Brine Interfaces
Figures 5 and 6 show calculated calcite surface speciation in non-carbonated and carbonated NaCl and CaCl2 brines. Calculation results are listed in Tables 4 and 5 in Supplementary Information. Note that the legends in Figs 5–8 refer to initial solution compositions. Final solution compositions are influenced by calcite dissolution and PCO2. Because in the CaCO3-H2O-CO2 system CO2, pH, and Ca2+ are coupled, ionic strength is particularly sensitive to pH and PCO2-dependent calcite dissolution reactions. So calcite dissolution in the pH < 4 for example causes calculated ionic strengths to be well above 1 M.

pH-dependent calcite surface speciation in non-carbonated brine.

pH-dependent calcite surface speciation in carbonated brine.

Bond Product Sum vs. pH in non-carbonated brine. The PHREEQC calculated pH of non-carbonated brine with calcite in equilibrium for 1 mol/L NaCl, CaCl2, and 0.01 mol/L NaCl, CaCl2, were 9.8, 8.2, 9.0, and 9.9 at PCO2 = 0 psi, and 25 °C. The initial pH of all fluids before equilibration with calcite was 7.

Bond Product Sum vs. pH for carbonated brine. The PHREEQC calculated pH of carbonated brine for 1 mol/L NaCl, CaCl2, and 0.01 mol/L NaCl, CaCl2, were 4.9, 4.1, 4.9, and 4.8 at PCO2 = 3000 psi, and 25 °C after equilibration with calcite. The calculated pre-calcite equilibration brine pH was 3.0, 2.6, 3.0, and 3.0 for 1 mol/L NaCl, CaCl2, and 0.01 mol/L NaCl, CaCl2, respectively, at PCO2 = 3000 psi, and 25 °C.
In both non-carbonated and carbonated solutions, low pH calcite surface charge is dominated by >CaOH2+. >CaOH2+ is the most abundant surface species at high pH as well in non-carbonated solutions. Increasing pH favors a decrease in >CaOH2+ and an increase in >CaCO3−, >CO3−, and >CO3Ca+ for a given available Ca+2. In carbonated solutions, high pHs and bicarbonate prompt appreciable formation of >CaCO3−, >CO3−, and >CO3Ca+.
Two shifts that stand out between the non-carbonated and carbonated cases are the conversion of >CaOH2+ to >CaCO3− and >CO3− to >CO3Ca+ with increasing CO2. These reactions are driven by respectively the higher bicarbonate and calcium levels in CO2-charged brine:
Calculation of Oil-on-Calcite Wetting
We combined the calculated oil and calcite speciation above into a bond product sum, BPS, the number of electrostatic bridges between the oil and calcite. Again, the BPS is a measure of electrostatic attraction between oil and calcite, is proportional to measured contact angles21,30, and is therefore a useful predictor of wetting. For our system, the bond product sum is the total of six concentration products that quantify the strength of six electrostatic bridges between oppositely charged oil and calcite species, as noted above. For natural systems containing sulphate the BPS would also include, for example, a [>CaSO4−][-NH+] term.
Figures 7 and 8 show the bond product sum for non-carbonated and carbonated conditions. Calculation outputs are listed in Table 6 and 7 of Supplementary Information. The most important electrostatic bridges are [>CaOH2+][-COO−], [>CO3−][-COOCa+], and [>CaCO3−][-COOCa+]; the first of these bridges provides most of the oil-calcite electrostatic linking.
The pH in non-carbonated brines increased from 7 to 10 after equilibration with calcite which decreases the BPS (Fig. 7) and the contact angle (Fig. 1). In contrast, in carbonated brines, the pH decreases to below 4 which decreases the BPS to almost three times than the non-carbonated brine (Fig. 8), accounting for the contact angle decrease in a range of 20 to 80° with various brines (Fig. 1) thus more hydrophilicity system. The pH difference between calcite-equilibrated carbonated and non-carbonated brine largely accounts for why Teklu et al.10 and we observed a dramatic contact angle decrease in carbonated brine (Fig. 1). Specifically, electrostatic adhesion decreases with carbonation because of a decrease in pH. In a carbonate reservoir the reduction in electrostatic adhesion with carbonation ultimately means greater oil recovery because it causes an increase in oil relative permeability31.
Although BPS prediction appears to be in line with contact angle measurements on calcite surfaces, to complement the BPS estimates and provide deeper thermodynamic insights to the nature the physics which controls wettability of brine-oil-carbonate, we computed surface potential of brine-oil and brine-calcite in light of diffuse double layer to calculate total disjoining pressure of oil-brine-carbonate system in non-carbonated and carbonate brine within DLVO framework32 as the sum
where IITotal is the disjoining pressure of the specific intermolecular interactions which reflects the interactive forces between the interfaces of brine-oil and brine-rock. IIelectrical is the electrostatic forces due to the development of the charges between interacting surfaces. A brief introduction of the forces and calculation procedures were presented elsewhere33. The Hamaker constant for oil-brine-rock in water is approximately 1 × 10−20 J34. Melrose35 used Hamaker constants ranging from 0.3 to 0.9 × 10−20 J. In this study, 0.81 × 10−20 J was used as the Hamaker constant34. We did not consider the structural forces to model the total disjoining pressure due to the fact that the structural forces are short-range interactions over a distance of less than 5 nm compared with long-range interactions36, e.g., London-van der Waals and electrical double layer forces.
We computed the brine chemistry and surface potential of fluid-fluid and fluid-rock with considering calcite dissolution and water uptake of CO2 for carbonate brine using PHREEQC, as shown in Table 9 and Table 10 listed in Supplementary Information. Constant charge (CC) and constant potential (CP) conditions based on the linear P-B expression37 were used to compute total disjoining pressure versus interfacial separation of the oil and calcite surfaces. The two conditions represent upper and lower bounding curves on the total disjoining pressure25,37.
Figure 9 shows the isotherms of total disjoining pressure versus separation distance between oil and calcite surfaces across various brines. Positive pressure indicates repulsion, and negative pressure implies attraction. In the presence of 0.01 mol/L CaCl2 and NaCl solution, carbonated brine gave a positive disjoining pressure which exhibits a progressively more repulsive barrier on approach, implying a strongly water-wet system in line with contact angle measurements. Non-carbonated brine gave a negative disjoining pressure indicates an oil-wet system except 0.01 mol/L NaCl at constant charge (CC). This however contradicts contact angle results, which show a water-wet system although contact angle is 20 to 30° more than the contact angle in carbonated brines. This is because non-carbonated brine gives a strongly negative surface potential, whereas the surface potential of brine-calcite remains positive, triggering attractive forces. We believe that both electrostatic and nonelectrostatic physisorption together with competitive ion chemisorption (ion exchange and surface complexation modelling)38 would be combined to better account for the total disjoining pressure.

Total disjoing pressure under the condition of constant charge (solid lines) and constant potential (dotted lines) versus film thickness in the presence of carbonated and non-carbonated brines with different ion type and salinity. (CC means constant charges condition, and CP represents constant potential condition).
In the presence of 1 mol/L CaCl2 and NaCl solution, carbonated brine yields a relatively lower adhesion compared to non-carbonated brine, indicating a lower contact angle in carbonated brine in line with experiments. However, the same contradiction remains, showing that both carbonated and non-carbonated brine gave negative disjoining pressure, signifying an oil-wet system. We believe that double layer collapse may be one of the main reasons to account for this negative disjoining pressure due to the high ionic strength39. Together, although the disjoining pressure does not completely predict the contact angle results, DLVO and surface complexation modelling predict the same trend over the contact angle results, showing that carbonated brine leads to less adhesion compared to non-carbonated brine thus hydrophilicity in line with contact angle results. In addition, our surface potential results also show that carbonation plays a minor role in surface potential of brine-calcite in the presence of CaCl2 (Table 10 in Supplementary Information), confirming that Ca level likely dominates brine-calcite surface potential thus zeta potential rather than pH in line with Mahrouqi et al.39.
Implications and Conclusions
To better predict and manage CO2 geological storage and enhanced oil recovery in carbonate oil reservoirs, we aimed to understand oil-CO2-brine-carbonate system wettability by measuring oil contact angle in carbonated and non-carbonated brines. We also coupled surface complexation/CO2 and calcite dissolution model, and accurately predicted measured oil-on-calcite contact angles in NaCl and CaCl2 solutions with and without CO2. To further complements surface complexation modelling, DLVO theory was used to calculate disjoining pressure at constant charge and constant potential conditions, confirming that DLVO and surface complexation modelling predict the same trend. Contact angle results show that carbonated water increases hydrophilicity. Reduced salinity increased hydrophilicity as did Ca2+. Our coupled surface complexation/CO2 and mineral dissolution model provides a mechanistic rationale for the CO2-induced wettability shift, and a means for coupling such observations into larger reservoir simulators. The latter might provide a path for more effectively tuning CO2 EOR to increase oil recovery from carbonate reservoirs. That being said, uncertainties remain. The surface complexation modelling might be improved by developing a more precise picture of the oil-water interface chemistry, specifically by verifying more closely the identities and surface acidity constants of surface polar groups e.g. through zeta potential measurements. The impact of salinity on oil and calcite surface complexation in high TDS solutions must be verified. Alternative calcite surface complexation stoichiometries than those in Table 1 exist27. Our preliminary calculations using the calcite surface stoichiometries of Song et al.27 predict the same trends seen above although the absolute values of the calculated BPS are different (Fig. 9 and Table 8 in Supplementary Information).
Methods
Substrates
Calcite minerals supplied by Ward’s Science were used in the contact angle tests. X-Ray Diffraction (XRD) tests confirmed that the composition of substrates were 100% calcite. To avoid any hysteresis and contamination the natural mineral surfaces (cleavage) were used as pendent spots.
Prior to experiments, substrates were cleaned with solvents (e.g., toluene and methanol) to remove any traces of organic and inorganic contaminants. Substrates were then rinsed with equilibrated deionised water to prevent undesired dissolution and dried in an oven at moderate temperature of 60 °C. Then, clean and dry substrates were exposed to air plasma for 10 min to remove organic surface contamination40. We also imaged the cleaved calcite substrate to obtain the surface roughnesusing atomic force microscopy (AFM) (WITec, ALPHA 300 RA for combined Raman-AFM imaging). Results show that the surface roughness was in a range of 0 to 4.8 nm30, implying that the surface roughness effect on contact angle should be negligible16,41.
Liquids Preparation
Texas crude oil from the United States was used in contact angle tests. Chemical analysis of crude oil indicated the acid and base number were 1.7 and 1.2 mg KOH/g, respectively. To prepare carbonated brines, 1.0 mole and 0.01 mole of NaCl and CaCl2 brines were prepared and individually loaded in a reactor. CO2 gas was injected in the reactor through a syringe pump with the aid of a compressor and mixed with the brine at 3000 psi and 25 °C until the brine was saturated with CO2 gas. Saturated brine was transferred into an accumulator and maintained under pressure until the experiment was carried out.
Experimental Procedure
Contact angle experiments were measured using a Vinci IFT apparatus (see Fig. 1 in Xie et al.28). All contact angles were measured at 3000 psi and 25 °C conditions. Calcite substrates were mounted on the apparatus turn table and placed inside the high pressure high temperature (HPHT) cell and sealed and vacuumed until state of vacuum was attained. The pressure cell was then filled with the desired brine and pressurised to 3000 psi. Subsequently, the experimental oil was slowly and steadily introduced into the cell through a capillary needle (0.64 mm diameter) until a droplet was formed. The droplet was then released on the substrate, and integrated software was utilised to measure left and right contact angles between substrate and the oil droplet. Contact angles were continuously recorded until equilibrium was achieved where contact angle became stable. This process was repeated for CO2-saturated brines. Throughout the experiment test pressure was closely monitored and maintained to prevent depressurisation of cell, and desaturation of the brine.
Simulation Methods
Surface complexation modelling (and DLVO theory) presumes an electrical double layer at each interface and the existence of charged surface species whose concentrations depend upon the chemical makeup of the water and the oil and mineral surface28. Surface equilibria and constants23,42,43,44 are listed in Table 1. The surface species concentrations were calculated using PHREEQC version 3.3.9 (Parkhurst and Appelo 2013) and a diffuse layer surface model. The calcite surface site density was assumed to be 5 sites/nm2,22. The oil/calcite surface area was set to 0.11 m2/g22.
References
Pierre Gadonneix A. S Liu,Tie’nan. 2013 World Energy Issues Monitor. In: Pierre Gadonneix (ed^(eds). World Energy Council (2013).
Klemme, H. & Ulmishek, G. F. Effective petroleum source rocks of the world: stratigraphic distribution and controlling depositional factors (1). AAPG Bulletin, 1809–1851 (1991).
Downs, H. H., Hoover, P. D. Enhaced recovery by wettability alteration. In Oil Field Chemistry: Enhanced recovery and production stimulation 396 ch. (1989).
Le Van, S & Chon, B. H. Effects of salinity and slug size in miscible CO2 water-alternating-gas core flooding experiments. Journal of Industrial and Engineering Chemistry (2017).
Zheng, S. & D. Yang. “Pressure Maintenance and Improving Oil Recovery by Means of Immiscible Water-Alternating-CO2 Processes in Thin Heavy-Oil Reservoirs.” SPE Reservoir Evaluation & Engineering 16(01), 60–71 (2013).
Mohd Shafian, S. R. et al. Enhancing the Efficiency of Immiscible Water Alternating Gas (WAG) Injection in a Matured, High Temperature and High CO2 Solution Gas Reservoir - A Laboratory Study. (ed^(eds). Society of Petroleum Engineers (2013).
Mosavat, N. & Torabi, F. Micro-optical analysis of carbonated water injection in irregular and heterogeneous pore geometry. Fuel 175, 191–201 (2016).
Zuloaga, P., Yu, W., Miao, J. & Sepehrnoori, K. Performance evaluation of CO2 Huff-n-Puff and continuous CO2 injection in tight oil reservoirs. Energy 134, 181–192 (2017).
Pu, W. et al. Experimental investigation of CO2 huff-n-puff process for enhancing oil recovery in tight reservoirs. Chemical Engineering Research and Design 111, 269–276 (2016).
Teklu, T. W., Alameri, W., Graves, R. M., Kazemi, H. & AlSumaiti, A. M. Low-salinity water-alternating-CO2 EOR. Journal of Petroleum Science and Engineering 142, 101–118 (2016).
Ramanathan, R., Shehata, A. M. & Nasr-El-Din, H. A. Water Alternating CO2 Injection Process - Does Modifying the Salinity of Injected Brine Improve Oil Recovery? (ed^(eds). Offshore Technology Conference (2015).
Wilson, A. “CO2 Low-Salinity Water Alternating Gas: A Promising New Approach for EOR.” Journal of Petroleum Technology 67(01), 84–86 (2015).
Kuuskraa, V. A., Van Leeuwen, T., Wallace, M. & DiPietro, P. Improving domestic energy security and lowering CO2 emissions with “next generation” CO2-enhanced oil recovery (CO2-EOR). National Energy Technology Laboratory, Pittsburgh, PA, USA (2011).
Ma, J. et al. Enhanced light oil recovery from tight formations through CO2 huff ‘n’ puff processes. Fuel 154, 35–44 (2015).
Iglauer, S. CO2–Water–Rock Wettability: Variability, Influencing Factors, and Implications for CO2 Geostorage. Accounts of Chemical Research 50, 1134–1142 (2017).
Arif, M., Lebedev, M., Barifcani, A. & Iglauer, S. CO2 storage in carbonates: Wettability of calcite. International Journal of Greenhouse Gas Control 62, 113–121 (2017).
Lake, L. W. Enhanced oil recovery, Prentice Hall. (1989).
Mahani, H. et al. Insights into the Mechanism of Wettability Alteration by Low-Salinity Flooding (LSF) in Carbonates. Energy & Fuels 29, 1352–1367 (2015).
Nasralla, R. A. & Nasr-El-Din, H. A. Double-Layer Expansion: Is It a Primary Mechanism of Improved Oil Recovery by Low-Salinity Waterflooding? SPE Reservoir Evaluation & Engineering 17, 49–59 (2014).
Drummond, C. & Israelachvili, J. Surface forces and wettability. Journal of Petroleum Science and Engineering 33, 123–133 (2002).
Chen, Y., Xie, Q., Sari, A., Brady, P. V. & Saeedi, A. Oil/water/rock wettability: Influencing factors and implications for low salinity water flooding in carbonate reservoirs. Fuel 215, 171–177 (2018).
Mahani, H., Keya, A. L., Berg, S. & Nasralla, R. Electrokinetics of Carbonate/Brine Interface in Low-Salinity Waterflooding: Effect of Brine Salinity, Composition, Rock Type, and pH on ζ-Potential and a Surface-Complexation Model. SPE Journal (2016).
Brady, P. V. & Thyne, G. Functional Wettability in Carbonate Reservoirs. Energy & Fuels 30, 9217–9225 (2016).
Venkatraman, A., Dindoruk, B., Elshahawi, H., Lake, L. W. & Johns, R. T. Modeling Effect of Geochemical Reactions on Real-Reservoir-Fluid Mixture During Carbon Dioxide Enhanced Oil Recovery. SPE Journal (2017).
Brady, P. V., Morrow, N. R., Fogden, A., Deniz, V. & Loahardjo, N. Winoto. Electrostatics and the Low Salinity Effect in Sandstone Reservoirs. Energy & Fuels 29, 666–677 (2015).
Brady, P. V. & Krumhansl, J. L. A surface complexation model of oil–brine–sandstone interfaces at 100 °C: Low salinity waterflooding. Journal of Petroleum Science and Engineering 81, 171–176 (2012).
Song, J. et al. Surface complexation modeling of calcite zeta potential measurements in brines with mixed potential determining ions (Ca2+, CO3 2−, Mg2+, SO4 2−) for characterizing carbonate wettability. Journal of Colloid and Interface Science 506, 169–179 (2017).
Xie, Q. et al. The low salinity effect at high temperatures. Fuel 200, 419–426 (2017).
Brady, P. V., Bryan, C. R., Thyne, G. & Li, H. Altering wettability to recover more oil from tight formations. Journal of Unconventional Oil and Gas Resources 15, 79–83 (2016).
Xie, Q. et al. pH effect on wettability of oil/brine/carbonate system: Implications for low salinity water flooding. Journal of Petroleum Science and Engineering 168, 419–425 (2018).
Hirasaki, G. & Zhang, D. L. Surface Chemistry of Oil Recovery From Fractured, Oil-Wet, Carbonate Formations. SPE Journal (2004).
Israelachvili, J. N. 17 - Adhesion and Wetting Phenomena. In: Intermolecular and Surface Forces (Third Edition) (ed^(ed. Israelachvili, J. N.). Academic Press (2011).
Xie, Q., Saeedi, A., Pooryousefy, E. & Liu, Y. Extended DLVO-based estimates of surface force in low salinity water flooding. Journal of Molecular Liquids 221, 658–665 (2016).
Hirasaki, G. J. Wettability:fundamentals and surface forces. SPE Formation Evaluation 6, 217–226 (1991).
Melrose, J. Interpretation of mixed wettability states in reservoir rocks. In: SPE Annual Technical Conference and Exhibition (ed^(eds) (1982).
Israelachvili, J. N. Chapter 13 - Van der Waals Forces between Particles and Surfaces. In: Intermolecular and Surface Forces (Third Edition) (ed^(ed. Israelachvili, J. N.). Academic Press (2011).
Gregory, J. Interaction of unequal double layers at constant charge. Journal of Colloid and Interface Science 51, 44–51 (1975).
Xie, Q. et al. A pH-Resolved Wettability Alteration: Implications for CO2-Assisted EOR in Carbonate Reservoirs. Energy & Fuels 31, 13593–13599 (2017).
Al Mahrouqi, D., Vinogradov, J. & Jackson, M. D. Zeta potential of artificial and natural calcite in aqueous solution. Advances in Colloid and Interface Science 240, 60–76 (2017).
Iglauer, S., Salamah, A., Sarmadivaleh, M., Liu, K. & Phan, C. Contamination of silica surfaces: Impact on water–CO2–quartz and glass contact angle measurements. International Journal of Greenhouse Gas Control 22, 325–328 (2014).
Pooryousefy, E., Xie, Q., Chen, Y., Sari, A. & Saeedi, A. Drivers of low salinity effect in sandstone reservoirs. Journal of Molecular Liquids (2017).
Hiorth, A., Cathles, L. M. & Madland, M. V. The Impact of Pore Water Chemistry on Carbonate Surface Charge and Oil Wettability. Transport in Porous Media 85, 1–21 (2010).
Pokrovsky, O. S., Schott, J. & Thomas, F. Dolomite surface speciation and reactivity in aquatic systems. Geochimica et Cosmochimica Acta 63, 3133–3143 (1999).
Van Cappellen, P., Charlet, L., Stumm, W. & Wersin, P. A surface complexation model of the carbonate mineral-aqueous solution interface. Geochimica et Cosmochimica Acta 57, 3505–3518 (1993).
Lager, A., Webb, K. J., Black, C. J. J., Singleton, M. & Sorbie, K. S. Low salinity oil recovery-an experimental investigation. SCA2006–36 (2006).
Dubey, S. & P. Doe. “Base number and wetting properties of crude oils.” SPE Reservoir Engineering 8(3), 195–200 (1993).
Buckley, J. S., K. Takamura & N. R. Morrow. “Influence of Electrical Surface Charges on the Wetting Properties of Crude Oils.” SPE Reservoir Engineering 4(03), (1989).
Brown, C. E. & E. L. Neustadter. “The Wettability of Oil/Water/Silica Systems With Reference to Oil Recovery.” Journal of Canadian Petroleum Technology 19(03), 12 (1980).
Buckley, J. S., Liu, Y. & Monsterleet, S. Mechanisms of Wetting Alteration by Crude Oils. SPE Journal (1998).
Buckley, J. S., Liu, Y., Xie, X. & Morrow, N. R. Asphaltenes and Crude Oil Wetting - The Effect of Oil Composition. SPE Journal (1997).
Suijkerbuijk, B. et al. Fundamental Investigations into Wettability and Low Salinity Flooding by Parameter Isolation. In: SPE Improved Oil Recovery Symposium (ed^(eds). Society of Petroleum Engineers (2012).
Tang, G.-Q. & Morrow, N. R. Influence of brine composition and fines migration on crude oil/brine/rock interactions and oil recovery. Journal of Petroleum Science and Engineering 24, 99–111 (1999).
Acknowledgements
This work was supported by a grant from Geoscience Australia under the China Australia Geological Storage of CO2 (CAGS) Project. We would like to extend our appreciation to Mr Bob Webb, our Technical Officer, for his help and support towards conducting the laboratory management and measurement. China Scholarship Council is thanked for providing Yongqiang Chen with scholarship for his PhD research. Sandia National Laboratories is a multimission laboratory managed and operated by National Technology and Engineering Solutions of Sandia, LLC., a wholly owned subsidiary of Honeywell International, Inc., for the U.S. Department of Energy’s National Nuclear Security Administration under contract DE-NA-0003525. This paper describes objective technical results and analysis. Any subjective views or opinions that might be expressed in the paper do not necessarily represent the views of the U.S. Department of Energy or the United States Government.
Author information
Authors and Affiliations
Contributions
Q. Xie and Y. Chen designed research, Y. Chen performed surface complexation modelling, A. Sari did contact angle tests. Q. Xie and P. Brady analysed data and wrote the paper with contribution from M. Hossain and A. Saeedi. All authors contributed to scientific discussions.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, Y., Sari, A., Xie, Q. et al. Electrostatic Origins of CO2-Increased Hydrophilicity in Carbonate Reservoirs. Sci Rep 8, 17691 (2018). https://doi.org/10.1038/s41598-018-35878-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-35878-3
Keywords
This article is cited by
-
Wettability alteration process at pore-scale during engineered waterflooding using computational fluid dynamics
Modeling Earth Systems and Environment (2022)
-
Molecular-scale origins of wettability at petroleum–brine–carbonate interfaces
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
