
Abstract
Electrochemical CO2 reduction (CO2R) using renewable electricity is a key pathway toward synthesizing fuels and chemicals. In this study, multi-physics modeling is used to interpret experimental data obtained for CO2R to CO using Ag catalysts in a membrane electrode assembly. The one-dimensional model is validated using measured CO2 crossover and product formation rates. The kinetics of CO formation are described by Marcus–Hush–Chidsey kinetics, which enables accurate prediction of the experimental data by accounting for the reorganization of the solvent during CO2R. The results show how the performance is dictated by competing phenomena including ion formation and transport, CO2 solubility, and water management. The model shows that increasing the ion-exchange capacity of the membrane and surface area of the catalyst increases CO formation rates by >100 mA cm–2 without negatively impacting CO2 utilization. Here we provide insights into how to manage the trade-off between productivity and CO2 utilization in CO2 electrolyzers.

Similar content being viewed by others

Main
Electrochemical CO2 reduction (CO2R) can be used to convert CO2 emitted from various stationary sources into valuable chemicals and fuels. Carbon monoxide (CO) is a valuable product of CO2R because of its utility as a reactant for the production of longer chain hydrocarbons via Fischer–Tropsch synthesis1. Techno-economic analyses of CO2R show that CO can be produced for as little as US$0.6–1.0 kg–1 (ref. 2); however, CO produced by steam reforming of natural gas currently costs ~US$0.15 kg–1 (ref. 3). The key to overcoming this price differential and making a CO2 electrolyzer economical is to minimize the energy costs by maximizing the partial current density for the CO evolution reaction (iCOER) and minimizing the cell voltage (Vcell)4,5.
Membrane electrode assemblies (MEAs) are the most efficient CO2 electrolyzer architectures for attaining high rates of CO formation6. MEA devices for CO2R are similar in construction to water electrolyzers and hydrogen fuel cells, consisting of porous electrodes separated by a thin ion-exchange membrane. The use of an anion-exchange membrane (AEM), a type of ion-exchange membrane composed of a polymer with fixed positive charges (typically quaternary amines), has been shown to enable high CO formation rates (iCO > 200 mA cm–2) in CO2 electrolyzers7. However, AEMs also permit the crossover of (bi)carbonate anions (that is, HCO3− and CO32−) from the cathode to the anode, where the (bi)carbonate anions are converted by a pH swing to reform CO2, which is lost from the anode8. The CO2 crossover rate is therefore another figure of merit that must be considered when designing a CO2 electrolyzer because it represents an efficiency loss that defines the costs associated with separating and recycling the CO2 reactant downstream of the electrolyzer9,10. Cation-exchange membranes and bipolar membranes can decrease the rate of CO2 crossover in CO2 electrolyzers by delivering protons to the cathode that reconvert (bi)carbonates back into CO2 before they cross the membrane11,12. However, these membranes yield lower CO partial current densities because of the acidic conditions furnished at the cathode, which promote the hydrogen evolution reaction (HER)13.
While the development of membranes and electrodes tailored for CO2R is an active field,14 relatively few theoretical studies have investigated how the properties of the materials used in electrolyzers impact the rate of CO2R in an MEA, and even fewer studies combine self-consistent experimental and theoretical studies. Several models of MEAs for CO2R exist that examine the physics of CO2 transport15,16,17,18. However, these models do not adequately account for the chemistry occurring at the anode, in the anolyte and in the membrane adjacent to the cathode, all of which impact the performance of an MEA. Consequently, these models cannot accurately reproduce experimental polarization and CO2 crossover measurements simultaneously. More sophisticated continuum models that correlate the performance of CO2R MEAs with material properties and operating conditions are therefore required to resolve the mechanisms of transport losses and optimize the properties of the membrane, catalyst layer and electrolyte. Consideration of electro-osmosis and water management is particularly important, but these phenomena are often neglected in CO2R studies.
In this Article, we describe experimental investigations of CO2R to form CO on Ag in MEA cells and use the crossover and partial current density data to inform and validate the development of a continuum one-dimensional (1D) multi-physics cell model. The model is then used to explore the effects of the thickness and electrochemically active surface area of the catalyst layer, ion-exchange capacity (IEC) of the membrane and anolyte concentration on CO2R performance metrics including CO2 crossover from cathode to anode, which is due to changes in the identity of the charge-carrying species. Key to predicting the rate of CO2R at high overpotentials is the adoption of Marcus–Hush–Chidsey (MHC) theory (which accounts for the reorganization of solvent molecules during electron transfer) as opposed to traditional Tafel kinetics. The experimentally validated model utilizes chemical engineering fundamentals to guide the design of next-generation CO2 electrolyzers.
Results
Model development and validation
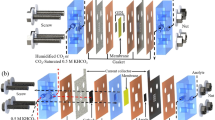
A 1D isothermal continuum model was developed in COMSOL Multiphysics 6.0 to simulate the partial current densities for H2, CO2 and O2 formation in a CO2 electrolyzer MEA and the crossover of carbon from cathode to anode (Fig. 1a,b). The model consists of five domains: a platinized titanium anode porous transport layer (thickness of 190 µm), an iridium oxide anode catalyst layer (10 µm), an ion-exchange membrane (50 µm), a cathode catalyst layer (10 µm) composed of silver nanoparticles on carbon mixed with ionomer and a cathode gas diffusion layer (GDL; 325 µm; Fig. 1c). Volume-averaged properties for each phase are used as inputs to solve the conservation equations for momentum, mass and charge transport (full model description in Methods). Model parameters are presented in Supplementary Tables 1–6.

a, Schematic diagram of a CO2 electrolyzer MEA with anodic and cathodic feedstocks of 1 M CsHCO3 and humidified CO2, respectively. b, Schematic diagram showing the processes of CO2 reduction, (bi)carbonate formation and crossover, and CO2 loss at the anode. c, The 1D continuum model domain showing the gas (g), liquid (aq), solid and ionomer phases.
Experimentally, the CO2 electrolyzer was operated at constant cell potentials ranging from 2.35 V to 4.00 V while measuring the partial current densities for CO and H2 formation19 (Fig. 2a,b). The CO formation rate increased to a peak value of 300 mA cm–2 as the potential was increased to 3.20 V. The CO partial current density then decreased as the potential was increased further to 4.00 V, and the H2 partial current density increased to 850 mA cm–2. The model was fit to the experimentally measured polarization data by adjusting the parameters in the MHC expression for the rate of CO2 reduction (that is, k0,CO and λreorg; equation (16) in Methods) and in the Tafel equation for HER (that is, i0,HER,base and αc,HER; equation (11) in Methods) while holding the other model parameters constant. Quantitative agreement between the model and experimental CO partial current densities is observed for the MHC model when λreorg = 1.34 eV is used (Fig. 2a). This value agrees with the range of fit values of λreorg (0.8–2.0 eV)20 for CO2R experiments performed with planar silver cathodes (see Supplementary Note 1 and Supplementary Fig. 1 for sensitivity analysis and more details on the physical interpretation of this value). Moreover, the charge transfer coefficient for H2 (αc,HER = 0.13) agrees well with a recent study using continuum modeling to resolve kinetic parameters of H2 formation on planar silver21. The applied voltage breakdown (Supplementary Note 2 and Supplementary Fig. 2) demonstrates that the kinetic overpotentials are the dominant contributors to the overall cell voltage. The high overpotential associated with the CO evolution reaction (COER) highlights the need to develop more efficient electrocatalysts. Moreover, the ionic and electrical resistances account for >300 mV of voltage loss at 1,000 mA cm–2. This result indicates that more conductive porous transport layers and membranes may also improve performance.

a,b, Experimental and modeled partial current densities for CO (a) and H2 (b) formation as a function of cell voltage. c, Experimental and modeled CO2 crossover fluxes. d, Comparison of experimental CO partial current densities with simulated results obtained with MHC and Tafel models.
To validate the mass-transport framework used in the model, the simulated and experimentally measured CO2 crossover fluxes were compared over a current density range of 0–1,050 mA cm–2 (Fig. 2c). Good agreement is observed between the model and experiment at high current densities; however, the model slightly overpredicts the CO2 crossover flux at lower current densities. This discrepancy exists because CO2 bubbles that form in the anode chamber are recirculated to the anolyte CsHCO3 reservoir by the peristaltic pump. This process provides the opportunity for CO2 to redissolve and buffer the anolyte, which decreases the amount of CO2 in the anolyte headspace measured experimentally. A model that encompasses the along-the-channel effects and anolyte reservoir is necessary to capture this effect.
MHC theory has been employed widely in the field of electrochemical engineering because it accounts for electron-transfer limitations at high overpotentials and the free-energy change associated with reorganizing the solvent after electron transfer22. By contrast, the Tafel model (equation (18) in Methods) assumes ion-transfer limits the overall rate of CO2R23. These two models both exhibit good agreement between the experimental data at low cell potentials where electron-transfer limitations are not expected (Fig. 2d). The agreement between the Tafel model and experiment at low potentials shows that the kinetic parameters obtained from experiments with planar silver electrodes are reasonably transferable to MEA experiments21. However, the Tafel model overpredicts the CO formation rate at high cell potentials, even when mass-transport effects are taken into account. We considered that agreement between the experimental data and Tafel model could be improved by incorporating a fit film resistance into the effective overpotential (equation (19) in Methods and Supplementary Fig. 3). The film resistance improves agreement between the Tafel model and experimental CO partial current densities at high cell potentials at the expense of agreement at low cell potentials. However, the fit film resistances (102 Ω cm2) are larger than the values expected from measurements of silver oxide films24. Collectively, these results suggest that both electron-transfer and mass-transfer limitations occur for CO2R in MEAs and that film resistances cannot explain curvature in the CO partial current density data. The need to use MHC to capture these electron-transfer limitations is consistent with the calculation of large barriers for solvent reorganization23, although further experimental investigation is needed to confirm this.
Because CO2 reduction involves bond breaking and reforming, there must be a contribution associated with ion transfer in addition to electron transfer. This mechanistic interpretation suggests the need for models beyond MHC, which purely accounts for electron transfer, toward models of coupled ion–electron transfer (CIET) that account for barriers associated with ion transfer and electron transfer. Work by Bazant23 has demonstrated that the rate law for an electron-transfer limited CIET is similar to the MHC kinetics utilized herein that provides the best fit to our experimental data (Supplementary Note 3), where the contribution associated with ion transfer is found in the pre-factor of the rate expression. Accordingly, the value of k0,CO fit to the MHC kinetics used in the present study can be considered a lumped parameter that implicitly includes the effect of ion transfer on the rate of CO2R. This analysis suggests that CO2R is probably governed by coupled ion–electron transfer kinetics in the limit of electron-transfer limited CIET. The use of electron-transfer limited CIET can also potentially explain the high value of the fit reorganization energy (λreorg = 1.34 eV) because this limit is most applicable when the effective ion-transfer free-energy barrier and the magnitude of the formal overpotential are much smaller than the reorganization energy. Thus, this higher value of the reorganization energy is consistent with the fact that ions are completely breaking and reforming solvation shells that couple to electron transfer and is perhaps exceeding ion-transfer energies for molecular bond breaking/reforming.
The assertion that CO2R occurs via an electron-transfer limited CIET is further supported by alternatively considering CIET in the ion-transfer limit. The work of Bazant on CIET also introduced a rate expression for ion-transfer-limited CIET23, which is similar to the BV kinetics (now with a pre-factor that accounts for barriers associated with electron transfer) that struggle to fit our experimental data. Hence, similar to how BV kinetics are shown to not be descriptive of our experimental data, ion-transfer limited CIET is also unlikely to explain polarization data observed in CO2R (Supplementary Note 4), suggesting that electron-transfer limitations, rather than ion-transfer limitations, dictate observed rates.
Transport of CO2 and ionic species
The validated model was used to investigate the transport of CO2 and ionic species in the CO2R MEA (Fig. 3). At 0 mA cm–2, the pH throughout the MEA resembles that of the 1 M CsHCO3 solution and the CO2 concentration profile is indicative of the gradient between the bulk concentration of CO2 present in the anode chamber and the maximum solubility of CO2 in 1 M CsHCO3 at 50 °C for a gas phase containing 88 wt% CO2 and 12 wt% H2O (14.7 mM; Fig. 3a). As the current density increases, the CO2 concentration increases in the anode catalyst layer as H+ produced by acidic oxygen evolution reaction (OER) decreases the local pH (Fig. 3b), shifting the (bi)carbonate equilibrium toward HCO3− (Fig. 3c)25. Concurrently, the pH in the cathode catalyst layer increases to >14 at 1,000 mA cm–2 due to hydroxide formation (Supplementary Fig. 4). This elevated current density decreases the CO2 concentration at the cathode by consuming CO2 to form CO, shifting the (bi)carbonate equilibrium from CO2 to CO32− (Fig. 3d) and increasing the total ionic concentration of the solution, thereby reducing the CO2 solubility. A main contributor to the reduced solubility of CO2 is the high concentration of cesium cations present in the cathode at high current densities (Supplementary Fig. 4), which originate from the anode and decrease the Henry’s constant from 14.7 mM atm–1 CO2 to 2 mM atm–1 CO2 (per equation (52) of Methods). Consistent with previous studies, the Donnan exclusion effect of the AEM is insufficient to block cation transport from anode to cathode26. The net result of these phenomena is a decrease in reactant CO2 available for the CO2R, and consequently, a decrease in CO formation rates at high cell potentials (Fig. 2a). Other possible effects of cesium on the rate of CO2R and the HER are discussed in Supplementary Note 5.

a–d, Modeled CO2 concentration (a), pH (b), HCO3– concentration (c) and CO32– (d) as functions of current density and position in the MEA. aCL, anode catalyst layer; cCL, cathode catalyst layer; aPTL, anode porous transport layer.
The partial pressure of CO in the cathode catalyst layer increases as the current density is increased from 0 to 200 mA cm–2 before decreasing at the onset of H2 evolution (Supplementary Fig. 5). The CO2 partial pressure decreases because CO2 dissolves and reacts in the cathode catalyst layer (Supplementary Fig. 5). The H2O partial pressure in the cathode catalyst layer decreases throughout the GDL as the current density is increased from 200 to 1,000 mA cm–2 because of the pressure drop through the GDL and consumption of water by CO2R and the HER. In the anode, the partial pressure of CO2 increases as the current density increases from 0 to 200 mA cm–2 because of the increase in CO2 concentration in the electrolyte (Supplementary Fig. 6). However, at 1,000 mA cm–2, the CO2 partial pressure in the anode decreases because the partial pressure of O2 increases (Supplementary Fig. 6) and the equilibrium between the CO2 in the gas and electrolyte phases is established. These nonintuitive results demonstrate the importance of modeling the dynamics of the gas phase and the CO2 phase equilibrium.
To understand the crossover of carbon from cathode to anode (Fig. 2c), we examined the transference numbers in the MEA. The CsHCO3 delivered to the anode generates a HCO3− concentration gradient that drives the transport of HCO3− from anode to cathode against the potential gradient. Consequently, a negative transference number for HCO3− is observed in the membrane at 10 mA cm–2 (Fig. 4a). To maintain charge conservation, CO32− is transported from cathode to anode with a transference number of ~2 in the membrane at 10 mA cm−2 (Fig. 4b). These opposing fluxes of carbon yield a proportional relationship between CO2 crossover flux and current density from 0 to 200 mA cm−2. The decrease and subsequent plateau in CO2 crossover at 200 mA cm−2 coincide with the appearance of CO32− as the sole charge-carrying species in the membrane. As the current density is increased to 1,000 mA cm−2, hydroxide emerges as the main charge carrier through the membrane (Fig. 4c), which reduces Ohmic losses due to the faster diffusion of OH− relative to CO32−. This transition in charge-carrying species from CO32− to OH− also decreases the CO2 crossover flux, however, CO2 is still measured in the anode effluent of the electrolyzer due to the continuous conversion of HCO3− from the anode feedstock into CO2. Consequently, HCO3− remains the dominant charge carrier in the anode at all current densities (Fig. 4a). The abrupt changes in transference numbers seen in the membrane are a result of the pH gradient through the membrane, which transforms HCO3− fed to the anode into CO32− and CO32− originating from the cathode into HCO3−. The flux of dissolved CO2 accounts for less than 1% of the total dissolved carbon that crosses the membrane. The results here underscore the opportunity to operate at high current densities where OH− is the charge carrier to reduce Ohmic losses and the need to model both the anode and cathode of a CO2 electrolyzer to describe electrolyzer performance accurately.

a–c, Modeled transference numbers for HCO3− (a), CO32− (b) and OH− (c) in the MEA as a function of current density. d, Modeled water fluxes across the membrane due to diffusion (dashed lines) and electro-osmosis (solid lines) and net water flux as a function of current density. e, Diagram showing the number of water molecules moved by electro-osmosis between the anode and cathode depending on the dominant charge-carrying species. aCL, anode catalyst layer; cCL, cathode catalyst layer.
Water transport and electro-osmosis
Water transport in the membrane is driven by chemical potential gradients and ionic fluxes, which lead to a decrease in water activity and liquid water pressure in the membrane as the current density is increased (Supplementary Fig. 7). The diffusional flux of water (from anode to cathode) is directly proportional to the water activity gradient in the MEA, whereas the flux of water due to electro-osmosis is dependent on the ionic charge carrier because the electro-osmotic coefficient is roughly equal to the number of water molecules in the hydration shell of each ion27 (equations (57) and (58) in Methods). As shown in Fig. 4d, the flux of water associated with diffusion and electro-osmosis both increase with increasing current density. At 10 mA cm–2, the diffusional flux of water is balanced by electro-osmosis. The average electro-osmotic coefficient in this current density regime is 12, because HCO3− and CO32− move in opposite directions along with 8 and 20 water molecules, respectively (Fig. 4e). At 200 mA cm–2, the flux of HCO3− approaches 0 (Fig. 4a), and therefore the average electro-osmotic coefficient corresponds to that of CO32− (that is, \({\zeta }_{\mathrm{CO}_{3}^{2-}}\) = 20). The flux of water associated with electro-osmosis at 200 mA cm–2 is higher than the flux of water due to diffusion as a result of the high electro-osmotic coefficient of CO32−, and therefore the net water flux is negative (from cathode to anode). At 1,000 mA cm–2, water transport by diffusion is greater than water transport by electro-osmosis because OH− (with an electro-osmotic coefficient of \({\zeta }_{{\rm{O}}{{\rm{H}}}^{-}}\) = 6) becomes the majority charge carrier, giving rise to a positive net water flux. This interplay points to intermediate current densities (200 mA cm–2) as being most susceptible to membrane dehydration due to the high electro-osmotic coefficient of CO32− relative to OH−, which is a nonintuitive result arising from the complicated coupling of interspecies transport and reaction rates.
The net electro-osmotic coefficient (denoted as β; equation (59) in Methods) describes how much water is transported through the membrane from anode to cathode relative to the amount of water consumed by the electrochemical reactions (Supplementary Fig. 8). At low current densities (that is, 10 mA cm–2), β is negative, which means that the net flux of water is directed away from the cathode, indicating that water must come from the humidified CO2 gas to supply the reactant for the CO2R and HER. As the total current density is increased to 500 mA cm–2 (at which point the CO formation rate is at its maximum), β becomes positive. At 1,000 mA cm−2, β increases to ~0.8. This result indicates that, when OH− is the dominant charge carrier, enough water is transported through the membrane to sustain the electrochemical reactions. Accordingly, the use of a humidified CO2 feed may not be necessary at high current densities when a liquid anolyte feed is used. These results show that water is not the limiting reagent in CO2R. Nonetheless, water management is still critical in CO2 electrolyzers because it impacts the Ohmic losses and transport of species in the membrane.
Effect of catalyst-layer thickness and surface area on CO2R
The validated model allows for the investigation of key properties and virtual experiments to provide design guidance by analysing the complex trade-offs endemic to this system. Herein the cathode catalyst-layer thickness and specific surface area (SSA) were explored. The model demonstrates that reducing the cathode catalyst-layer thickness from 10 to 1 µm decreases the partial current density for CO (Fig. 5a) and H2 formation (Supplementary Fig. 9). The H2 partial current density decreases less than the CO partial current density, which leads to a lower faradaic efficiency for CO (FECO) at high current densities (Supplementary Fig. 9). The lower H2 partial current density with thinner catalyst layers comes as a result of the lower surface area relative to the thicker catalyst layers. Moreover, at a constant current density, the pH is higher in the smaller volume of the thinner catalyst layers for the same flux of OH− from CO2R and the HER (Supplementary Fig. 9). The higher pH in the thinner catalyst layer reduces the CO2 concentration (Supplementary Fig. 9). The CO partial current density is therefore more sensitive to increasing the catalyst-layer thickness than the partial current density for the HER because of the compounding effects of increased surface area and CO2 reactant concentration. While these phenomena lead to a higher CO partial current density with thicker catalyst layers, the CO2 utilization (that is, the amount of CO formed divided by the amount of CO2 that is reacted to form HCO3− and CO32− and subsequently released at the anode; equation (77) in Methods) follows the opposite trend (Fig. 5b). This trade-off suggests an optimum catalyst layer thickness of 5 µm to simultaneously enable a CO partial current density >200 mA cm–2 and CO2 utilization efficiency >60 %.

a,b, Partial current density for CO formation as a function of cell voltage (a) and CO2 utilization as a function of total current density (b) for cathode catalyst layers with thicknesses of 10, 5, 2.5 and 1 µm at a constant SSA of 3 × 107 m−1. c,d, Partial current density for CO formation as a function of cell voltage (c) and CO2 utilization as a function of total current density (d) for catalyst layers with a SSA of 1 × 107, 3 × 107 and 7 × 107 m−1 at a constant cathode catalyst-layer (cCL) thickness of 10 µm. The base-case simulation results are shown with solid lines.
Catalyst SSA is another important characteristic of CO2 electrolyzers that is inversely proportional to the size of the catalyst particles as per equation (21) in Methods. The model shows that increasing the SSA (that is, reducing the particle size while maintaining the catalyst mass) increases both the CO partial current density (Fig. 5c) and the H2 partial current density (Supplementary Fig. 10) owing to a higher availability of surface sites for electron transfer. The lower SSA catalyst layers achieve a higher peak FECO (Supplementary Fig. 10) owing to the lower onset potential for H2 formation, but this peak diminishes at the onset of CO evolution. The higher CO partial current density with higher SSA catalyst layers is enabled by a higher surface area for transport of CO2 across the gas/ionomer interface, as per equation (51) of Methods. The faster transport of CO2 across the ionomer layer yields a higher CO2 concentration relative to the maximum solubility (Supplementary Fig. 10). Consequently, the increased CO formation rates enabled by decreasing particle size come at the expense of increased crossover of CO2 across the membrane (Supplementary Fig. 10). At high current densities, however, the crossover of CO2 is not strongly affected by catalyst particle size. Therefore, a higher CO2 utilization is observed with smaller catalyst particles at high current densities (Fig. 5d). The implication of these results is that smaller catalyst particles enable higher CO formation rates than larger catalyst particles without decreasing the CO2 utilization at high current densities.
The thickness of the GDL was found to have less of an effect on CO formation in MEAs than catalyst-layer thickness and SSA (Supplementary Fig. 11). Decreasing the GDL thickness from the nominal value for Sigracet 39BC (325 µm) to 100 µm led to a smaller gas-phase pressure drop between the flow channel and cathode catalyst layer. Consequently, a higher partial pressure for CO2 is observed in the cathode catalyst layer for thinner GDLs (Supplementary Fig. 11), which increases the concentration of CO2 in the electrolyte. This higher CO2 concentration modestly increases the peak CO partial current density (Supplementary Fig. 11).
Effect of anolyte pH and membrane IEC on CO2R and the HER
HCO3− is commonly used as a buffering anion for CO2R because it slows the pH rise at the cathode to reduce the rate of conversion of CO2 to (bi)carbonates28,29. However, Sustainion is much more conductive in its OH− form than in its HCO3− form7. To examine these trade-offs, we simulated a CsOH anolyte and compared the results to those obtained with CsHCO3. Switching the anolyte from CsHCO3 to CsOH increases the average conductivity of the membrane from ~12 mS cm−1 to ~25 mS cm−1 (Supplementary Fig. 12), which reduces the ohmic resistance associated with ion transport. These results are consistent with independent conductivity measurements documented in a previous study30. Accordingly, the cell voltage for maximum CO formation is shifted to 3 V with CsOH, compared with 3.3 V with CsHCO3 (Supplementary Fig. 12). The lower peak in the CO formation rate with CsOH compared to CsHCO3 is caused by the higher pH in the cathode catalyst layer (Supplementary Fig. 12). This higher pH with CsOH decreases the CO formation rate by decreasing the concentration of CO2 in the cathode relative to CsHCO3 (Supplementary Fig. 13). Additionally, the CsOH anolyte increases the net electro-osmotic coefficient (β) relative to CsHCO3 (Supplementary Fig. 13) because of the larger flux of OH− relative to HCO3− and CO32−, which increases the water activity at the cathode, thereby giving rise to faster HER kinetics.
The conductivity of the membrane is directly correlated with the concentration of fixed-charge groups (that is, the IEC). Although commonly used, Sustainion has a notably low IEC compared with other AEMs14. The model shows that increasing the IEC of the membrane from 1.2 to 2.0 mmol g−1 leads to a dramatic increase in the CO partial current density (Fig. 6a). By contrast, the H2 partial current density is not strongly impacted by an increase in IEC (Supplementary Fig. 14). The higher IEC membranes expectedly exhibit higher conductivities (Fig. 6b). However, the high IEC membranes also decrease the pH (Fig. 6c) in the cathode catalyst layer by excluding co-ions (for example, H+ and K+) via Donnan exclusion, trapping them in the cathode catalyst layer, while also more efficiently transporting alkaline counter-ion species (that is, CO32− and OH−) away from the cathode catalyst layer, which reduces the overpotential that drives HER. The lower HER overpotential has a larger impact on the H2 partial current density than the increased conductivity, therefore, a slightly lower H2 partial current density is observed for membranes with a higher IEC (Supplementary Fig. 14). It is important to note that the model does not account for the effect of IEC on the swelling behavior and mechanical integrity of the ionomer, which could lead to a decrease in performance as the IEC is increased.

a, CO partial current density as a function of cell voltage for simulated IECs of 1.2, 1.5 and 2.0 mmol g−1. b, Ionic conductivity of the different IEC membranes at a cell potential of −2.3 V. c, pH in the cathode catalyst of the different IEC membranes at a constant total current density of 100 mA cm−2. d, CO2 utilization as a function of current density and membrane IEC. The base-case simulation results are shown with solid lines.
The lower pH in the cathode catalyst layer observed in the higher IEC membranes reduces the extent of CO2 conversion into CO32−, thereby enabling a higher CO2 concentration (Supplementary Fig. 14). This effect, combined with the higher conductivity of the higher IEC membranes, explains why a higher CO partial current density is observed. The higher IEC membranes also increase the crossover of carbon from cathode to anode because of the higher affinity for CO32− transport over OH− transport (Supplementary Fig. 15). However, the increase in CO formation rate enabled by higher IEC membranes is larger than the increase in CO2 crossover rate, which results in higher CO2 utilization (Fig. 6d). Collectively, these results show that increasing the IEC of the membrane provides a means for increasing CO formation rates without enhancing the rate of the HER or reducing CO2 utilization.
Hydroxides can neutralize the positively-charged imidazolium groups in ionomer membranes, leading to ‘current induced membrane discharge’ (CIMD)31. Our simulation shows that CIMD decreases the CO partial current density at all potentials relative to the base case, which assumes a unity fraction of protonated imidazolium groups (that is, no CIMD occurs; Supplementary Fig. 16). The decrease in CO formation caused by CIMD is more pronounced for more weakly basic fixed-charge sites (that is, sites with higher pKb values; Supplementary Note 6). For a pKb of −2, the simulation results are nearly identical to the basecase. The CO2 crossover flux is shown to decrease with increasing pKb values due to a lower uptake of HCO3− and CO32− in the membrane (Supplementary Fig. 16). This phenomenon occurs because of the reduced fraction of protonated imidazolium groups in the membrane (Supplementary Fig. 16). The reduced concentration of protonated imidazolium increases the concentration of Cs+ in the membrane, thereby increasing the propensity for precipitation in CO2 electrolyzers (Supplementary Fig. 16). These results indicate that increasing AEMs must furnish highly concentrated and basic fixed-charge groups to enable high CO formation rates.
Discussion
In this study, we used a 1D continuum model validated against measured experimental data to investigate the myriad of coupled phenomena occurring in MEAs undergoing CO2 reduction. The model reveals that both mass- and electron-transfer limitations dictate limiting current densities for CO2R. The latter effect necessitates the use of MHC theory to reproduce experimental behavior and highlights the need for further studies investigating the nature of electron transfer and solvent reorganization in CO2R. This study demonstrates how continuum modeling can be used to link quantum mechanical theories of electron transfer with device-scale performance.
The simulations show that CO2 and water transport across the MEA are defined by the principal charge-carrying species in the membrane, which varies depending on the current-density regime. At low current densities (10 mA cm−2), the transport of HCO3− from anode to cathode against the potential gradient decreases the amount of CO2 emitted at the anode and reduces the net flux of water from anode to cathode. At intermediate current densities (200 mA cm−2), CO32− is the sole charge carrier, which increases the flux of CO2 and water from cathode to anode, thereby reducing the concentration and activity of water and CO2 at the cathode, respectively. Finally, at high current densities (1,000 mA cm−2), the high pH in the MEA gives rise to OH− as the principal charge-carrying species. The transport of OH− reduces the crossover of carbon and water from cathode to anode, which leads to improved hydration and conductivity of the membrane. Anion-exchange membranes with strongly basic fixed-charge groups are key to transporting OH− efficiently and mitigating current-induced membrane discharge; thus, with less stable or charged membranes this could become limiting.
Model sensitivity analyses performed on the anolyte, catalyst layer and membrane properties demonstrate the trade-off between CO formation rates and carbon crossover in CO2 electrolyzers that arise from the coupling between interspecies ionic and water transport and various chemical and electrochemical kinetics. Thicker catalyst layers yield higher CO formation rates, but also lower CO2 utilizations. Catalyst layers with higher SSAs (that is, catalysts with smaller particle sizes) increase the CO formation rate without negatively impacting CO2 utilization at high current densities. Membranes with higher IEC reduce Ohmic resistances and lower the pH at the cathode, which benefits CO formation and increases CO2 utilization without enhancing deleterious HER. Collectively, these results provide insights into the dynamics of water and ion transport in CO2 electrolyzers and present different materials optimization strategies that inform future experiments. Furthermore, this study highlights the importance of using self-consistent (electro)chemical engineering fundamentals to explore and unravel such complex systems.
Methods
Governing equations for charge, mass and momentum transport
Electron transport in the conductive solid phases (Ti anode transport layer, Ir-oxide anode catalyst layer, Ag cathode catalyst layer and carbon GDL) was modeled using Ohm’s law
where is and ɸs represent the current density and electric potential in the solid electron-conducting phases. The effective conductivity (σeff) of the porous electron-conducting phases was determined using a Bruggeman relation
where εS and σs are the volume fraction and nominal conductivity of the solid conductor, respectively (see Supplementary Table 1 for values). The governing equations for mass transfer of each chemical species j in the gas phase (j of CO2(g), CO(g), H2(g) and H2O(g)) and i in the electrolyte phase (i of H+(aq), OH– (aq), HCO3– (aq), CO32– (aq), CO2(aq) and Cs+(aq)) are
where Jj and Mj are the mass flux and molecular weight of gaseous species j, respectively, ρG is the density of the gas phase, uG is the mass-averaged velocity of the gas phase, ni is the molar flux of dissolved species i, εG is the gas volume fraction and Rk,i/j is the volumetric rate of mole generation/consumption by process k of species i or j. The volume fraction of ionomer (εI) in the catalyst layers was determined on the basis of the ionomer-to-catalyst mass ratio of 3 used in the precursor catalyst ink. The volume fractions of gas and liquid (εL) in the remainder of the void space were determined on the basis of the capillary pressure (pcap) in the catalyst layer using experimental water saturation data for Pt/C from Zenyuk et al.32 (Supplementary Fig. 17), which was assumed to hold for Ag/C, where
The implicit assumption of equation (4) is that the buffer reaction kinetics are the same in the ionomer and bulk electrolyte. While these kinetics may in fact vary between the two phases, previous work has shown that the kinetic parameters obtained from bulk electrolyte measurements provide accurate approximations of the (bi)carbonate buffer dynamics in AEMs33.
The momentum balances on the liquid and gas phases are given as
where QG is the net rate of mass generation in the gas phase. ρL, uL and QL are the density, mass-averaged velocity and net rate of mass generation in the liquid phase.
Reaction chemistry
At the anode, iridium oxide catalyzes the OER under acid and basic conditions as
The silver cathode catalyst catalyzes the COER and the HER, which occurs through both H+ and H2O reduction as
The electrochemical formation of H2 through water and proton reduction is described by the following concentration-dependent Tafel expressions:
where i0,HER,base and i0,HER,acid are the exchange current densities for the HER in base and acid, respectively. αc,HER and ηHER are the cathodic transfer coefficient and overpotential of HER, respectively. F is Faraday’s constant (96,485 C mol−1), R is the ideal gas constant (8.314 J mol−1 K−1) and T is the temperature (323 K). These reactions were assumed to exhibit a first-order dependence on the reactant concentration (\({c}_{\mathrm{H}^{+}}\)) or water activity (aw). Nonidealities were not considered for the ionic species (that is, H+) owing to the increased computational complexity and lack of available parameters for activity correlations for concentrated multi-component electrolytes. Therefore, the fit exchange current densities employed in the model can be considered lumped parameters that encompass the nonidealities. The overpotential of each electrochemical reaction k (ηk) was calculated according to the reduction potential of reaction k at standard conditions (\({U}_{k}^{\,0}\)), accounting for changes in the local pH using the Nernst equation
The partial current densities for OER in acid (iOER, acid) and base (iOER, base) were modeled using concentration-dependent Tafel expressions similar to those used for the HER at the cathode34
where i0,OER,base and i0,OER,acid are the exchange current densities for the OER in base and acid, respectively. αa,OER and ηOER are the anodic transfer coefficient and overpotential of the OER, respectively.
While the HER and OER have been shown to follow a simple Tafel relationship35, recent work has pointed to the need to account for the reorganization energy (λreorg) of the products, reactants and solvent when modeling the CO2R reaction due to observed limitations in current density that cannot be solely attributed to mass transfer20. Therefore, a simplified asymptotic approximation of the indefinite integral in MHC theory is used to relate the partial current density of inner-sphere electron transfer to the overpotential and change in free energy of the solvent, products and reactants in the absence of electron transfer36,37,
where k0,CO is a constant pre-exponential factor that relates to the attempt frequency for CO2R and erfc is the complementary error function. The rate order of 1.5 for CO formation with respect to CO2 concentration was deduced on the basis of experiments performed at different partial pressures of CO238. In MHC, λreorg is the energy barrier associated with the reorganization of the water network in the catalytic reaction environment to accommodate the quantum tunnelling of electrons from the electrode to CO2 (see Supplementary Fig. 18 for a schematic depiction of solvent reorganization). \({\widetilde{\eta }}_{\rm{CO}}\) is the dimensionless overpotential, which is defined as
For the sake of comparison with the MHC kinetics above, CO2R was also modeled using Tafel kinetics as
where i0,COER, αc,COER and ηCOER are the exchange current density, cathodic transfer coefficient and overpotential for the CO evolution reaction, respectively. Kinetic parameters are given in Supplementary Tables 2 and 3. There is precedent to account for a film resistance (Rfilm) within the overpotential term of Tafel expressions. This empirical kinetic formulation was also considered
The molar consumption of reactants and generation of products in the electrolyte by the charge transfer (CT) reactions follows Faraday’s Law
where nk is the number of electrons transferred in the reaction and si,k is the stoichiometric coefficient for species i in reaction k (that is, the OER, HER or CO2R reaction). This source term applies to the generation and consumption of H+, H2O, OH− and CO2. The catalyst-layer specific surface area (av) was determined using the geometric relationship from Weng et al.17
where rp is the average radii of the catalyst particles, which was estimated as 25 nm for the basecase simulation based on transmission electron microscopy images (Supplementary Fig. 19).
Within the electrolyte phase, the following homogeneous reactions between CO2, OH−, CO32−, HCO3− and H+ occur:
The consumption and generation of chemical species i (including water) by these buffer (B) reactions is governed by the law of mass action
where Kn is the equilibrium constant of buffer reaction n, si,n is the stoichiometric coefficient of species i the corresponding bulk reaction n (si,n < 0 for reactants and si,n > 0 for products). kn and k–n are the forward and reverse reaction rate constants for reaction n, respectively. Parameters for these reactions are given in Supplementary Table 4.
Ion transport
The molar ionic fluxes in the electrolyte (ni) were taken to occur via diffusion, migration and convection under conditions where dilute solution theory applies
Here zi is the charge of the mobile ionic species and ɸI is the ionic potential. Electroneutrality was assumed with consideration of the fixed charges present in the catalyst-layer ionomers and membrane
where ρionomer and IECionomer,effective are the density and effective ion-exchange capacity (mol g−1 wet polymer) of the ionomer. The chemical equilibrium between the imidazolium groups in the Sustainion ionomer and hydroxides is
where −NH2 and \(-{\rm{N{H}}}_{3}^{+}\) are the deprotonated and protonated imidazolium groups. This equilibrium reaction results in neutralization of fixed charge groups in the ionomer (that is, current-induced membrane discharge) that reduces the effective ion-exchange capacity of the ionomer31. The isotherm describing this phenomenon is given defined by the pKB of the imidazolium groups (pKB,ionomer) as
where αCIMD is the fraction of protonated imidazolium groups. The effective ion-exchange capacity of the ionomer is therefore given as
The base-case simulation assumes αCIMD = 1, and the pKB,ionomer was varied for sensitivity analysis.
Within the catalyst layers, ion transport was assumed to occur through both liquid electrolyte and ionomer. To model this situation, we assumed a single electrolyte phase composed of a mixture of electrolyte and ionomer with a density of fixed charges equal to the volume fraction of ionomer in the catalyst layer multiplied by the nominal fixed-charge density of the ionomer. Physically, this formulation suggests that that the fixed-charges present in the ionomer are homogeneously dispersed in the electrolyte volume of the catalyst layer. The effective diffusion coefficients were determined based on the water mole fraction (xi) and volume fraction of water in the ionomer Φw
where Di,w is the diffusion coefficient of species i in water (Supplementary Table 5), q is a tortuosity parameter that was fit to experimentally measured conductivity data of Sustainion measured in bicarbonate and hydroxide forms7, ψi is the ratio between species–solvent and species–membrane interaction effects, which depend on the reduced molecular weight of species i in the membrane (Mi,M) and in water (Mi,w),
where MM is the molecular weight of the membrane, which is set to an arbitrarily large value of 10,000 g mol−1 and Vw is the molar volume of water. The molar volume of the membrane (VM) was calculated on the basis of the ion-exchange capacity and the dry density of the ionomer
The mole fraction (xw) and volume fraction of water (Φw) in the ionomer were calculated using the water uptake (λ) assuming free swelling
The value of λ was determined as a function of water activity and the fraction of imidazolium groups exchanged with carbonates or hydroxides based on constant temperature water-uptake measurements for Sustainion39 (Supplementary Fig. 20). While the co-ion (Cs+) is known to also impact the water uptake, this data is not available in the literature and therefore is neglected in the model. Donnan equilibrium was imposed at each interface to maintain charge neutrality in the catalyst and membrane layers
where \({\phi }_{i,{{\rm{x}}}^{+}}\) is the electrolyte potential on the right side of the interface and \({\phi }_{i,{{\rm{x}}}^{-}}\) represents the same quantity on the left side of the interface. Similarly, \({c}_{i,{{\rm{x}}}^{+}}\) and \({c}_{i,{{\rm{x}}}^{-}}\) are the concentrations on the right and left side, respectively. The convective velocity of the liquid phase (uL) in the catalyst layer pores was determined using Darcy’s law
where μL is the liquid viscosity, pL is the pressure of the liquid phase and \({\kappa }_{\rm{sat}}^{0}\) is the bulk saturated permeability (Supplementary Table 1). The relative permeability (κrL) of the liquid phase was estimated on the basis of a cubic dependency on water saturation
Gas transport
The mass flux of each gas-phase species (Jj) was calculated using the Stefan–Maxwell equations for multi-component mass transport with consideration of convection
where ωj is the mass fraction of gas species j, ρj is the mass concentration of gas species j and Mn is the average molecular weight. The effective diffusion coefficients (\({D}_{j}^{{\rm{eff}}}\)) were calculated based on the molecular diffusion coefficients (\({D}_{j}^{\rm{m}}\)) and Knudsen diffusion coefficients (\({D}_{j}^{\rm{k}}\)) while accounting for the porosity of a porous medium using the Bruggeman correlation
The molecular diffusion coefficients were determined on the basis of the formulation reported by Fuller et al.40
where νp,j is the diffusion volume of species j. The convective velocity of the gas phase (uG) was determined using Darcy’s law
The relative permeability (κrG) of the gas phase was estimated on the basis of a cubic dependency on water saturation
The final equation used to solve the governing mass balances for the gas phase is the summation of mass fractions
CO2 phase transfer and solubility
The rate of phase transfer (PT) for CO2 (\({R}_{\rm{PT},{\rm{C}}{{\rm{O}}}_{2}}\)) occurring at the gas/ionomer interface was calculated as follows:
where \({{\rm{H}}}_{{\rm{C}}{{\rm{O}}}_{2}}\) is the Henry’s Law coefficient for CO2 dissolved in the electrolyte, \({c}_{{\rm{C}}{{\rm{O}}}_{2}}\) is the concentration of dissolved CO2, \({M}_{{\rm{C}}{{\rm{O}}}_{2}}\) is the molar mass of CO2, \({k}_{\rm{GL},{\rm{C}}{{\rm{O}}}_{2}}\) is the mass-transfer coefficient, which was obtained from experimental data by Yang et al.41 and \({y}_{{\rm{C}}{{\rm{O}}}_{2}}\) is the mole fraction of CO2 in the gas phase. The reduction in CO2 solubility observed at high salt concentrations (that is, the ‘salting-out’ effect) was accounted for using the Sechenov formulation with parameters obtained from Weisenberger and Schumpe42 (Supplementary Table 6)
where hi represents the solubility coefficient for the different salt species in solution. The nominal Henry’s Law coefficient for CO2 dissolved in water (in units mM atm–1) is given as
CO and H2 were assumed to be insoluble in the ionomer and generated directly in the gas phase, therefore the rate of CO and H2 generation in the gas phase were calculated using Faraday’s law
Water chemical potential and transport
The ionomer phase in the MEA is considered to be continuous between the anode and cathode catalyst layers. Therefore, liquid water is transported across the membrane by dissolving in the ionomer phase of the anode catalyst layer and transporting across the membrane to the cathode catalyst layer according to the chemical potential gradient, which is defined as
where aw is the activity of water in the ionomer, pL,M is the pressure of liquid water in the membrane, Vm,w is the molar volume of water (18 ml mol−1) and pL,ref is the reference liquid pressure (1 atm). Concentrated solution theory was considered for water transport owing to the need to account for electro-osmosis. Therefore, the equations that describes water conservation and flux are given by
where nw is the molar flux of water, μw is the water chemical potential and Rk,w is the net molar rate of water generation by electrochemical and buffer reactions and phase transfer phenomena. αw is the water transport coefficient, which was implemented as a function of water activity as per experimental measurements by Petrovick et al.43 (Supplementary Fig. 21). The first term in equation (57) describes the transport of water under chemical-potential-gradient driving force, analogous to the relationship between the diffusion coefficient and concentration gradient for Fickian diffusion processes. ζi is the electro-osmotic coefficient for species i, which was determined experimentally43
Correspondingly, the second term in equation (57) is associated with electro-osmotic driving forces that couple water transport to the transport of ions in the electrolyte.
The net electro-osmotic coefficient (β) relates the amount of water transported relative to the amount of water consumed by electrochemical reactions and is given as
The rate at which liquid water transfers from the liquid to the ionomer phase (RLI,w) is governed by the difference in pressure between the two phases
where kMT,L is the interfacial mass-transfer coefficient for liquid water (Supplementary Table 6). The rate of water transport from the gas phase to the ionomer phase is given by
where RH is the relative humidity and kMT,V is the interfacial mass-transfer coefficient for water vapor (Supplementary Table 6). These phase transfer source terms are used in the momentum balances for the liquid and gas phases
The opposite signs for the mass source terms ensures that the amount of water that leaves one phase is exactly equal to the water that enters the other phase.
Boundary conditions, Faradaic efficiency and CO2 utilization
As seen in Fig. 1b, the far left of the simulation domain represents the interface between the anode porous transport layer and the flow field/current collector. Here Dirichlet boundary conditions are used to describe the solid-phase potential, water chemical potential and pressure of liquid water and gas in the channel
The boundary condition described by equation (64) indicates that the anode is at a reference potential of 0 V versus the standard hydrogen electrode (SHE). The boundary conditions described by equations (65) and (66) indicate that the water activity is 1 and that the electrolyte is at ambient pressure, respectively (that is, a liquid electrolyte feed is used).
The flux of ions and dissolved CO2 to and from the anode flow field is given by a mass-transfer correlation
where the mass-transfer coefficients are given by a flat-plate correlation describing convective mass transfer
where Rei and Sci are the Reynolds and Schmidt numbers for species i and Lelect. is the characteristic electrode length. Convective transport of gas across the membrane is assumed to be negligible. Water transport across the membrane is driven only by diffusion and electro-osmosis within the hydrophilic domains of the polymer because there is no forced convection within these mesoscale channels. Therefore, the velocity of both gas and liquid phases at the membrane–electrode interfaces is assumed to be 0 m s–1 in the momentum balance equations
Dirichlet boundary conditions are used for the gas-phase mass conservation equations to represent the use of 100% humidified CO2 at the interface between the carbon GDL/cathode flow field and the use of N2 as a carrier gas at the anode porous transport layer/anode flow field
A no-flux boundary condition was used for the chemical potential of water at the cathode CL/GDL interface
Finally, chronoamperometry experiments were simulated by setting the Vcell at the catalyst layer/gas diffusion layer interface,
The boundary condition described by equation (75) indicates that the imposed potential at the cathode boundary in the model is equivalent to the electrolytic cell potential applied in the experiment, whereas equation (64) specifies the anode as the ground.
The Faradaic efficiency for CO (FECO) and CO2 utilization efficiency are
where the numerator of equation (77) represents the amount of CO formed and the denominator represents the amount of CO2 that reacts to form (bi)carbonates and crosses the membrane to the anode where it reconverts and is released as CO2. The equations that comprise the model were solved using the MUMPS solver in COMSOL with 14,000 elements and a relative error of 1 × 10−4. Nomenclature is defined in the Supplementary Information.
Data availability
The experimental data are available as an excel file in the Supplementary Information.
References
Izadi, P. & Harnisch, F. Microbial vertical bar electrochemical CO2 reduction: to integrate or not to integrate? Joule 6, 935–940 (2022).
De Luna, P. et al. What would it take for renewably powered electrosynthesis to displace petrochemical processes? Science 364, eaav3506 (2019).
Somoza-Tornos, A., Guerra, O. J., Crow, A. M., Smith, W. A. & Hodge, B. M. Process modeling, techno-economic assessment, and life cycle assessment of the electrochemical reduction of CO2: a review. Iscience 24, 102813 (2021).
Salvatore, D. & Berlinguette, C. P. Voltage matters when reducing CO2 in an electrochemical flow cell. ACS Energy Lett. 5, 215–220 (2020).
Jouny, M., Luc, W. & Jiao, F. General techno-economic analysis of CO2 electrolysis systems. Ind. Eng. Chem. Res. 57, 2165–2177 (2018).
Endrodi, B. et al. Operando cathode activation with alkali metal cations for high current density operation of water-fed zero-gap carbon dioxide electrolysers. Nat. Energy 6, 439–448 (2021).
Kutz, R. B. et al. Sustainion imidazolium-functionalized polymers for carbon dioxide electrolysis. Energy Technol. Ger. 5, 929–936 (2017).
Jeng, E. & Jiao, F. Investigation of CO2 single-pass conversion in a flow electrolyzer. React. Chem. Eng. 5, 1768–1775 (2020).
Blommaert, M. A., Subramanian, S., Yang, K., Smith, W. A. & Vermaas, D. A. High indirect energy consumption in AEM-based CO2 electrolyzers demonstrates the potential of bipolar membranes. ACS Appl. Mater. Interfaces 14, 557–563 (2022).
da Cunha, S. & Resasco, J. Maximizing single-pass conversion does not result in practical readiness for CO2 reduction electrolyzers. Nat. Commun. 14, 5513 (2023).
Xie, K. et al. Bipolar membrane electrolyzers enable high single-pass CO2 electroreduction to multicarbon products. Nat. Commun. 13, 3609 (2022).
Disch, J., Ingenhoven, S. & Vierrath, S. Bipolar membrane with porous anion exchange layer for efficient and long-term stable electrochemical reduction of CO2 to CO. Adv. Energy Mater. 13, 2301614 (2023).
Lees, E. W., Mowbray, B. A. W., Parlane, F. G. L. & Berlinguette, C. P. Gas diffusion electrodes and membranes for CO2 reduction electrolysers. Nat. Rev. Mater. 7, 55–64 (2022).
Salvatore, D. A. et al. Designing anion exchange membranes for CO2 electrolysers. Nat. Energy 6, 339–348 (2021).
Weng, L. C., Bell, A. T. & Weber, A. Z. Towards membrane–electrode assembly systems for CO2 reduction: a modeling study. Energ. Environ. Sci. 12, 1950–1968 (2019).
Blake, J. W., Padding, J. T. & Haverkort, J. W. Analytical modelling of CO2 reduction in gas-diffusion electrode catalyst layers. Electrochim. Acta 393, 138987 (2021).
Weng, L. C., Bell, A. T. & Weber, A. Z. Modeling gas-diffusion electrodes for CO2 reduction. Phys. Chem. Chem. Phys. 20, 16973–16984 (2018).
Loffelholz M. et al Modeling electrochemical CO2 reduction at silver gas diffusion electrodes using a TFFA approach. Chem. Eng. J. 435, 134920 (2022).
Romiluyi, O., Danilovic, N., Bell, A. T. & Weber, A. Z. Membrane-electrode assembly design parameters for optimal CO2 reduction. Electrochem. Sci. Adv. 3, e2100186 (2023).
Brown, S. M. et al Electron transfer limitation in carbon dioxide reduction revealed by data-driven Tafel analysis. Preprint at https://doi.org/10.26434/chemrxiv.13244906.v1 (2020).
Corpus, K. R. M. et al. Coupling covariance matrix adaptation with continuum modeling for determination of kinetic parameters associated with electrochemical CO2 reduction. Joule https://doi.org/10.1016/j.joule.2023.05.007 (2023).
Henstridge, M. C., Laborda, E., Rees, N. V. & Compton, R. G. Marcus–Hush–Chidsey theory of electron transfer applied to voltammetry: a review. Electrochim. Acta 84, 12–20 (2012).
Bazant, M. Z. Unified quantum theory of electrochemical kinetics by coupled ion-electron transfer. Faraday Discuss. 246, 60–124 (2023).
Reddy, P. N., Reddy, M. H. P., Pierson, J. F. & Uthanna, S. Characterization of silver oxide films formed by reactive RF sputtering at different substrate temperatures. ISRN Opt. 2014, 684317 (2014).
Vass, A. et al. Local chemical environment governs anode processes in CO2 electrolyzers. ACS Energy Lett. 6, 3801–3808 (2021).
Wheeler, D. G. et al. Quantification of water transport in a CO2 electrolyzer. Energ. Environ. Sci. 13, 5126–5134 (2020).
Petrovick, J. G. et al. Electrochemical measurement of water transport numbers in anion-exchange membranes. J. Electrochem. Soc. 170, 114519 (2023).
Dunwell, M. et al. The central role of bicarbonate in the electrochemical reduction of carbon dioxide on gold. J. Am. Chem. Soc. 139, 3774–3783 (2017).
Hashiba, H. et al. Effects of electrolyte buffer capacity on surface reactant species and the reaction rate of CO in electrochemical CO2 reduction. J. Phys. Chem. C 122, 3719–3726 (2018).
Luo X. Y., Kushner D. I., Kusoglu A. Anion exchange membranes: the effect of reinforcement in water and electrolyte. J. Membrane Sci. https://doi.org/10.1016/j.memsci.2023.121945 (2023).
Andersen, M. B. et al. Current-induced membrane discharge. Phys. Rev. Lett. 109, 108301 (2012).
Zenyuk, I. V., Das, P. K. & Weber, A. Z. Understanding impacts of catalyst-layer thickness on fuel-cell performance via mathematical modeling. J. Electrochem. Soc. 163, F691–F703 (2016).
Bui, J. C. et al. Analysis of bipolar membranes for electrochemical CO2 capture from air and oceanwater. Energ. Environ. Sci. 16, 5076–5095 (2023).
Weng, L. C., Bell, A. T. & Weber, A. Z. A systematic analysis of Cu-based membrane-electrode assemblies for CO2 reduction through multiphysics simulation. Energ. Environ. Sci. 13, 3592–3606 (2020).
Khan, S. U. M. & Bockris, J. O. Electronic states in solution and charge-transfer. J. Phys. Chem. US 87, 2599–2603 (1983).
Zeng, Y., Smith, R. B., Bai, P. & Bazant, M. Z. Simple formula for Marcus–Hush–Chidsey kinetics. J. Electroanal. Chem. 735, 77–83 (2014).
Smith, R. B. & Bazant, M. Z. Multiphase porous electrode theory. J. Electrochem. Soc. 164, E3291 (2017).
Singh, M. R., Goodpaster, J. D., Weber, A. Z., Head-Gordon, M. & Bell, A. T. Mechanistic insights into electrochemical reduction of CO2 over Ag using density functional theory and transport models. Proc. Natl Acad. Sci. USA 114, E8812–E8821 (2017).
Peng, J., Roy, A. L., Greenbaum, S. G. & Zawodzinski, T. A. Effect of CO2 absorption on ion and water mobility in an anion exchange membrane. J. Power Sources 380, 64–75 (2018).
Fuller, E. N., Schettle, P. D. & Giddings, J. C. A new method for prediction of binary gas-phase diffusion coeffecients. Ind. Eng. Chem. 58, 19-+ (1966).
Yang, Z. M. et al. Modeling and upscaling analysis of gas diffusion electrode-based electrochemical carbon dioxide reduction systems. ACS Sustain. Chem. Eng. 9, 351–361 (2021).
Weisenberger, S. & Schumpe, A. Estimation of gas solubilities in salt solutions at temperatures from 273 K to 363 K. Aiche J. 42, 298–300 (1996).
Petrovick, J. G. et al. Electrochemical measurement of water transport numbers in anion-exchange membranes. J. Electrochem. Soc. 170, 114519 (2023).
Acknowledgements
The authors acknowledge A. J. King for graphic design and interpretation of the CO2R reorganization energy, and J. G. Petrovick for assistance with implementing the electro-osmotic coefficients in the model. Transmission electron microscopy and powder X-ray diffraction were performed by D. Lee in H. Zheng’s group, supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences (BES), Materials Sciences and Engineering Division under contract no. DE-AC02-05-CH11231 within the in situ TEM programme (KC22ZH). The authors gratefully acknowledge Lawrence Berkeley National Laboratory’s Laboratory Directed Research and Development (LDRD) grant for funding. This material is also partially based upon work performed by support from the DOE EERE Bioenergy Technologies Office under contract no. DE-AC02-05CH11231 (A.Z.W). E.W.L. acknowledges support from the National Science and Engineering Research Council (NSERC) postdoctoral fellowship. J.C.B. was supported in part by a fellowship award under contract FA9550-21-F-0003 through the National Defense Science and Engineering Graduate (NDSEG) Fellowship Program, sponsored by the Army Research Office (ARO).
Author information
Authors and Affiliations
Contributions
E.W.L. conceived the study, developed the model and wrote the initial manuscript draft. J.C.B. helped implement the Marcus–Hush–Chidsey kinetics and provided modeling support. O.R. performed all the experiments in the study. A.T.B. and A.Z.W. supervised the study, contributed to theory and model development, helped analyze the experimental data and managed the project. All authors contributed to writing and editing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Chemical Engineering thanks Haotian Wang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Notes 1–6, Figs. 1–23, model parameters and nomenclature.
Supplementary Data.
Experimental CO and H2 partial current densities and CO2 crossover measurements.
Source data
Source Data Fig. 2
Data in Fig. 2 of main text.
Source Data Fig. 3
Data in Fig. 3 of main text.
Source Data Fig. 4
Data in Fig. 4 of main text.
Source Data Fig. 5
Data in Fig. 5 of main text.
Source Data Fig. 6
Data in Fig. 6 of main text.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Lees, E.W., Bui, J.C., Romiluyi, O. et al. Exploring CO2 reduction and crossover in membrane electrode assemblies. Nat Chem Eng 1, 340–353 (2024). https://doi.org/10.1038/s44286-024-00062-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s44286-024-00062-0
This article is cited by
-
Solvent reorganization model takes the lead
Nature Chemical Engineering (2024)
