
Abstract
Transforming growth factor-β1 (TGF-β1) is a potent inducer of fibroblast to myofibroblast differentiation and contributes to the pro-fibrotic microenvironment during cardiac remodeling. Fibroblast growth factor-2 (FGF-2) is a growth factor secreted by adipose tissue-derived stromal cells (ASC) which can antagonize TGF-β1 signaling. We hypothesized that TGF-β1-induced cardiac fibroblast to myofibroblast differentiation is abrogated by FGF-2 and ASC conditioned medium (ASC-CMed). Our experiments demonstrated that TGF-β1 treatment-induced cardiac fibroblast differentiation into myofibroblasts, as evidenced by the formation of contractile stress fibers rich in αSMA. FGF-2 blocked the differentiation, as evidenced by the reduction in gene (TAGLN, p < 0.0001; ACTA2, p = 0.0056) and protein (αSMA, p = 0.0338) expression of mesenchymal markers and extracellular matrix components gene expression (COL1A1, p < 0.0001; COL3A1, p = 0.0029). ASC-CMed did not block myofibroblast differentiation. The treatment with FGF-2 increased matrix metalloproteinases gene expression (MMP1, p < 0.0001; MMP14, p = 0.0027) and decreased the expression of tissue inhibitor of metalloproteinase gene TIMP2 (p = 0.0023). ASC-CMed did not influence these genes. The proliferation of TGF-β1-induced human cardiac fibroblasts was restored by both FGF-2 (p = 0.0002) and ASC-CMed (p = 0.0121). The present study supports the anti-fibrotic effects of FGF-2 through the blockage of cardiac fibroblast differentiation into myofibroblasts. ASC-CMed, however, did not replicate the anti-fibrotic effects of FGF-2 in vitro.
Similar content being viewed by others
Introduction
Fibroblasts are the most abundant cell type in the heart and regulate the homeostasis of the extracellular matrix (ECM). The ECM provides the architecture of cardiac tissue, it supports the structural integrity and it regulates cellular communication and function. A major component of the ECM is collagen which is deposited primarily by cardiac fibroblasts1. Pathological stimuli, such as myocardial infarction, disrupt the cardiac tissue homeostasis2 which predisposes the onset and progression of fibrosis3,4 by the activation and differentiation of fibroblasts into myofibroblasts1,5. Cardiac fibrosis features the production and deposition of excessive amounts of extracellular matrix by cardiac myofibroblasts. Fibroblast activation and subsequent differentiation into myofibroblasts are primarily driven by transforming growth factor-β (TGF-β) and contributes to the pro-fibrotic cardiac microenvironment6,7,8,9,10.
To date, no therapies exist that prevent or reverse fibrosis in vivo, yet it is possible to antagonize TGF-β signaling with specific growth factors such as FGF-2. Fibroblast growth factor 2 (FGF-2) is relevant in wound healing processes in vivo11,12,13 and in vitro14,15,16,17 to stimulate proliferation of tissue cells, connective tissue fibroblasts and promote angiogenesis, while it suppresses apoptosis. Moreover, FGF-2 antagonizes TGF-β signaling and thus affects fibrosis, albeit in early stages18. In fibroblasts of various origins, FGF-2 suppressed the expression of TGFB119 and its protein16,20, that also suppressed the deposition of collagen21,22. All these studies suggest that FGF-2 is an attractive molecule to target in TGF-β regulated fibrotic disease.
Mesenchymal stromal cells (MSC), such as adipose tissue-derived stromal cells (ASC), as well as their conditioned medium, have been shown to improve cardiac remodeling and thus to modulate cardiac fibrosis23,24,25,26,27,28,29,30. Because ASC release a series of anti-fibrotic factors, among which are FGF, IGF and HGF31,32,33,34, a possible mechanism underlying their anti-fibrotic effect could be to antagonize TGF-β signaling and thus the inhibition of the transformation of fibroblasts into myofibroblasts as well as the reduction of extracellular matrix production.
In a previous study, we demonstrated that TGF-β1-induced differentiation of dermal fibroblasts to myofibroblasts could be modulated by adipose tissue-derived stromal cells’ conditioned medium (ASC-CMed)35. Therefore, we hypothesized that cardiac fibroblast differentiation into myofibroblast could also be abrogated by ASC-CMed.
Results
Formation of F-actin stress fibers in differentiating NHCF-V is refractory to suppression by ASC-CMed
After five days of pro-fibrotic stimulus, NHCF-V undergo myofibroblast differentiation, phalloidin detected F-actin and cells had an increase in αSMA expression (Fig. 1). Human cardiac fibroblast without TGF-β1 stimuli had a mild phalloidin staining distributed all over the cytoplasm, on the other hand, the TGF-β1 stimulated cells had the detection of phalloidin in the contractile fibers which had colocalization with the αSMA staining. FGF-2, but not ASC-CMed, could inhibit the phalloidin and αSMA in the contractile fibers. The expression of αSMA seems to be more evident in the TGF-β1 stimulated cells. As expected, TGF-β1 induced myofibroblast differentiation of cardiac fibroblasts which leads to the formation of smooth actin fibers at cytoskeletal level. Only FGF-2 inhibited the formation of these fibers.

Formation of F-actin stress fibers in differentiating NHCF-V is refractory to suppression by ASC-CMed. Representative immunofluorescence micrographs of human cardiac fibroblasts (NHCF-V) under stimulation with TGF-β1 or co-stimulation with TGF-β1 and FGF-2, both in FBM and ASC-CMed, for 5 days. Upon TGF-β1 stimulation, cells developed transcellular αSMA-expressing stress fibers (arrows). ASC-CMed did not inhibit the development of stress fibers. Blue: DAPI; Green: phalloidin; Red: αSMA. Scale reference: 100 μm.
FGF-2, but not ASC-CMed, inhibits the expression of mesenchymal markers in NHCF-V stimulated with TGF-β1
The expression of mesenchymal genes TAGLN, encoding SM22α, and ACTA2, encoding αSMA, was increased TGF-β1 stimulated fibroblasts, irrespective of ASC-CMed (Fig. 2). FGF-2 suppressed the expression of TAGLN (One-way ANOVA, p < 0.0001; Sidak’s multiple comparison test, p < 0.0001) and ACTA2 in cardiac fibroblasts stimulated with TGF-β1 (One-way ANOVA, p = 0.0007; Sidak’s multiple comparison test, p = 0.0056). The influence of ASC-CMed no more than tended to decrease TAGLN expression (One-way ANOVA, p < 0.0001; Sidak’s multiple comparison test, p = 0.0820) but not ACTA2 expression.

FGF-2, but not ASC-CMed, inhibits the gene expression of mesenchymal markers. (A) TAGLN, and (B) ACTA2, by RT-qPCR of NHCF-V after stimulation with TGFβ1 or co-stimulation with TGF-β1 and FGF-2, both in FBM and ASC-CMed, for five days. Data were analyzed by One-way ANOVA with Sidak’s multiple comparison test for the groups FBM/TGF-β1 vs. FBM/TGF-β1/FGF-2 and FBM/TGF-β1 vs. ASC-CMed/TGF-β1; p-values for the Sidak’s multiple comparison test are shown in the figure. Values represent mean ± SEM of 3 independent experiments in duplicate.
TGF-β1 upregulated the expression of αSMA in human cardiac fibroblasts which was blocked by FGF-2 (Fig. 3) (One-way ANOVA, p = 0.0413; Sidak’s multiple comparison test, p = 0.0338). ASC-CMed did not alter the upregulation of αSMA by TGF-β1 (Fig. 3). Under these culture conditions, cardiac fibroblasts had a basal expression of SM22α which increased 1.4-fold after TGF-β1 stimulation. Thus, although FGF-2 inhibits the production of αSMA, it could not reverse the expression of the already formed SM22α.

FGF-2, but not ASC-CMed, inhibits the protein expression of mesenchymal markers. (A) SM22α, and (B) αSMA, by immunoblotting of NHCF-V after stimulation with TGFβ1 or co-stimulation with TGF-β1 and FGF-2, both in FBM and ASC-CMed, for five days. Data were analyzed by One-way ANOVA with Sidak’s multiple comparison test for the groups FBM/TGF-β1 vs. FBM/TGF-β1/FGF-2 and FBM/TGF-β1 vs. ASC-CMed/TGF-β1; p-values for the Sidak’s multiple comparison test are shown in the figure. Values represent mean ± SEM of 3 independent experiments.
FGF2, but not ASC-CMed, modulates extracellular matrix production in NHCF-V stimulated with TGF-β1
The gene expression of collagens, as well as of matrix metalloproteinases (MMPs) - enzymes responsible for ECM degradation - and the tissue inhibitors of metalloproteinases (TIMPs), was analyzed. The stimulation with TGF-β1 upregulated the transcription of both COL1A1 and COL3A1, genes responsible for the protein synthesis of the alpha-1 chain of collagens I and III respectively (Fig. 4A,B). This increase was more pronounced for COL1A1. Treatment with FGF-2 in samples stimulated by TGF-β1 was responsible for a statistically significant downregulation in COL1A1 (One-way ANOVA, p < 0.0001; Sidak’s multiple comparison test, p < 0.0001) and COL3A1 (One-way ANOVA, p = 0.0572; Sidak’s multiple comparison test, p = 0.0290), demonstrating the strong inhibition of NHCF-V towards the myofibroblast phenotype.

FGF2, but not ASC-CMed, modulates the expression of extracellular matrix-related genes. (A) COL1A1, (B) COL3A1, (C) MMP1, (D) MMP2, (E) MMP14, (F) TIMP1, and (G) TIMP2 by RT-qPCR of NHCF-V after stimulation with TGF-β1 or co-stimulation with TGF-β1 and FGF-2, both in FBM and ASC-CMed, for five days. Data were analyzed by One-way ANOVA with Sidak’s multiple comparison test for the groups FBM/TGF-β1 vs. FBM/TGF-β1/FGF-2 and FBM/TGF-β1 vs. ASC-CMed/TGF-β1; p-values for the Sidak’s multiple comparison test are shown in the figure. Values represent mean ± SEM of 3 independent experiments in duplicate.
The gene expression of MMPs and TIMPs did not change irrespective of treatment, except for FGF-2 (Fig. 4C–G). Expression of MMP1, encoding matrix metalloproteinase1 i.e. collagenase, was downregulated by TGF-β1. In contrast, FGF-2 upregulated MMP1 expression compared to control groups and TGF-β1 stimulation (One-way ANOVA, p < 0.0001; Sidak’s multiple comparison test, p < 0.0001). Treatment with ASC-CMed did not affect the TGF-ß-induced downregulation of MMP1 in NHCF-V. The expression of MMP2 gene was not regulated by TGF-ß, FGF or ASC-CMed (co)stimulation of NHCF-V. Although TGF-β1 did not affect the expression of MMP14, FGF-2 upregulated its expression (One-way ANOVA, p < 0.0001; Sidak’s multiple comparison test, p = 0.0027). Treatment with ASC-CMed tended to increase the expression of MMP14.
Expression of TIMPs genes TIMP1 and TIMP2 which are regulators of MMP activity had differential expression. Expression of TIMP1, remained unchanged irrespective of TGF-ß1 or ASC-CMed treatment. Expression of TIMP2 was decreased by FGF-2 (One-way ANOVA, p = 0.0005; Sidak’s multiple comparison test, p = 0.0023), while neither TGF-ß1 nor ASC-CMed affected its expression. Interestingly, TIMP2 is a co-factor of membrane-bound MMP14, which is e.g. responsible for activation of ECM-bound MMPs.
Immunofluorescence microscopy of intracellular collagen I demonstrated an increase in the protein content for all the groups stimulated with TGF-β1 (Fig. 5). FGF-2 could not decrease the intracellular collagen I at the microscopical level, so that virtually all the TGF-β1 stimulated cells and ASC-CMed/TGF-β1 expressed the protein in their cytoplasm. Both groups without TGF-β1 stimulation showed a very limited expression of collagen I.

Pro-collagen production in cardiac fibroblasts. Immunofluorescence analysis of expression of pro-collagen in human cardiac fibroblasts undergoing myofibroblast differentiation for five days. Collagen I was upregulated upon TGF-β1 stimuli and neither ASC-CMed nor FGF-2 inhibited the process. Minor expression of collagen I was showed in cultured cells without TGF-β1 stimuli. Blue: DAPI; Yellow: collagen I. Scale reference: 200 μm.
FGF2 and ASC-CMed restore the TGF-ß1-inhibited proliferation of NHCF-V
NHCF-V proliferation was measured after five days of stimulation with TGF-β1. The pro-fibrotic stimulus led to a decrease in cell proliferation, as detected by Ki-67 staining. TGF-β1 suppressed the proliferation of human cardiac fibroblasts; control cells had 40.8% of proliferating cells compared to 22.3% in TGF-β1 stimulated cells (Fig. 6). Treatment with FGF-2, recovered the proliferation to 37.8% (One-way ANOVA, p < 0.0001; Sidak’s multiple comparison test, p = 0.0002) while the proliferation of fibroblasts cells stimulated with TGF-β1 was virtually rescued by ASC-CMed (32.6%) (One-way ANOVA, p < 0.0001; Sidak’s multiple comparison test, p = 0.0121). Treatment of nonstimulated cardiac fibroblasts with ASC-CMed increased proliferation to 47.3%.

FGF2 and ASC-CMed restore the TGF-ß1-inhibited proliferation of NHCF-V. Immunofluorescence proliferation analysis of human cardiac fibroblasts undergoing myofibroblast differentiation for five days. TGF-β1 stimuli downregulated the proliferation of cardiac fibroblasts. ASC-CMed and FGF-2 upregulated the proliferation. Blue: DAPI; Red: Ki-67. Scale reference: 200 μm. Data were analyzed by One-way ANOVA with Sidak’s multiple comparison test for the groups FBM/TGF-β1 vs. FBM/TGF-β1/FGF-2 and FBM/TGF-β1 vs. ASC-CMed/TGF-β1; p-values for the Sidak’s multiple comparison test are shown in the figure. Values represent mean ± SEM of 3 independent experiments in quadruplicate.
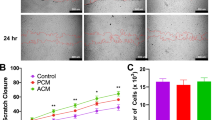
Wound healing is not affected by NHCF-V stimulation with TGF-β1
Wound healing was examined with a scratch assay and assessed after 24 hours. The percentage of gap closure did not differ among all groups (One-way ANOVA, p = 0.3716). Still, the treatment with FGF-2 or ASC-CMed tended to increase wound healing potential (Fig. 7).

Wound healing is not affected by NHCF-V stimulation with TGF-β1. Wound healing assay (24 h) in NHCF-V under stimulation with TGF-β1 or co-stimulation with TGF-β1 and FGF-2, both in FBM and ASC-CMed. Original magnification: 4x. Values represent mean ± SEM of 3 independent experiments in triplicate.
Discussion
We demonstrated that FGF-2 - but not the secreted factors of adipose tissue-derived stromal cells – downmodulate TGF-ß1-induced fibroblast into myofibroblast differentiation. Our results show that FGF-2 reduced differentiation via the reduction of mesenchymal gene expression (TAGLN and ACTA2), and the reduction of αSMA expression subsequently. Concomitantly, we demonstrated that FGF-2 downregulated expression of collagens (COL1A1 and COL3A1), upregulate expression of matrix metalloproteinases (MMP1 and MMP14), and downregulated expression of inhibitor of metalloproteinase TIMP2. Finally, we showed that not only FGF-2 but also ASC-CMed restored the proliferation of TGF-β1-impaired fibroblasts.
TGF-β1 signaling is a pivotal mechanism of the activation of fibroblasts into myofibroblasts and, thus, fibrosis6,7,8,9,10. The ability of FGF-2 to antagonize TGF-β signaling, in turn, has been demonstrated in a series of studies17,36,37,38,39, each of these suggesting different mechanisms of interference on the TGF-β pathway. Among these suggested mechanisms are: the activation of ERK and JNK pathways17; the expression of let-7 miRNA, TGFβ receptor suppressor38,39; the expression of miRNA-20a, another repressor of the TGFβ receptor complex36; and 4 the inhibition of the transcriptional regulator of muscle differentiation myf-540.
We hypothesized that, because ASC are known to secrete pro-regenerative growth factors such as FGF-2, treatment of human cardiac fibroblasts with ASC-CMed would abrogate TGF-β1-induced differentiation into myofibroblasts. Both gene and protein expression of mesenchymal and extracellular matrix-related markers showed that, differently than expected, ASC-CMed did not exert a noticeable effect on TGF-β1-induced myofibroblast activation. We did find, however, that ASC-CMed restored fibroblast proliferation impaired by TGF-β1 stimulation. This finding might point to potential differences between the FGF-2 thresholds required for restoring proliferation and blocking differentiation. However, another possibility is that ASC produce other growth factors that also impact proliferation but are not involved in the differentiation process. In this regard, it has been previously shown that epidermal growth factor (EGF) from ASC was capable to enhance the proliferation of skin fibroblasts in vitro41.
To our knowledge, this is the first report to investigate the effects of conditioned medium from mesenchymal stromal cells - here ASC - in the TGF-β1-induced differentiation of cardiac fibroblasts into myofibroblasts. Several other studies, however, have investigated the effects of MSC secretome in the spontaneous differentiation of cardiac fibroblasts42,43,44,45,46. Thus, although our findings oppose to many of the results described in these studies, there are important differences - which deserve attention - between published data and our current work (Table 1).
Three out of the five studies reported anti-fibrotic effects of MSC conditioned-medium on cardiac fibroblasts, including a reduction in expression of αSMA, collagens and TIMPs and increased MMPs expression and/or activity43,45,46. These studies were all performed with rat - not human - cardiac fibroblasts and none of these included a control group treated with Fibroblast Growth Medium (FGM), a medium capable to inhibit the spontaneous differentiation of fibroblasts, as we did. Thus, in this respect our results are not comparable to these studies for several reasons, the main being that the induction of cardiac fibroblast differentiation with TGF-β1 is a much more effective and powerful mechanism than the spontaneous differentiation, being consequently much more difficult to be blocked or reversed. Furthermore, both other studies which investigated the effects of MSC conditioned-medium on the spontaneous differentiation of cardiac fibroblasts reported either no differences42 or even a trend towards the pro-fibrotic phenotype, with reduced MMP2 and MMP9 protein expression and activity and increased TIMP1 protein expression44. An important observation is that the concentration of FGF-2 in the ASC-CMed could be too low to counteract TGF-β1 action, while it is enough to block spontaneous differentiation. Previous studies demonstrated that the concentration of FGF-2 in ASC-CMed was less than 150 pg/mL33,34, much lower than the FGF-2 concentration necessary to block the TGF-β1-induced differentiation in this study. Another point which deserves attention is the variety of factors secreted by ASC which could interfere with FGF-2 activity. Among the factors that might be involved in the FGF-2 blockage of TGF-β1 signaling are inflammatory factors, as INFα, TNFα, and IL-1β, which block the FGF receptor and, thus, do not allow FGF-2 to inhibit TGF-β1 action39. In fact, studies demonstrated that ASC may release TNFα and IL-1β, besides many other interleukins33,47,48. These factors, together, could abrogate the effect of FGF-2 in the low doses it is found in ASC-CMed. Still, it is essential to highlight the fact that ASC also produce TGF-β, as demonstrated by several studies33,34,49,50. In this regard, Rehman et al. showed that ASC produce around ten times more TGF-β than FGF-234. The conjoint action of all these factors, which are contained in the ASC secretome, could explain, in part, the insufficiency of ASC-CMed to block the fibrotic stimule.
Still, in our view, the partial recapitulation in vitro of the fibroblast-related component of cardiac fibrosis, with the use of primary human cardiac fibroblast cultured in FGM and induced with TGF-β1 is a suitable in vitro model of the natural conditions in the fibrotic heart. Thus, considering the controversial findings between the studies using the spontaneous differentiation in addition to the results of the present study, we consider that in TGF-β-driven cardiac fibrosis ASC-CMed may not suffice as a sole therapeutic modality to block or reverse ongoing fibrosis. FGF-2, however, corroborating with the findings of two previous studies21,22, did block TGF-β1-induced pro-fibrotic conversion of cardiac human fibroblasts.
The present study supports the anti-fibrotic effects of FGF-2 through the blockage of NHCF-V differentiation into myofibroblasts, as demonstrated by the modulation of gene and/or protein expression in a series of mesenchymal and extracellular matrix-related markers. ASC-CMed, however, could not demonstrate the same effects found with FGF-2.
Methods
Experimental groups
Ventricular normal human cardiac fibroblasts (NHCF-V) were allocated into 5 groups with different induction/FGF-2/ASC-CMed combinations, as described in Table 2.
Cell sources, cell culture, and conditioned medium
Ventricular normal human cardiac fibroblasts were purchased from Lonza (NHCF-V; #CC-2904, Lonza, Basel, Switzerland) and cultured with Fibroblast Growth Medium-3 with 10% of fetal bovine serum (FBS) (FGM3; #CC-4526, Lonza, Basel, Switzerland) added with 500 ng/mL Amphotericin B (#15290018, Gibco Invitrogen, Carlsbad, USA) at 37 °C in a humidified incubator with 5% CO2. The medium was refreshed every 3 days. Cells were passed at a ratio of 1:3 after reaching confluency. NHCF-V were used between passages 3–5.
At day 0 of the experiment, NHCF-V were seeded at a 10,000 cells/cm2 density. After 24 hours, NHCF-V were allocated into the 5 distinct groups and cultured for 5 days. Fibroblast growth factor 2 (FGF-2; #100-18C, PeproTech, Inc., Rocky Hill, N.J.) and human transforming growth factor-beta 1 (TGF-β1; #100-21, PeproTech, New Jersey, USA) were used at a concentration of 10 ng/mL and 5 ng/mL, respectively, in all experiments.
Human ASC were isolated as described previously51. Briefly, human abdominal fat was obtained by liposuction, washed with phosphate-buffered saline (PBS) and digested enzymatically with 0.1% collagenase A (#11088793001, Roche Diagnostic, Mannheim, Germany) in PBS with 1% bovine serum albumin (BSA; #A9647, Sigma-Aldrich, Boston, USA). The tissue was shaken constantly at 37 °C for 2 h. After this, the digested tissue was mixed with 1% PBS/BSA, filtered, centrifuged and the cell pellet was resuspended in Dulbecco’s Modified Eagle’s Medium (DMEM; #12-604F, Lonza, Basel, Switzerland) with 10% fetal bovine serum (FBS; #F0804, Sigma-Aldrich, Missouri, United States), 1% penicillin/streptomycin (#15140122, Gibco Invitrogen, Carlsbad, USA) and 1% L-glutamine (#17-605E, Lonza Biowhittaker, Verviers, Belgium). Cells were cultured at 37 °C in a humidified incubator with 5% CO2. The medium was refreshed every 2 days. Cells were passed at a ratio of 1:3 after reached confluency.
ASC conditioned medium (ASC-CMed) was obtained from confluent cultures of ASC between passages 3 and 6 from at least 3 different donors. For ASC-CMed, cells were cultured in DMEM without serum. Cells were kept at 37 °C with a minimum relative humidity of 95% and an atmosphere of 5% CO2 in the air. The conditioned medium was harvested after 24 hours, passed through a 0.22 µm filter and stored t −20 °C until use. Before the experiment, the conditioned medium was concentrated 30 times using Amicon® Ultra 15 mL filters (UFC900324, Merck, Darmstadt, Germany) and resuspended to the initial volume using Fibroblast Growth Medium-2 (FGM2; #CC-3132, Lonza, Basel, Switzerland).
Immunofluorescence, gene expression, and immunoblotting
Immunofluorescence
NHCF-V were cultured in 48 well tissue culture plates. After 5 and 21 days of induction, cells were fixed at room temperature with 2% paraformaldehyde (PFA) for 30 minutes. Cells were permeabilized with 1% Triton-X100 in PBS for 15 minutes at room temperature and blocked with 5% donkey serum in PBS and 1% BSA for another 15 minutes at room temperature. Subsequently, cells were incubated with primary antibodies diluted in 5% donkey serum in PBS for 2 hours at room temperature. The following primary antibodies were used: mouse anti-αSMA (1:400; #ab5694, Abcam, Cambridge, UK), rabbit anti-collagen I (1:200, #ab34710, Abcam, Cambridge, UK), rabbit anti-collagen III (1:200, #ab34710, Abcam, Cambridge, UK) and rabbit anti Ki-67 (1:400; #ab15580, Abcam, Cambridge, UK). Controls were incubated with 5% donkey serum in PBS. Next, cells were washed with 0.05% Tween-20 in PBS and incubated with secondary antibodies in 5% donkey serum in PBS with 4′,6-diamidino-2-phenylindole (DAPI; 1:5000; #D9542-5MG, Sigma-Aldrich, Missouri, United States) and Alexa Fluor® 488 phalloidin (1:400; #A12379, Life Technologies, Carlsbad, United States) for 1 hour at room temperature. The following secondary antibodies were used: donkey anti-rabbit IgG (H + L) Alexa Fluor® 594 (1:400; #A-21207, Life Technologies, Carlsbad, United States) and donkey anti-mouse IgG (H + L) Alexa Fluor® 647 (1:400; #A-31571, Life Technologies, Carlsbad, United States). Finally, cells were washed 3 times with PBS and the plates were imaged with Evos FL System (Thermo Fisher Scientific, Waltham, United States) using Texas Red (TXR), Cy5, DAPI and Green Fluorescent Protein (GFP) channels with 20x magnification.
Gene expression analysis
NHCF-V were cultured in 25 cm2 flasks. After 5 days of induction, RNA isolation was performed using TRIzol reagent (#15596018, Invitrogen Corp, Carlsbad, United States) according to the manufacturer’s protocol. RNA concentration and purity were determined using NanoDrop technology (Thermo Scientific, Hemel Hempstead, United Kingdom). Between 300 ng and 500 ng of total RNA was used for cDNA synthesis, which was performed using RevertAidTM First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, United States) according to the manufacturer’s protocol. The cDNA-equivalent of 5 ng total RNA was used per single qPCR reaction. PCR was performed using SYBR Green (Bio-Rad, Hercules, United States) with the ViiA7 Real-Time PCR system (Applied Biosystems, Foster City, United States). Each analysis was done in duplicate for each one of the independent experiments. The primers used are listed in the Supplementary Table S1. Data were analyzed using ViiA7 software (Applied Biosystems, Foster City, United States) and normalized with the ∆Ct method, using the geometrical mean of 18S ribosomal RNA (18S RNA) cycle threshold (CT) values. The fold-change in gene expression versus the no treatment control group (ECM) was calculated using the ∆∆CT method.
Immunoblotting analysis
NHCF-V were cultured in 25 cm2 flasks. After 5 days of induction, cells were rinsed with cold PBS and lysed in 100 µL of cold lysis buffer (RIPA; #89900, Thermo Fisher Scientific, Waltham, United States) containing 1% protease inhibitor cocktail (PIC; #P8340, Sigma Aldrich, St. Louis, United States) and 1% Halt™ phosphatase inhibitor cocktail (#78420, Thermo Fisher Scientific, Waltham, United States). The lysed cells were collected in 2 mL microcentrifuge tubes and the contents homogenized by sonication at 30 W for 30 seconds and centrifuged at 7,500 g at 4 °C for 5 minutes. The supernatant was collected for the protein concentration determination using the Bio-Rad DC protein assay (#5000112; Bio-Rad, Hercules, United States) according to the manufacturer’s protocol. Gels (12%) were loaded with 4–10 μg of protein. After electrophoresis, gels were blotted onto nitrocellulose membranes (#170-4270; Bio-Rad, Hercules, United States). Blots were blocked with Odyssey® Blocking Buffer (#927-40000, LI-COR, Lincoln, USA) in a dilution of 1:1 with PBS overnight at 4 °C. Afterward, blots were incubated with the primary antibodies overnight. The following primary antibodies were used: rabbit anti-SM22α (1:1000; #ab14106, Abcam, Cambridge, UK), mouse anti-αSMA (1:1000; #ab5694, Abcam, Cambridge, UK) and rabbit anti-GAPDH (1:1000; #ab9485, Abcam, Cambridge, UK). Then, the membranes were washed during 30 minutes with Tris-buffered saline with 0.1% Tween-20 (TBST) and incubated for 1 hour with the Odyssey® secondary antibodies goat anti-rabbit IRDye 680LT (1:10000; #926-68021, LI-COR, Lincoln, USA) and goat anti-mouse IRDye 800CW (1:10,000; #926-32210, LI-COR, Lincoln, USA). Non-bound secondary antibodies were removed by washing with TBST for 30 minutes. Then, blots were washed for 5 minutes with Tris-buffered saline (TBS) and scanned with Odyssey® Infrared Imaging System (LI-COR, Lincoln, USA).
Wound healing assay
NHCF-V were seeded in 48-well tissue culture plates at a density of 15,000 cells/cm2 and cultured in FGM3, 37 °C, and 5% CO2 until 70% confluence was reached. Them, cells were divided into 5 groups and stimulated for 5 days, as prescribed previously. After this period, the confluent monolayers were scored with a 10 µl sterile pipette tip to leave a scratch of approximately 0.4–0.5 mm in width. Culture medium was, then, immediately removed and cells were carefully washed with PBS - to remove any dislodged cells - and the medium was replaced with fresh FGM2. Pictures were taken immediately after the scratch and, again, after 24 hours. The percentage of wound reduction was evaluated in ImageJ.
Statistical analysis
All data were obtained from at least three independent experiments, performed in duplicate or triplicate. Data are presented as mean ± standard error of the mean (SEM). Graphs and statistical analysis were done using GraphPad Prism (Version 6.01; GraphPad Software, Inc., La Jolla, United States). Differences among multiple groups were analyzed by One-way ANOVA with Sidak’s multiple comparison test for the following groups: FGM2/TGF-β1 vs. FGM2/TGF-β1/FGF-2 and FGM2/TGF-β1 vs. ASC-CMed/TGF-β1.
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Kong, P., Christia, P. & Frangogiannis, N. G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 71, 549–574 (2014).
Edgley, A. J., Krum, H. & Kelly, D. J. Targeting fibrosis for the treatment of heart failure: a role for transforming growth factor-β. Cardiovasc. Ther. 30, e30–40 (2012).
Piera-Velazquez, S., Li, Z. & Jimenez, S. A. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am. J. Pathol. 179, 1074–1080 (2011).
Zeisberg, E. M. & Kalluri, R. Origins of cardiac fibroblasts. Circ. Res. 107, 1304–1312 (2010).
Travers, J. G., Kamal, F. A., Robbins, J., Yutzey, K. E. & Blaxall, B. C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 118, 1021–1040 (2016).
Biernacka, A., Dobaczewski, M. & Frangogiannis, N. G. TGF-β signaling in fibrosis. Growth Factors 29, 196–202 (2011).
Piersma, B., Bank, R. A. & Boersema, M. Signaling in Fibrosis: TGF-β, WNT, and YAP/TAZ Converge. Front. Med. 2, 59 (2015).
Dobaczewski, M., Chen, W. & Frangogiannis, N. G. Transforming growth factor (TGF)-β signaling in cardiac remodeling. J. Mol. Cell. Cardiol. 51, 600–606 (2011).
Pohlers, D. et al. TGF-β and fibrosis in different organs — molecular pathway imprints. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1792, 746–756 (2009).
Leask, A. TGFβ, cardiac fibroblasts, and the fibrotic response. Cardiovasc. Res. 74, 207–212 (2007).
Spyrou, G. E. & Naylor, I. L. The effect of basic fibroblast growth factor on scarring. Br. J. Plast. Surg. 55, 275–282 (2002).
Akasaka, Y., Ono, I., Yamashita, T., Jimbow, K. & Ishii, T. Basic fibroblast growth factor promotes apoptosis and suppresses granulation tissue formation in acute incisional wounds. J. Pathol. 203, 710–720 (2004).
Nunes, Q. M., Li, Y., Sun, C., Kinnunen, T. K. & Fernig, D. G. Fibroblast growth factors as tissue repair and regeneration therapeutics, https://doi.org/10.7287/peerj.preprints.1378v1 (2015).
Gallego-Muñoz, P. et al. Effects of TGFβ1, PDGF-BB, and bFGF, on human corneal fibroblasts proliferation and differentiation during stromal repair. Cytokine 96, 94–101 (2017).
Song, Q. H., Klepeis, V. E., Nugent, M. A. & Trinkaus-Randall, V. TGF-beta1 regulates TGF-beta1 and FGF-2 mRNA expression during fibroblast wound healing. Mol. Pathol. 55, 164–176 (2002).
Maltseva, O., Folger, P., Zekaria, D., Petridou, S. & Masur, S. K. Fibroblast growth factor reversal of the corneal myofibroblast phenotype. Invest. Ophthalmol. Vis. Sci. 42, 2490–2495 (2001).
Dolivo, D. M., Larson, S. A. & Dominko, T. FGF2-mediated attenuation of myofibroblast activation is modulated by distinct MAPK signaling pathways in human dermal fibroblasts. J. Dermatol. Sci. 88, 339–348 (2017).
Dolivo, D. M., Larson, S. A. & Dominko, T. Fibroblast Growth Factor 2 as an Antifibrotic: Antagonism of Myofibroblast Differentiation and Suppression of Pro-Fibrotic Gene Expression. Cytokine Growth Factor Rev. 38, 49–58 (2017).
Song, Q. H. TGF-beta1 regulates TGF-beta1 and FGF-2 mRNA expression during fibroblast wound healing. Mol. Pathol. 55, 164–176 (2002).
Tiede, S. et al. Basic fibroblast growth factor: a potential new therapeutic tool for the treatment of hypertrophic and keloid scars. Ann. Anat. 191, 33–44 (2009).
Svystonyuk, D. A. et al. Fibroblast growth factor-2 regulates human cardiac myofibroblast-mediated extracellular matrix remodeling. J. Transl. Med. 13 (2015).
Park, D. S. J. et al. Heparin Augmentation Enhances Bioactive Properties of Acellular Extracellular Matrix Scaffold. Tissue Eng. Part A 24, 128–134 (2018).
Mazo, M. et al. Treatment of Reperfused Ischemia With Adipose-Derived Stem Cells in a Preclinical Swine Model of Myocardial Infarction. Cell Transplant. 21, 2723–2733 (2012).
Yu, L. H. et al. Improvement of cardiac function and remodeling by transplanting adipose tissue-derived stromal cells into a mouse model of acute myocardial infarction. Int. J. Cardiol. 139, 166–172 (2010).
Perin, E. C. et al. Adipose-derived regenerative cells in patients with ischemic cardiomyopathy: The PRECISE Trial. Am. Heart J. 168, 88–95.e2 (2014).
Mazo, M. et al. Transplantation of adipose derived stromal cells is associated with functional improvement in a rat model of chronic myocardial infarction. Eur. J. Heart Fail. 10, 454–462 (2008).
Tano, N. et al. Epicardial placement of mesenchymal stromal cell-sheets for the treatment of ischemic cardiomyopathy; in vivo proof-of-concept study. Mol. Ther. 22, 1864–1871 (2014).
Araña, M. et al. Epicardial delivery of collagen patches with adipose-derived stem cells in rat and minipig models of chronic myocardial infarction. Biomaterials 35, 143–151 (2014).
He, J., Cai, Y., Luo, L. M. & Liu, H. B. Hypoxic adipose mesenchymal stem cells derived conditioned medium protects myocardial infarct in rat. Eur. Rev. Med. Pharmacol. Sci. 19, 4397–4406 (2015).
Jiang, Y. et al. Intramyocardial injection of hypoxia-preconditioned adipose-derived stromal cells treats acute myocardial infarction: an in vivo study in swine. Cell Tissue Res. 358, 417–432 (2014).
Kapur, S. K. & Katz, A. J. Review of the adipose derived stem cell secretome. Biochimie 95, 2222–2228 (2013).
Salgado, A. J. B. O. G., Reis, R. L. G., Sousa, N. J. C. & Gimble, J. M. Adipose tissue derived stem cells secretome: soluble factors and their roles in regenerative medicine. Curr. Stem Cell Res. Ther. 5, 103–110 (2010).
Du, W. J. et al. Heterogeneity of proangiogenic features in mesenchymal stem cells derived from bone marrow, adipose tissue, umbilical cord, and placenta. Stem Cell Res. Ther. 7, 163 (2016).
Rehman, J. et al. Secretion of angiogenic and antiapoptotic factors by human adipose stromal cells. Circulation 109, 1292–1298 (2004).
Spiekman, M. et al. Adipose tissue-derived stromal cells inhibit TGF-β1-induced differentiation of human dermal fibroblasts and keloid scar-derived fibroblasts in a paracrine fashion. Plast. Reconstr. Surg. 134, 699–712 (2014).
Correia, A. C. P., Moonen, J.-R. A. J., Brinker, M. G. L. & Krenning, G. FGF2 inhibits endothelial-mesenchymal transition through microRNA-20a-mediated repression of canonical TGF-β signaling. J. Cell Sci. 129, 569–579 (2016).
Papetti, M., Shujath, J., Riley, K. N. & Herman, I. M. FGF-2 antagonizes the TGF-beta1-mediated induction of pericyte alpha-smooth muscle actin expression: a role for myf-5 and Smad-mediated signaling pathways. Invest. Ophthalmol. Vis. Sci. 44, 4994–5005 (2003).
Chen, P.-Y., Qin, L., Li, G., Tellides, G. & Simons, M. Smooth muscle FGF/TGFβ cross talk regulates atherosclerosis progression. EMBO Mol. Med. 8, 712–728 (2016).
Chen, P.-Y. et al. FGF regulates TGF-β signaling and endothelial-to-mesenchymal transition via control of let-7 miRNA expression. Cell Rep. 2, 1684–1696 (2012).
Papetti, M., Shujath, J., Riley, K. N. & Herman, I. M. FGF-2 Antagonizes the TGF-β1-Mediated Induction of Pericyte α-Smooth Muscle Actin Expression: A Role for Myf-5 and Smad-Mediated SignalingPathways. Investigative Opthalmology & Visual Science 44, 4994 (2003).
Zhao, J., Hu, L., Liu, J., Gong, N. & Chen, L. The effects of cytokines in adipose stem cell-conditioned medium on the migration and proliferation of skin fibroblasts in vitro. Biomed Res. Int. 2013, 578479 (2013).
Ohnishi, S., Sumiyoshi, H., Kitamura, S. & Nagaya, N. Mesenchymal stem cells attenuate cardiac fibroblast proliferation and collagen synthesis through paracrine actions. FEBS Lett. 581, 3961–3966 (2007).
Mias, C. et al. Mesenchymal stem cells promote matrix metalloproteinase secretion by cardiac fibroblasts and reduce cardiac ventricular fibrosis after myocardial infarction. Stem Cells 27, 2734–2743 (2009).
Wang, Y. et al. Effects of mesenchymal stem cells on matrix metalloproteinase synthesis in cardiac fibroblasts. Exp. Biol. Med. 236, 1197–1204 (2011).
Mao, Q., Lin, C.-X., Liang, X.-L., Gao, J.-S. & Xu, B. Mesenchymal stem cells overexpressing integrin-linked kinase attenuate cardiac fibroblast proliferation and collagen synthesis through paracrine actions. Mol. Med. Rep. 7, 1617–1623 (2013).
Li, X. et al. Direct intercellular communications dominate the interaction between adipose-derived MSCs and myofibroblasts against cardiac fibrosis. Protein Cell 6, 735–745 (2015).
Kilroy, G. E. et al. Cytokine profile of human adipose-derived stem cells: expression of angiogenic, hematopoietic, and pro-inflammatory factors. J. Cell. Physiol. 212, 702–709 (2007).
Prichard, H. L., Reichert, W. & Klitzman, B. IFATS collection: Adipose-derived stromal cells improve the foreign body response. Stem Cells 26, 2691–2695 (2008).
Nakagami, H. et al. Novel autologous cell therapy in ischemic limb disease through growth factor secretion by cultured adipose tissue-derived stromal cells. Arterioscler. Thromb. Vasc. Biol. 25, 2542–2547 (2005).
Cui, L. et al. Expanded Adipose-Derived Stem Cells Suppress Mixed Lymphocyte Reaction by Secretion of Prostaglandin E2. Tissue Eng. 13, 1185–1195 (2007).
Tuin, A., Kluijtmans, S. G., Bouwstra, J. B., Harmsen, M. C. & Van Luyn, M. J. A. Recombinant gelatin microspheres: novel formulations for tissue repair? Tissue Eng. Part A 16, 1811–1821 (2010).
Author information
Authors and Affiliations
Contributions
T.T.A.L. has contributed to the conception and design of the work, data collection, data analysis and interpretation, drafting the article, critical revision of the article and final approval of the version to be published. G.R.L. has contributed to the conception and design of the work, data collection, data analysis and interpretation, drafting the article, critical revision of the article and final approval of the version to be published. L.F.P.M. has contributed to the data analysis and interpretation, critical revision of the article and final approval of the version to be published. M.C.H. has contributed to the conception and design of the work, data analysis and interpretation, critical revision of the article and final approval of the version to be published.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liguori, T.T.A., Liguori, G.R., Moreira, L.F.P. et al. Fibroblast growth factor-2, but not the adipose tissue-derived stromal cells secretome, inhibits TGF-β1-induced differentiation of human cardiac fibroblasts into myofibroblasts. Sci Rep 8, 16633 (2018). https://doi.org/10.1038/s41598-018-34747-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-34747-3
Keywords
This article is cited by
-
Adipose-derived stromal cells increase the formation of collagens through paracrine and juxtacrine mechanisms in a fibroblast co-culture model utilizing macromolecular crowding
Stem Cell Research & Therapy (2022)
-
Transcriptome analysis of a dog model of congestive heart failure shows that collagen-related 2-oxoglutarate-dependent dioxygenases contribute to heart failure
Scientific Reports (2022)
-
Anti-fibrotic effects of pharmacologic FGF-2: a review of recent literature
Journal of Molecular Medicine (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
