
Abstract
Developing wheat–alien chromosome introgression lines to improve bread wheat’s resistance to stresses, such as drought, salinity stress and diseases, requires reliable markers to identify and characterize the alien chromatins. Leymus mollis is a wild relative of bread wheat resistant to salinity and economically important diseases of wheat, but its genome sequence and cytological markers are not available. We devised a molecular marker-assisted strategy for L. mollis chromosome identification and applied it to produce 10 wheat–L. mollis chromosome addition lines. Using 47 L. racemosus genome polymorphic PCR markers and DArTseq genotyping, we distinguished the L. mollis chromosomes and differentiated disomic and monosomic lines by progeny test. DArTseq genotyping generated 14,530 L. mollis SNP markers and the chromosome-specific SNP markers were used to determine the homoeologous groups of L. mollis chromosomes in the addition lines. To validate the marker-based results, genomic in situ hybridization was applied to confirm the presence and cytological status of L. mollis chromosomes in the lines. This study demonstrates that adequate molecular markers allow the production and characterization of wheat–alien addition lines without in situ hybridization, which saves considerable time and effort.
Similar content being viewed by others
Introduction
Leymus species are prominent in Triticeae studies as pasture plants and gene sources for improving bread wheat1,2,3,4,5,6. They are reported to have the Ns genomes from Psathyrostachys and Xm genomes from an unknown source5,7,8. However, recent molecular analyses indicate that both genomes are from Psathyrostachys and that tetraploid Leymus species are segmental polyploids (2n = 4x = 28, Ns1Ns1Ns2Ns2)4,9. This genomic representation of tetraploid Leymus species as segmental polyploids is consistently used in this article.
Reports show that Leymus species are resistant to salinity and economically important wheat diseases6,10,11. Their chromosome segments in bread wheat genetic background improve wheat’s tolerance to biotic and abiotic stresses, such as Fusarium head blight, stripe rust and powdery mildew diseases3,6,12,13,14,15,16,17, heat stress18, aluminium toxicity19, and salinity stress20. The recognition of the potentials of Leymus species as valuable gene sources for the improvement of wheat dates back to the 1960s when Tsitsin reported the production of different combinations of wheat–Leymus amphidiploids21. Subsequent studies in this direction have demonstrated high cytogenetic stability in wheat–L. mollis octoploids and varying segregation and transmission rates of alien (L. mollis) chromosomes in different backcross generations of wheat–L. mollis backcross populations22,23,24,25. The segregation rates are usually narrower in BC1F1 as compared to F2, while alien transmission rates are higher in disomic lines, especially disomic substitution lines, than monosomic lines24,25. Different types (whole-arm or Robertsonian, intercalary and distal) of wheat–Leymus translocation lines have also been developed3,12,26,27,28,29, and Li et al.29 recorded average translocation frequency of 7.55% for L. mollis chromosomes, while Kishii28 found that the translocation frequencies of L. racemosus chromosomes ranged between 0 and 8%, with higher translocation frequencies in the short arms. However, the unavailability of L. mollis genome sequence information and known polymorphic cytological markers to differentiate its chromosomes constitute obstacles to rapid development and adequate characterization of wheat–L. mollis chromosome introgression lines (CILs).
Leymus racemosus, a close relative of L. mollis, has reliable variable cytological markers which enables the application of fluorescence in situ hybridization (FISH) to differentiate its chromosomes in wheat–L. racemosus addition lines30. Using these lines and other wheat–L. racemosus CILs for marker validation, we recently developed many L. racemosus genome-based DNA markers and successfully transferred a good proportion of the PCR-based markers to L. mollis genome31. Therefore, in this study, we applied the transferred markers to develop a methodology for producing wheat–L. mollis addition lines without in situ hybridization. The PCR markers were used for marker-assisted selection, and DArTseq was applied to further genotype the selected lines, allowing development of L. mollis chromosome-specific SNP markers to complement the PCR-based markers. In order to validate the marker-based results, genomic in situ hybridization (GISH) was applied to confirm the status (disomic or monosomic) of the added chromosomes. The L. mollis chromosome-specific markers developed in this study enabled clear differentiation of the wheat–L. mollis addition lines, and SNP markers among the two Leymus species highlighted their genomic relationship. Also, preliminary phenotypic data summarized the varying effects of the different L. mollis chromosomes.
Methods
Plant materials
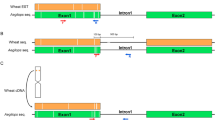
Bread wheat cv. Chinese Spring (CS) (2n = 6x = 42, AABBDD; recipient) was crossed with L. mollis (2n = 4x = 28, Ns1Ns1Ns2Ns2; donor) to produce wheat–L. mollis F1 hybrids (2n = 5x = 35; ABDNs1Ns2). To ensure the survival of the hybrids, embryo rescue was used32. The hybrids were backcrossed with CS, and the populations were characterized by chromosome counting and chromosome-specific DNA markers (Fig. 1). The two parents are maintained in the genebank of Tottori Chromosome Bank of Wheat (TACBOW), Arid Land Research Center, Tottori University, with accession numbers KT020-003 (CS) and TACBOW0113 (L. mollis). The L. mollis accession was originally collected from the Japan seashore (37°5′38″N 140°59′6″E) close to Iwaki, Fukushima, Japan.

Pedigree of 10 wheat–L. mollis chromosome addition lines CS, Chinese Spring; Lm, L. mollis; single letters in brackets, monosomic lines; double letters in brackets, disomic lines.
Molecular marker-assisted selection of monosomic and disomic lines
Genomic DNA samples of seedlings of the backcross populations were isolated and purified using the cetyl trimethyl ammonium bromide (CTAB) method and genotyped with L. racemosus PCR-based markers transferred to L. mollis31. Non-carrier segregants and duplicated carriers were discarded, and the remaining plants were advanced to BC3F4 under a temperature-controlled (22 °C day/18 °C night) greenhouse. In each generation, disomic plants were distinguished using progeny test. In this test, we assumed that monosomic addition lines produce non-carrier (2n = 42, AABBDD), monosomic addition (2n = 43, AABBDD + 1′ [Ns]), and disomic addition (2n = 44, AABBDD + 1′′ [Ns]) segregants, whereas genetically stable disomic plants do not segregate: they produce only disomic addition lines (2n = 44, AABBDD + 1′′ [Ns]). This is because, in meiosis, the monosomic addition lines are expected to produce two different gametes: ABD and ABDNs, while the disomic addition lines should produce one gamete, ABDNs.
DArTseq genotyping
In order to confirm the presence of L. mollis chromosomes in the selected addition lines and develop more L. mollis chromosome-specific markers, DArTseq genotyping was applied to rescreen three replicates of each of the lines alongside the two parents and L. racemosus. The latter was included to estimate the genomic relationship between the two Leymus species because L. racemosus genome PCR markers were used to genotype the wheat–L. mollis addition lines. All the DNA samples were sent to Diversity Arrays Technology Pty Ltd, Australia (http://www.diversityarrays.com/) for sequencing and marker identification. The genomic representations were sequenced on HiSeq 2500 and the sequences were aligned to wheat_ChineseSpring04 reference and wheat_ConsensusMap_version_4, as our analysis was based on DArTseq platform optimized for hexaploid wheat. DArTseq data in SNP 1 Row Mapping Format, which we used in this study, was scored “0” for a reference allele homozygote (wheat allele only), “1” for an SNP allele homozygote (L. mollis allele only), and “2” for a heterozygote (both wheat and L. mollis alleles). SNP markers with call rate of 100% (definite scores across all the samples: bread wheat, L. mollis and wheat–L. mollis genomes) were used for the analysis. Markers with SNP alleles in the wheat genome were discarded. Genetic mapping-related statistics were not considered because our objective was to identify polymorphic markers to differentiate between the wheat, Leymus and wheat–L. mollis genomes. The data were analyzed for polymorphism between the wheat and L. mollis genomes, and polymorphic markers were used to identify L. mollis segments in the wheat–L. mollis lines. Possible substitutions of wheat chromosomes were analyzed by filtering the SNP markers specific to the L. mollis segment (markers with score of “1”) in each line. It should be understood that wheat DArTseq platform grouped all the markers (including L. mollis-specific) into the 21 chromosomes of wheat, hence the presence of L. mollis chromosome in a wheat–L. mollis line would be indicated by a score of “1” if the wheat homoeolog is substituted, or 2 if the homoeolog is not substituted. The former case indicates a substitution line, while the latter indicates an addition line.
On the basis of the correspondence between the SNP alleles and reference alleles in each CS chromosome provided by DArTseq, L. mollis chromosome-specific SNP markers were used to determine the homoeologous groups (HGs) of L. mollis chromosomes in the wheat background. Genomic relationship between the genomes of the two Leymus species was roughly estimated by marker polymorphism between the genomes, whereas markers consistently called (call rate of 100%) among the two genomes, wheat–L. mollis and wheat–L. racemosus addition lines were used to estimate the relationship between the chromosomes of the two Leymus species. This latter set of markers was used for cluster analysis (http://genomes.urv.cat/UPGMA/) to reveal the associations among the chromosomes of the two Leymus species.
Identification of L. mollis chromosomes by GISH
L. mollis genomic DNA was labeled with fluorescein-12-dUTP (Thermo Scientific) using Random Primers DNA Labeling System (Invitrogen). With the labeled L. mollis genomic DNA as probe, GISH was performed for the 10 addition lines following a protocol described for Triticeae species33, with slight modifications: steps 3–9 of slide pre-hybridization were skipped and the probe was denatured at 100 °C for 5 min instead of 75 °C for 10 min. After hybridization, the slides were viewed and photographed with an Olympus BX61 automated fluorescence microscope (Olympus).
Preliminary phenotypic evaluation of the CALs
The 10 addition lines alongside the background wheat cultivar (CS) were laid out in a completely randomized design (CRD) with six replicates in a greenhouse. Seeds of all the genotypes were sown in petri dishes under the same condition, and uniform seedlings of each genotype were transplanted to plastic pots, one plant in each pot. DNA samples of seedlings from monosomic lines were genotyped by PCR to ensure that only alien carriers were transplanted. All the plants grew under a temperature-controlled (22 °C day/18 °C night) condition. Adequate cultural practices necessary for optimum crop performance were observed. Data were taken on number of days to heading and physiological maturity, plant height, spike length, number of spikes per plant, grain yield per spike and grain yield per plant. Two-tailed t-test was applied to compare the mean values of traits between CS and each addition line. At this preliminary stage, differences among the addition lines were not considered, as all the lines are intended to be collectively evaluated under different stress conditions for further selection and production of translocation and recombination lines with desired segments of L. mollis chromosomes.
Results
Production of wheat–L. mollis chromosome addition lines
We developed 10 distinct wheat–L. mollis addition lines (Fig. 1; Table 1). The wheat × L. mollis F1 hybrids exhibited perennial growth, flowered, but failed to set seed. Repeated attempts to double the chromosomes of the hybrids by colchicine treatment were unsuccessful, so we crossed the hybrids with CS pollens. This approach also failed, but when we crossed in a reciprocal direction, two seeds were harvested. These seeds germinated and grew into fertile annual octoploids (2n = 8x = 56, AABBDDNs1Ns2). One of them was used to generate backcross populations, from which the addition lines were produced. In the third backcross generation, we selected, by chromosome counting, plants with 43 chromosomes—assumed to be monosomic addition lines—and arbitrarily assigned alphabetical tags (LmA–N) to them (Fig. 1). Leymus markers were then used for L. mollis chromosome identification and differentiation. Chromosome-specific markers (Table 1) were used to differentiate the lines, while progeny test was used to distinguish between disomic (6) and monosomic (4) lines as we advanced the populations to BC3F4.
Development of L. mollis polymorphic markers and identification of alien chromosomes in the wheat background
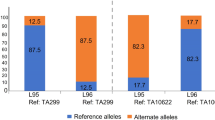
From 95 PCR markers transferred from L. racemosus to L. mollis31 and 15,426 SNP markers in the genomes of L. mollis and CS, we developed 14,577 L. mollis polymorphic markers (47 PCR-based and 14,530 SNP markers) (Table 1 and online Supplementary Table S1). The PCR analysis indicated 49% marker polymorphism between CS and L. mollis genomes31, whereas the SNP markers revealed a higher polymorphism (~94%) (see online Supplementary Table S1). Chromosomes of L. mollis in the wheat background were efficiently identified by 27 PCR markers and 6,317 SNP markers (Table 1). A total of 5,957 L. mollis chromosome-specific markers, ranging from 185 in LmL to 796 in LmI (Table 1), enabled unambiguous differentiation of the 10 lines. The number of SNP markers with a genotypic score of 1 (representing only the L. mollis allele) in each line ranged between 3 (0.05%) and 15 (0.2%), which lie within genotyping error range. All the lines retained almost 100% of the reference (wheat) alleles in addition to the SNP alleles, showing that none of the CS chromosomes was substituted. Therefore, all the introgressions were confirmed to be addition lines.
Homoeologous groups of L. mollis chromosomes added to wheat
The HGs of all the L. mollis chromosomes added to wheat were determined from the correspondence of L. mollis chromosome-specific SNP markers to the HGs of CS. The 10 L. mollis chromosomes fitted well into seven homoeologous groups, with six chromosomes falling into three HGs (Table 2). Chromosomes in the same HG obviously belong to different sub-genomes, granted that L. mollis is a tetraploid species.
Confirmation of the cytological status of each L. mollis chromosome in the addition lines GISH
Because the use of marker-assisted selection in differentiating monosomic and disomic lines is not a common practice, we validated the status (monosomic or disomic) of L. mollis chromosomes in the addition lines by GISH, which confirmed the marker-based results (Fig. 2). All the disomic addition lines were confirmed to have a pair of L. mollis chromosomes, whereas the monosomic addition lines had single L. mollis chromosomes.

Identification of L. mollis chromosomes added to wheat using genomic in situ hybridization (GISH). (A–N), L. mollis chromosomes; double letters, disomic lines; single letters, monosomic lines; arrows point to the added chromosomes detected with fluorescein-12-dUTP (green).
Relationship between L. mollis and L. racemosus genomes
Out of 8,653 SNP markers consistently scored in the two Leymus genomes, 75% were monomorphic (see online Supplementary Table S2). A cluster analysis using 579 SNP markers among the 10 wheat-L. mollis lines produced in this study and 8 wheat–L. racemosus addition lines reported previously30 revealed an interesting relationship between the chromosomes of the two Leymus species. Seven chromosomes each of L. mollis and L. racemosus in six HGs clustered in pairs: L. mollis chromosome H (LmH) and L. racemosus chromosome N (LrN); L. mollis chromosome F (LmF) and L. racemosus chromosome H (LrH); L. mollis chromosome C (LmC) and L. racemosus chromosome J (LrJ); L. mollis chromosome D (LmD) and L. racemosus chromosome K (LrK); L. mollis chromosome N (LmN) and L. racemosus chromosome F (LrF); L. mollis chromosome I (LmI) and L. racemosus chromosome I (LrI); L. mollis chromosome A (LmA) and L. racemosus chromosome A (LrA) (Fig. 3). In HG3, L. mollis chromosomes LmF and LmH were associated with L. racemosus chromosomes LrH and LrN, respectively. Apart from LmL in HG1, whose homoeolog in L. racemosus was not produced, every other HG included at least two chromosomes, one from each Leymus species. In each HG, the genomic distance between homoeologous chromosomes of the two species was narrower than that within the same species (Table 3). In HG3 for instance, the distance indices between chromosomes LmF and LrH (18) and LmH and LrN (16) are clearly lower than those between LmF and LmH (90) and LrH and LrN (88). Using the genetic association among the chromosomes (Fig. 3; Table 3), we tentatively named the chromosomes (Table 4). For each of the species, we numbered the chromosomes according to their HGs and arbitrarily assigned each chromosome of a homoeologous pair to different sub-genomes, designated as Ns1 and Ns2; the superscripts “m” and “r” indicate L. mollis and L. racemosus, respectively. All non-homoeologous chromosomes in each species were assumed to be in the same sub-genome, as homoeology between two chromosomes should not exist in one sub-genome.

Relationship between L. mollis and L. racemosus chromosomes LmA–N, L. mollis chromosomes; LrA–N, L. racemosus chromosomes; numbers at the nodes are bootstrap values; clustering method: Unweighted pair group method with arithmetic mean (UPGMA) (http://genomes.urv.cat/UPGMA/).
Phenotypic variation between CS and the addition lines
From the results of the preliminary evaluation we conducted (Fig. 4; Supplementary Table S3), all the addition lines were significantly different from CS in at least one of the seven traits measured, indicating the effects of L. mollis chromosomes in the lines. Number of days to heading (DH) was significantly reduced in six of the lines, significantly increased in one, while three lines were not significantly different from CS (Fig. 4a). Five of the six lines with significantly reduced DH also reached physiological maturity significantly earlier than CS, while the other five lines were not significantly different from CS (Fig. 4b). Plant height was significantly reduced in five lines, significantly increased in one line, while four lines were not different from CS (Fig. 4c). All the lines were significantly different from CS in at least one yield component (Fig. 4d–g). Although spikes were significantly longer in some of the lines (Fig. 4d), number of spikes per plant, grain yield per spike and grain yield per plant were significantly lower in all the lines, except LmG and LmI, which were not significantly different from CS in number of spikes per plant and grain yield per spike, respectively.

Variation in quantitative traits between wheat (cv. CS) and wheat-L. mollis addition lines. CS, Chinese Spring; LmA–N, L. mollis chromosomes; PM, physiological maturity; *, p < 0.05; **, p < 0.01; ***, p < 0.001; error bars, standard errors of the means. Mean values (n = 6) between CS and each addition line were compared using two-tailed t-test. Differences among the lines were not considered at this preliminary stage, as all the lines will be subjected to different conditions of stress for further selection and production of translocation and recombination lines with desired segments of L. mollis chromosomes.
Discussion
In our experience30,34, production of wheat–alien CILs from intergeneric F1 hybrid plants requires at least eight years of laborious cytogenetic and/or phenotypic screening. Here, we have shown that marker-assisted selection can considerably reduce the time and effort. In this study, marker development, genotyping, selection and confirmation of the status of each alien chromosome in the 10 addition lines were completed within three years. Our marker-based approach of identifying disomic lines was supported by GISH results. Therefore, the use of molecular markers from the genomes of important wild relatives to track their chromosomes in wheat is a reliable method to facilitate speedy expansion of diversity in wheat germplasm31,35,36. This approach would incentivize wheat breeders to develop cultivars with improved adaptability to keep pace with the projected global increase in wheat demand37.
We avoided spurious duplication of different segments of the same chromosome in different addition lines. The chromosome-specific markers developed in this study clearly differentiated the 10 lines. The duplicates we initially selected by chromosome counting were clarified by molecular markers. Identical PCR amplifications and DArTseq genotypic data revealed lines carrying the same L. mollis chromosome. This would not be possible with GISH because GISH uses genomic DNA as probe, which can only identify the alien chromosomes but cannot show differences between them. Using phenotypic data to identify plants that carry different segments of the same chromosome is difficult; hence, they may be mistaken as carriers of different chromosomes. Such situations can be effectively managed with appropriate molecular markers.
Determination of the HG of each L. mollis chromosome using the chromosome-specific SNP markers was consistent with the chromosome clustering using shared SNP markers. The HGs of the remaining L. mollis chromosomes, if introgressed in wheat, can be easily determined with this approach. Other methods of HG determination can result in assignment of different HGs to the same chromosome30,38. To determine the HGs of alien chromosomes, their ability to substitute wheat chromosomes in interspecific or intergeneric hybrids and functionally compensate the substituted chromosomes in substitution lines is usually applied39,40,41,42. This approach is difficult and its accuracy relies, to a large extent, on the morphological similarities between the substitution line and the wild type, which is under environmental influences.
The high proportion of monomorphic markers in the two genomes of Leymus species strongly indicates high genomic similarity between the genomes, and the association of their chromosomes suggests that the genomic distance between the sub-genomes of the two species is smaller than the distance between the sub-genomes within each of the species. This gives the impression that the two Leymus species may have evolved from independent hybridization events of the same diploid species with partially differentiated genomes, for instance Ns1 and Ns2. However, at the moment, we cannot draw a convincing conclusion on the evolutionary relationship between the two Leymus species or their sub-genomes, as this would require the application of the genomic resources of their diploid progenitors as analyzers of the polyploid genomes. Diploid analyzers would also enable reliable discrimination of the sub-genomes and help to clarify the sources of the elementary genomes of the Leymus species. Noteworthy is that the chromosomes of hybrids from Leymus species form complete meiotic pairs43,44,45,46, which is a rare phenomenon in interspecific hybrids. This and the high transferability of DNA markers between L. racemosus and L. mollis suggest that the genomic resources of any of the two species should be interchangeably deployed to analyze their genomes. But we could not characterize L. mollis chromosomes with L. racemosus cytological markers, so the genomic difference between these species still needs to be clarified. The tentative nomenclature for Leymus chromosomes proposed here can easily accommodate all the chromosomes of Leymus species, irrespective of ploidy level. Considering the consistency of HGs determined independently using the chromosome-specific and common SNP markers, we are certain about the HGs of the L. mollis chromosomes, but the accuracy of the assignment of the chromosomes to the sub-genomes needs to be confirmed using diploid analyzers.
Interestingly, the preliminary phenotypic results obtained in this study have shown that L. mollis chromosome segments can be used to develop early maturing cultivars which can be cultivated in areas with short periods of favourable wheat growing conditions. However, the exact segment of the chromosome(s) should be identified and used to produce translocation or recombination lines, to reduce the effects of deleterious genes which may have caused the reduction in the yield components observed in the addition lines. At the moment, since all the genotypes were evaluated under normal growth conditions, we cannot conclude that the relative superior yield components of CS will be sustained under stress conditions. Therefore, all the genotypes will be evaluated under different simulated and actual stress conditions to explore the possibility of selecting lines with the sturdy traits of Leymus species6,10,11,12,13.
Essentially, we have proven that wheat–alien addition lines can be rapidly developed and reliably characterized using DNA markers for effective selection of carriers and recognition of disomic and monosomic lines. This approach requires the development of chromosome-specific DNA markers from the genomes of potential gene donors or closely related sequenced genomes. The integration of DArTseq genotyping allows confirmation of the PCR results, development of more chromosome-specific markers, and further characterization of the addition lines. Allocation of all the markers on the DArTseq platform to wheat chromosomes enables effective analysis of wheat–alien complexes. The application of genotyping-by-sequencing approaches, including DArTseq, in the analysis of germplasm of wheat and other plants has gained reasonable popularity47,48,49,50. This is not the case with the characterization of wheat-alien introgression lines involving distant relatives of wheat, possibly because it may be thought that wide genetic distance and differences in the ploidy levels of bread wheat and these relatives would not allow identification of homoeology between their genomes. Our previous report on wheat–L. racemosus31 and the results of this study have shown clearly that alien segment-specific markers can easily be isolated and the correspondence of alien chromosome-specific SNP markers with bread wheat reference alleles can be used to determine the HGs of the alien chromosomes. This study has clearly demonstrated that DArTseq SNPs can be integrated with PCR markers to produce and characterize wheat–alien addition lines without necessarily applying in situ hybridization for alien chromosome identification. Given its reliability and savings in time and efforts, we recommend the use of this simple methodology to accelerate introgression breeding of wheat.
References
Anamthawat-Jonsson, K. Genetic and genomic relationships in Leymus Hochst. Hereditas 135, 247–253 (2001).
Anamthawat-Jonsson, K. et al. Evolutionary diversification of satellite DNA sequences from Leymus (Poaceae: Triticeae). Genome 52, 381–390, https://doi.org/10.1139/g09-013 (2009).
Chen, P. et al. Development and characterization of wheat–Leymus racemosus translocation lines with resistance to Fusarium Head Blight. Theor Appl Genet 111, 941–948, https://doi.org/10.1007/s00122-005-0026-z (2005).
Fan, X. et al. Evolutionary pattern of rDNA following polyploidy in Leymus (Triticeae: Poaceae). Mol Phylogenet Evol 77, 296–306, https://doi.org/10.1016/j.ympev.2014.04.016 (2014).
Fan, X. et al. Phylogeny and evolutionary history of Leymus (Triticeae; Poaceae) based on a single-copy nuclear gene encoding plastid acetyl-CoA carboxylase. BMC Evol Biol 9, 247, https://doi.org/10.1186/1471-2148-9-247 (2009).
Yang, X. et al. Development and molecular cytogenetic identification of a novel wheat–Leymus mollis Lm#7Ns (7D) disomic substitution line with stripe rust resistance. PLoS One 10, e0140227, https://doi.org/10.1371/journal.pone.0140227 (2015).
Zhang, H. & Dvorak, J. The genome origin of tetraploid species of Leymus (Poaceae: Triticeae) inferred from variation in repeated nucleotide sequences. Am J Bot 78, 871–884 (1991).
Wang, R. R. & Jensen, K. B. Absence of the J genome in Leymus species (Poaceae: Triticeae): evidence from DNA hybridization and meiotic pairing. Genome 37, 231–235 (1994).
Anamthawat-Jonsson, K. Molecular cytogenetics of Leymus: Mapping the Ns genome-specific repetitive sequences. J Syst Evol 52, 716–721, https://doi.org/10.1111/jse.12106 (2014).
McGuire, P. & Dvorak, J. High salt-tolerance potential in wheatgrasses. Crop Science 21, 702–705 (1981).
Xiao, X., Sha, L., Fan, X. & Zhou, Y. Salt tolerance on seed germination of five Leymus Species. World Journal of Agricultural Sciences 8, 585–589, https://doi.org/10.5829/idosi.wjas.2012.8.6.1691 (2012).
Bao, Y., Wang, J., He, F., Ma, H. & Wang, H. Molecular cytogenetic identification of a wheat (Triticum aestivum) –American dune grass (Leymus mollis) translocation line resistant to stripe rust. Genet Mol Res 11, 3198–3206, https://doi.org/10.4238/2012.May.22.2 (2012).
Zhang, A. et al. Molecular cytogenetics identification of a wheat–Leymus mollis double disomic addition line with stripe rust resistance. Genome 60, 375–383, https://doi.org/10.1139/gen-2016-0151 (2017).
Wang, L. & Chen, P. Development of Triticum aestivum–Leymus racemosus ditelosomic substitution line 7Lr#1S(7A) with resistance to wheat scab and its meiotic behavior analysis. Chin Sci Bull 53, 3522–3529 (2008).
Qi, L. L., Pumphrey, M. O., Friebe, B., Chen, P. D. & Gill, B. S. Molecular cytogenetic characterization of alien introgressions with gene Fhb3 for resistance to Fusarium head blight disease of wheat. Theor Appl Genet 117, 1155–1166, https://doi.org/10.1007/s00122-008-0853-9 (2008).
Yang, X. F. et al. Chromosome constitution and origin analysis in three derivatives of Triticum aestivum–Leymus mollis by molecular cytogenetic identification. Genome 57, 583–591, https://doi.org/10.1139/gen-2014-0161 (2014).
Yang, X. F. et al. Isolation and molecular cytogenetic characterization of a wheat–Leymus mollis double monosomic addition line and its progenies with resistance to stripe rust. Genome 60, 1029–1036, https://doi.org/10.1139/gen-2017-0078 (2017).
Mohammed, Y. S. A. et al. Impact of wheat–Leymus racemosus added chromosomes on wheat adaptation and tolerance to heat stress. Breeding Sci 63, 450–460, https://doi.org/10.1270/jsbbs.63.450 (2014).
Mohammed, Y. S., Eltayeb, A. E. & Tsujimoto, H. Enhancement of aluminum tolerance in wheat by addition of chromosomes from the wild relative Leymus racemosus. Breed Sci 63, 407–416, https://doi.org/10.1270/jsbbs.63.407 (2013).
Liu, X., Shi, J., Zhang, X. Y., Ma, Y. S. & Jia, J. Z. Screening salt tolerance germplasms and tagging the tolerance gene(s) using microsatellite (SSR) markers in wheat. Acta Bot Sin 43, 948–954 (2001).
Tsitsin, N. V. Remote hybridization as a method of creating new species and varieties of plants. Euphytica 14, 326–330, https://doi.org/10.1007/BF00149519 (1965).
Wang, J. et al. Morphological and molecular cytogenetic characterization of partial octoploid. Tritileymus. Genet Resour Crop Ev 60, 1453–1462, https://doi.org/10.1007/s10722-012-9933-4 (2013).
Fu, J., Chen, S. & Zhang, A. Studies of the formation and cytogenetics of octoploid Tritileymus. Acta Genet Sin 20, 317–323 (1993).
Fu, J., Chen, S., Zhang, A., Hou, W. & Yang, Q. Cytogenetic studies on the cross progenies between octoploid Tritileymus and Triticum aestivum. Acta Genet Sin 23, 24–31 (1996).
Fu, J. et al. Cytogenetic studies on the cross between octoploid Tritileymus and nullisomic wheat. Acta Genet Sin 24, 350–357 (1997).
Pang, Y. H. et al. Molecular cytogenetic characterization of a wheat–Leymus mollis 3D(3Ns) substitution line with resistance to leaf rust. J Genet Genomics 41, 205–214, https://doi.org/10.1016/j.jgg.2013.11.008 (2014).
Pang, Y. H. et al. Cytogenetic and molecular identification of a wheat–Leymus mollis alien multiple substitution line from octoploid Tritileymus x Triticum durum. Genet Mol Res 13, 3903–3913, https://doi.org/10.4238/2014.May.23.1 (2014).
Kishii, M. Production of wheat–Leymus racemosus translocation lines. eWIS 111, 11–13 (2011).
Li, H. et al. Development of Triticum aestivum–Leymus mollis translocation lines and identification of resistance to stripe rust. J Genet Genomics 42, 129–132, https://doi.org/10.1016/j.jgg.2014.11.008 (2015).
Kishii, M., Yamada, T., Sasakuma, T. & Tsujimoto, H. Production of wheat–Leymus racemosus chromosome addition lines. Theor Appl Genet 109, 255–260, https://doi.org/10.1007/s00122-004-1631-y (2004).
Edet, O. U. et al. Efficient anchoring of alien chromosome segments introgressed into bread wheat by new Leymus racemosus genome-based markers. BMC Genet 19, 18, https://doi.org/10.1186/s12863-018-0603-1 (2018).
Cisneros, A. & Tel-Zur, N. Embryo rescue and plant regeneration following interspecific crosses in the genus Hylocereus (Cactaceae). Euphytica 174, 73–82, https://doi.org/10.1007/s10681-010-0135-x (2010).
Brammer, S. P., Vasconcelos, S., Poersch, L. B., Oliveira, A. R. & Brasileiro-Vidal, A. C. Genomic in situ hybridization in Triticeae: A methodological approach. In: Plant Breeding from Laboratories to Fields (ed Andersen S. B.) 1–22 (InTech, Rijeka, 2013).
Kishii, M. et al. Production of wheat–Psathyrostachys huashanica chromosome addition lines. Genes Genet Syst 85, 281–286 (2010).
Zhang, J. et al. A resource of large-scale molecular markers for monitoring Agropyron cristatum chromatin introgression in wheat background based on transcriptome sequences. Sci Rep 7, 11942, https://doi.org/10.1038/s41598-017-12219-4 (2017).
Gong, W. et al. Agronomic traits and molecular marker identification of Wheat–Aegilops caudata addition lines. Front Plant Sci 8, 1743, https://doi.org/10.3389/fpls.2017.01743 (2017).
Shiferaw, B. et al. Crops that feed the world 10. Past successes and future challenges to the role played by wheat in global food security. Food Secur 5, 291–317, https://doi.org/10.1007/s12571-013-0263-y (2013).
Larson, S. R. et al. Leymus EST linkage maps identify 4NsL-5NsL reciprocal translocation, wheat–Leymus chromosome introgressions, and functionally important gene loci. Theor Appl Genet 124, 189–206, https://doi.org/10.1007/s00122-011-1698-1 (2012).
Morris, R. & Sears, E. R. The cytogenetics of wheat and its relatives. In: Wheat and wheat improvement (eds Quisenberry, R. S. & Reitz, L. P.) 19–87 (Madison, 1967).
Molnar-Lang, M., Molnar, I., Szakacs, E., Linc, G. & Bedo, Z. Production and molecular cytogenetic identification of wheat-alien hybrids and introgression lines. In: Genomics of plant genetic resources: managing sequencing and mining genetic resources Vol. 1 (eds Tuberosa, R., Graner, A. & Frison, E.) 255–284 (Springer, 2014).
Calderon, M. D., Ramirez, M. D., Martin, A. & Prieto, P. Development of Hordeum chilense 4Hch introgression lines in durum wheat: a tool for breeders and complex trait analysis. Plant Breeding 131, 733–738, https://doi.org/10.1111/j.1439-0523.2012.02010.x (2012).
Badaeva, E. D., Budashkina, E. B., Badaev, N. S., Kalinina, N. P. & Shkutina, F. M. General features of chromosome substitutions in Triticum aestivum x T. timopheevii hybrids. Theor Appl Genet 82, 227–232, https://doi.org/10.1007/BF00226218 (1991).
Dewey, D. Cytogenetics of tetraploid Elymus cinereus, E. triticoides, E. multicaulis, E. karatviensis, and their F1 hybrids. Bot Gaz 133, 51–57 (1972).
Dewey, D. Genome relations among diploid Elymus junceus and certain tetraploid and octoploid Elymus species. Am J Bot 57, 633–639 (1970).
Dewey, D. R. The genomic system of classification as a guide to intergeneric hybridization with the perennial Triticeae. In: Gene manipulation in plant improvement (ed Gustafson, J. P.) 209–279 (Plenum Publishing Corporation, 1984).
Kishii, M., Wang, R. R. & Tsujimoto, H. Characteristics and behaviour of the chromosomes of Leymus mollis and L. racemosus (Triticeae, Poaceae) during mitosis and meiosis. Chromosome Res 11, 741–748 (2003).
Li, H. et al. A high density GBS map of bread wheat and its application for dissecting complex disease resistance traits. BMC Genomics 16, 216, https://doi.org/10.1186/s12864-015-1424-5 (2015).
Baloch, F. S. et al. A whole genome DArTseq and SNP analysis for genetic diversity assessment in durum wheat from Central Fertile Crescent. Plos One 12, doi:ARTN e016782110.1371/journal.pone.0167821 (2017).
Davey, J. W. et al. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat Rev Genet 12, 499–510, https://doi.org/10.1038/nrg3012 (2011).
Andrews, K. R., Good, J. M., Miller, M. R., Luikart, G. & Hohenlohe, P. A. Harnessing the power of RADseq for ecological and evolutionary genomics. Nat Rev Genet 17, 81–92, https://doi.org/10.1038/nrg.2015.28 (2016).
Acknowledgements
This study was supported by a Japan Society for the Promotion of Science KAKENHI Grant (No. 25292039). The authors thank Mses. I. Higashida and K. Inagaki of Molecular Breeding Laboratory, Arid Land Research Center, and Mses. A. Hashinaga and K. Kubota of the Faculty of Agriculture, Tottori University, Japan for providing helpful technical assistance.
Author information
Authors and Affiliations
Contributions
All the authors conceived and designed the experiments. H.T. and M.K. produced the F1 hybrids and backcross populations. O.U.E. and S.C. conducted chromosome observations and GISH. O.U.E. and Y.S.A.G. developed molecular markers, genotyped, and selected the addition lines. O.U.E. analyzed the genotypic data and wrote the manuscript with help from co-authors. All the authors read and approved the final version.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Edet, O.U., Gorafi, Y.S.A., Cho, Sw. et al. Novel molecular marker-assisted strategy for production of wheat–Leymus mollis chromosome addition lines. Sci Rep 8, 16117 (2018). https://doi.org/10.1038/s41598-018-34545-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-34545-x
Keywords
This article is cited by
-
In-depth genome diversity, population structure and linkage disequilibrium analysis of worldwide diverse safflower (Carthamus tinctorius L.) accessions using NGS data generated by DArTseq technology
Molecular Biology Reports (2020)
-
Whole-genome diversity, population structure and linkage disequilibrium analysis of globally diverse wheat genotypes using genotyping-by-sequencing DArTseq platform
3 Biotech (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
