
Abstract
We examined the transcriptome/post-transcriptome for persistent gene expression changes after radiation exposure in a baboon model. Eighteen baboons were irradiated with a whole body equivalent dose of 2.5 or 5 Gy. Blood samples were taken before, 7, 28 and 75–106 days after radiation exposure. Stage I was a whole genome screening for mRNA combined with a qRT-PCR platform for detection of 667 miRNAs. Candidate mRNAs and miRNAs differentially up- or down-regulated in stage I were chosen for validation in stage II using the remaining samples. Only 12 of 32 candidate genes provided analyzable results with two mRNAs showing significant 3–5-fold differences in gene expression over the reference (p < 0.0001). From 667 candidate miRNAs, 290 miRNA were eligible for analysis with 21 miRNAs independently validated using qRT-PCR. These miRNAs showed persistent expression changes on each day and over days 7–106 days after exposure (n = 7). In particular miR-212 involved in radiosensitivity and immune modulation appeared persistently and 48–77-fold up-regulated over the entire time period. We are finally trying to put our results into a context of clinical implications and provide possible hints on underlying molecular mechanisms to be examined in future studies.
Similar content being viewed by others


Introduction
Taking advantage of a non-human primate model with high genetic homology to humans, we wondered whether radiation exposure in healthy animals might result in gene expression changes in the peripheral blood persisting over weeks and months after radiation exposure. Clearly, those examinations cannot be performed in humans and in patients treated with radiation therapy it is impossible to discriminate whether changes in gene expression origin from the disease they are suffering from or the radiation exposure. A literature review identified two areas where persistent gene expression might be of clinical interest.
Patients treated for a first cancer are at risk for a second tumor after radiation or chemotherapy1,2,3,4. Small RNAs such as miRNAs are recognized to play an important role in cancer diagnosis and even therapy5,6,7,8,9. Regarding the classification of different tumor types, miRNAs might be better than protein coding mRNA10. For example, it was reported that a signature miRNA was downregulated 1–2 years prior to the diagnosis of lung cancer, suggesting the prediction potential using miRNAs5. Provided these changes in post-transcriptional miRNAs are causally related to tumor development, these could serve as therapeutic targets. Prerequisite of a sufficient diagnosis or prediction of the later occurring tumor entity are persistent radiation-related gene expression changes after exposure.
Radiation-related or cancer-associated behavioral symptoms such as fatigue, depression and sleep disturbances are reported after treatment of tumor entities such as breast cancer11,12,13, prostate cancer14 or lung cancers15. In particular fatigue is difficult to manage medically since no single effective intervention exists for severe fatigue other than for e.g. pain16. Fatigue is reported to last over months or even years after cancer therapy15,17. Inflammatory processes are reported to represent a possible mechanism for the fatigue18,19 as well as other mechanisms such as disrupted metabolism of adenosine triphosphate16. However, currently, there is no accepted pathophysiological evidence to explain the development of fatigue16 and it is challenging to discriminate whether fatigue is cancer- or radiation-related. Examinations of persistent gene expression changes might contribute to understanding long-term post-treatment sequelae and might provide clues about an underlying molecular basis for it.
In collaboration with the French Army Biomedical Research Institute, we assessed blood samples obtained from irradiated healthy baboons taken before (day 0), 7 days, 28 days and 75–106 days after partial or total body exposure. On a subset of the blood samples, we performed a whole genome screening and identified protein coding mRNA genes (screening stage I). These mRNAs were then validated (stage II) using qRT-PCR with the remaining samples. In stage I we also screened for 667 miRNAs using a qRT-PCR platform. The selected candidate miRNAs were validated on the remaining samples in stage II using the same qRT-PCR platform but restricting the analysis to the candidate miRNAs from stage I.
With this design we searched for persistent gene expression changes on the transcriptional (protein coding mRNAs) and the post-transcriptional level (miRNAs) so that we might add information on the significance of mRNA or miRNAs for cancer diagnosis. The irradiation of healthy baboons enables the identification of radiation-related gene expression changes which might represent the molecular basis for post-treatment fatigue and the discrimination from a cancer-related fatigue.
Materials and Methods
Animals and ethical approval
Eighteen baboons were bred by the Centre National de la Recherche Scientifique (Rousset sur Arc, France) for the purpose of biomedical research. In the nonhuman primate facility of the French Army Biomedical Research Institute, the baboons were placed in individual cages at 21 °C, with a relative humidity of 55% and a 12 h/12 h light-dark schedule. The animals received fresh fruit and solid food twice a day, and had access to water ad libitum. The male baboons had an average age of 8.1 years (±3.3 years) and weighed 23.7 (±5.2 kg). The experiment was approved by the French Army Animal Ethics Committee (No 2010/12.0). All baboons were treated in compliance with the European legislation related to animal care and protection in order to minimize discomfort. All applicable international, national and institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted.
Irradiation
The animals were anesthetized with a combination of tiletamine and zolazepam (6 mg.kg−1 intramuscularly, Zoletil 100; Virbac, Carros, France) before irradiation. Then, the baboons were placed in restraint chairs, sitting orthogonally, front to a horizontal and homogeneous field of gamma rays delivered by a 60Co source (IRDI 4000; Alsthom, Levallois, France) to perform either total body irradiation (TBI) or partial body irradiation (PBI). In order to attain different patterns of PBI, a 20 cm thick lead screen was used to shield different parts of the body (for details see20,21. Two baboons were exposed to 5 Gy TBI and two others to 2.5 Gy TBI. Eight different exposure patterns were simulated and two baboons were exposed per pattern which summed up to 14 baboons receiving PBI (for details see20,21) corresponding to an equivalent TBI dose of 2.5 or 5 Gy. Two dose rates were used (8 cGy/min for 5 Gy TBI and 5 Gy 50% PBI, and 32 cGy/min for all other situations) because the Cobalt 60 source was changed during this study. Moreover, to achieve the same homogeneous radiation field whatever the dose rate, all baboons were irradiated at the same distance from the source. Consequently, radiation exposures lasted between 8 min and 62 min. The mid-line tissue (right anterior iliac crest) dose in air was measured with an ionization chamber. Delivered doses were monitored by alumina powder thermoluminescent dosimeters placed on different cutaneous areas (thorax, thoracic and lumbar vertebrae, head, tibia, femur, femoral head, for details see20,21).
A full-time attending veterinarian (AV) was employed in the nonhuman primate facility. A written program of veterinary care was applied with regularly scheduled visits. The AV monitored the baboons during the whole study. Specifically, the veterinary surgeon in charge of animal welfare fulfilled a mission of council and inspection to ensure that nonhuman primates were provided supportive care in ways that minimize fear, pain, stress, and suffering. Accordingly, two baboons showing signs of pain received buprenorphine (Vetergesic Multidose, Sogeval, Sheriff Hutton, UK). A transient weight loss of 4–10% was observed in 3 baboons, but in general no change of behaviour and activity was observed and animals moved in their cages as usually.
RNA extraction and quality control
Whole blood samples (2.5 ml) were processed following the PAXgene Blood RNA system (BD Diagnostics, PreAnalytiX GmbH, Hombrechtikon, Switzerland). In brief, blood was drawn into a PAXgene Blood RNA tube at the French Army Biomedical Research Institute. The tube was gently inverted (10 times), stored at room temperature overnight then at −20 °C. After all samples were collected, the PAXGene tubes were sent to Germany for further processing. After thawing, washing and centrifugation, cells in the supernatant were lysed (Proteinase K, PAXgene Blood RNA protocol) followed by addition of Lysis/Binding Solution taken from the mirVana kit (Life Technologies, Darmstadt, Germany). With the mirVana kit total RNA, including small RNA species, was isolated by combining a Phenol-Chloroform RNA precipitation with further processing using a silica membrane. After several washing procedures, DNA residuals became digested on the membrane (RNAse free DNAse Set, Qiagen, Hilden, Germany). RNA was eluted in a collection tube and frozen at −20 °C. Quality and quantity of isolated total RNA were measured spectrophotometrically (NanoDrop, PeqLab Biotechnology, Erlangen, Germany). RNA integrity was assessed by the 2100 Agilent Bioanalyzer (Life Science Group, Penzberg, Germany) and DNA contamination was controlled by conventional PCR using an actin primer. We used only RNA specimens with a ratio of A260/A280 ≥ 2.0 (Nanodrop) and RNA integrity number (RIN) ≥7.5 for whole genome microarray (IMGM Laboratories, Martinsried, Germany) or RIN ≥7.3 for qRT-PCR analyses.
Stage I screening for mRNAs and miRNAs
The whole genome screening for differentially expressed genes (DEG) (protein coding mRNAs) was performed on 26 RNA samples taken from about one third of the baboons for each of the four time points which was before (pre-exposure samples, n = 5), 7 days (n = 7), 28 days (n = 7) and 75–106 days (n = 7) after radiation exposure. In order to increase the robustness we used pre-exposure RNA samples from other baboons than the RNA samples taken after irradiation (Table 1). The sample number for screening purposes taken after irradiation was higher (seven instead of five), because of their higher yield of RNA per sample, which were not used in previous examinations and, therefore, more RNA remained for this analysis on persistent gene expression over time. We used the Agilent oligo microarray GE 8 × 60 K (Agilent Technologies, Waldbronn, Germany) combined with a one-color based hybridization protocol of GeneSpring GX12 software for data analysis as described in detail elsewhere22. We analyzed gene expression by quantile normalized log2-transformed probe signals as an outcome. We used the non-parametric Mann Whitney (MW) test to compare gene expression across the different time points using the unexposed group as the reference (control). Only those gene transcripts that had a call “present” in at least 60% of RNA specimens were included in the analysis of gene expression and only genes with MW p-values ≤0.05 and a ≥2-fold gene expression difference among compared groups were considered to represent a candidate gene for validation in stage II. In parallel, a commercially available 384-well low density arrays (LDA) high throughput qRT-PCR platform was used that provided the simultaneous detection of 380 different miRNAs. Two different LDAs (type A and B) were combined so that the detection of 667 miRNA species (partly spotted in duplicate to completely fill the LDA) was possible. For miRNA screening purposes we used the same samples as used for the mRNAs. Due to the explorative nature of this study, the low sample size and the nonparametric statistics employed, we did not correct for multiple comparisons on the screening stage I of the study, but considered this within the validation stage II of our study where the numbers of hypotheses tested in parallel was reduced from about 20,000 (stage I) to 32 mRNAs and 21 miRNAs in stage II (see below). Gene expression data presented in this publication have been deposited at the NCBI’s Gene Expression Omnibus (GEO accession number GSE77254).
Stage II: Validation of stage I candidate genes via qRT-PCR
For validating the mRNA candidate genes from stage I (screening) using remaining RNA samples, we used a custom LDA and TaqMan chemistry (order IDs for the examined genes are provided in Table 2). A 1 µg RNA aliquot of each RNA sample was reverse transcribed using a two-step PCR protocol (High Capacity Kit). 400 µl cDNA (1 µg RNA equivalent) was mixed with 400 µl 2 x RT-PCR master mix and pipetted into the 8 fill ports of the LDA. Cards were centrifuged twice (1,200 rpm, 1 min, Multifuge3S-R, Heraeus, Germany), sealed, and transferred into the 7900 qRT-PCR instrument. The qRT-PCR was run for two hours following the qRT-PCR protocol for 384-well LDA format. All measurements were run in duplicate.
Two commercially available 384-well LDAs (type A and B) were used for detection of 667 miRNA species in stage I (see above), but only those candidate miRNAs showing significant changes in miRNA expression over the unexposed group were independently validated in stage II. Aliquots from each RNA sample (in general 2 µg total RNA/LDA type A/B) were reversely transcribed without preamplification over three hours using “Megaplex pools without preamplification l for microRNA expression analysis protocol”. Using different sets of primers, two kinds of cDNAs suitable for each of both LDAs were created. In a second step, the whole template cDNA and 450 µl 2x RT-PCR master mix were adjusted to a total volume of 900 µl by adding nuclease free water, and aliquots of 100 µl were pipetted into each fill port of a 384-well human LDA. Cards were centrifuged twice (see above), sealed, transferred into the 7900 RTQ-PCR instrument and again the 384-well LDA RTQ-PCR protocol was run over two hours.
All technical procedures for qRT-PCR were performed in accordance with standard operating procedures implemented in our laboratory in 2008 when the Bundeswehr Institute of Radiobiology became certified according to DIN EN ISO 9001/2008. All chemicals for qRT-PCR using TaqMan chemistry were provided by Life Technologies, Darmstadt, Germany.
For the custom LDA, threshold cycle (CT) values were normalized relative to the 18S rRNA measured in an aliquot of the RNA samples using a 96-well-format TaqMan qRT-PCR platform. We have found that this approach to normalization was more robust compared to the use of the internal control (GAPDH and 18 S rRNA) spotted on the LDA. For the commercial miRNA LDAs type A and B we used the median miRNA expression on each LDA for normalization purposes, because this proved to be more robust and a slightly more precise method compared to a normalization approach using a housekeeping miRNA species provided on the LDA (data not shown). The CT-values of the housekeeping gene was subtracted from the CT-value of each of the spotted genes, following the ∆CT-quantitative approach for normalization purposes.
Statistical analysis
During screening for potential candidate mRNAs or miRNAs for persistent changes in gene expression over time we used the same biosamples consisting of 5–7 samples per time point and used the unexposed pre-exposure samples as the reference. The filter (criteria) that we used was statistically significant (p ≤ 0.05) and ≥2-fold mRNA and miRNA expression changes over time after radiation exposure in those RNA species that had a call “present” in at least 60% of the samples. The validation was performed using the qRT-PCR gene expression results. During validation we assessed the assumptions of normality (Kolmogorov-Smirnov) and equal variance and if required utilized either the pooled (equal variance) or the Satterthwaite variant of the t-test (unequal variance). Normality of mRNA and miRNA expression values was achieved after log transformation where necessary. For these RNA species we used a Bonferroni correction to adjust the p-values for multiple comparisons. Descriptive statistics (n, mean, standard deviation, min, max) and p-values (t-test and the nonparametric Kruskal-Wallis (KW) test, where applicable) were calculated for each of the RNA species and per time point. After the validation we performed logistic regression analysis for each of the variables (genes) of interest separately (univariate) and using all samples together to increase the power. Odds ratios (OR), 95% confidence intervals (95% CI) and corresponding p-values (Wald Chi-Square) were calculated. We also determined the area under a receiver-operator characteristic (ROC) curve providing a reasonable indication of overall diagnostic accuracy. ROC areas of 1.0 indicate complete distinction between pre-exposure samples and time points after exposure. All calculations were performed using SAS (release 9.2, Cary NC, USA).
Results
Stage I: screening for candidate mRNAs and miRNAs
On average, we isolated 10 (stdev +/−2.9), 5.3 (+/−3.7), 13.4 (+/−9.3) and 8.1 (+/−2.2) µg total RNA from 2.5 ml whole blood before irradiation and 7, 28 and 75–106 days after irradiation, respectively. RNA integrity (RIN) with mean values of 8.5 (stdev +/−0.5), 9.0 (+/−0.5) 9.0 (+/−0.3) and 8.6 (+/−0.5) on these time points suggested high quality RNA sufficient for running both methods.
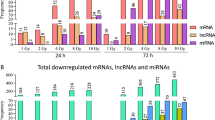
From about 20,000 protein coding mRNAs, 46% on average over all time points (range: 41–49%) appeared expressed. An about equal number of 324 up-regulated and 366 down-regulated DEG was observed on days 7–106 after irradiation, but less than 10 genes showed an overlap in up- or down-regulated gene expression changes over all time points (Fig. 1). Also, a 2–3-fold reduction in the number of DEG was observed over time (Fig. 1).

Venn diagrams showing the number of up- (left side) and down-regulated (right side) protein coding genes (mRNA transcripts) observed at 7, 28 and 75–106 days after radiation exposure. The number of differentially expressed genes (DEG) observed on one, two or all three time points after exposure is shown in the corresponding overlapping circles. Numbers in parenthesis represent the total number of differentially expressed genes.
Based on the fold-difference, the p-value and sustained changed mRNA expression over the days after irradiation, we selected 32 candidate mRNAs for validation at stage II (Table 2, left part).
During the screening of 667 miRNAs, we identified 290 miRNAs eligible for analysis. In total, 70 miRNAs revealed significant expression changes after radiation exposure and were forwarded for validation in stage II (data not shown).
Stage II: validation using qRT-PCR measurements
During stage II, validation of the 32 candidate mRNAs from stage I, we found 20 mRNAs showed either no amplification plot or amplification plots in a minority of all samples (n ≤ 3). Those were excluded from further analysis. There remained 12 genes for validation, but a validation of gene expression changes in the same direction as seen during stage I screening was either not detected (n = 4), or only in a part of the indicated time points after irradiation (n = 4, Table 2, right side). Another three mRNAs showed an inverse regulation in gene expression among the screening and the validation samples. For instance, during stage I screening, CACNB3 appeared about 3-fold up-regulated on days 7 and 28 after exposure, but during validation at stage II a statistically significant (p = 0.002) 3-fold down-regulation of the remaining samples using qRT-PCR was observed (Table 2, Fig. 2F). Only CA2 showed a 2–5-fold up-regulation on days 7 and 28 during screening and validation with a borderline significance at day 7 (p = 0.07) and a high significance at day 28 (p < 0.0001, Table 2, Fig. 2F).

Differential gene expression changes over time are depicted for miRNAs (A–E) and mRNAs (F). Up to three examples for genes either differentially expressed only on days 7 (A) or 28 (B) and on two consecutive days (7–28, (C) or 28–75, (D) or over the whole period of time (E) are shown. Candidate mRNAs differentially expressed on days 7–28 are shown in panel F. Symbols represent geometric mean values and error bars reflect the standard error of mean. Numbers of measurements per group are shown in Supplement Table 2.
Independent validation of the 70 miRNAs from the screening stage resulted in 21 miRNAs which were significantly differentially expressed only on days 7 (n = 1, Fig. 2A) or 28 (n = 4, Fig. 2B) or over two time points such as 7 and 28 (n = 4, Fig. 2C) or 28 and 75–106 days after irradiation (n = 7, Fig. 2D, Supplement Table 2). Another five miRNAs revealed persistent DEG at all three time points (Fig. 2E, Supplement Table 2). For these 21 miRNAs, all data from stage I and stage II were merged and analyzed together resulting in highly significant DEG that survived Bonferroni correction for multiple comparisons (p < 0.002 [0.05/21]) and ROC between 0.9–1.0 were calculated for most genes suggesting a complete or almost complete separation of gene expression relative to the pre-exposure reference (bold p-values, Supplement Table 2, right part). A 2-6-fold up- or downregulation was observed in 17 of the 21 miRNAs, but 10-28-fold (e.g. miR-378-1, miR-1305, miR-331-5p, miR454) or even up to 77-fold changes in miRNA expression (miR-212) were observed in the remaining 5 miRNAs. In particular miR212 revealed a persistent 48-77-fold DEG over all three time points after irradiation (Supplement Table 2, Fig. 2E).
Discussion
Taking advantage of a non-human primate model with high genetic homology to humans, we examined the whole genome for radiation-related transcriptional (protein coding mRNAs) and post-transcriptional (non-coding miRNAs) gene expression changes in the peripheral blood persisting over weeks and months after radiation exposure. At this time frame after exposure, the baboons’ peripheral blood had recovered from the radiation effects and showed normal blood cell counts. In other words, does radiation cause a long lasting altered transcriptome/post-transcriptome in previously irradiated individuals? This examination was also challenged by employing different partial and whole body exposure patterns in order to scope with different exposure scenarios. However, it was the goal of this research to examine persistent changes in gene expression which could be found independently of partial and whole body exposure patterns resulting in either 2.5 Gy or 5 Gy whole body equivalent doses. Examinations such as that cannot be performed in healthy humans, but might provide insights into radiation effects occurring delayed after exposure (see below).
During the transcriptome screening, several hundred up- and down-regulated mRNAs were identified with 14 mRNAs consistently differing from the reference for the three time points after exposure. Due to methodological reasons, most of the 32 selected mRNAs could not be validated, except for CA2 which appeared 2-5-fold up-regulated during screening as well as the validation on the remaining samples (Fig. 2F). CA2 (carbonic anhydrase 2) catalyzes reversible hydration of carbon dioxide. Defects in this enzyme are associated with e.g. osteopetrosis (marble bone disease). Another gene, CACNB3 (calcium voltage-gated channel auxiliary subunit beta 3, with a role in the regulation of transcription factors and calcium transport) appeared about 3-fold up-regulated during screening and about 3-fold down-regulated during validation (Fig. 2F). An inverse regulation during the validation process using another method and other biological samples is a very rare event, however the p-value (p = 0.002) supports the robustness of the findings and argues against a result by chance. Since the microarray targeted another exon than the one used for validation by the Taqman assay, it is more likely that exons of the same gene might show inverse gene expression values after radiation exposure when considering mechanisms such as alternative splicing and cassette exons23. So, the interpretation of some mRNA findings is challenging considering these methodological issues.
Regarding our post-transcriptomic analysis, we identified substantial changes in the expression of 21 successfully validated miRNAs (Fig. 2A–E). Some of these altered miRNAs showed a transient alteration e.g. observed only on the seventh or the 28th day after exposure, but not later (Fig. 2A,B). However, we found that six miRNAs persisted over the whole period of time (Supplement Table 2, Fig. 2E). For instance, 48 to 77 fold-differences relative to unexposed blood samples taken before radiation exposure were observed for miR-212 and persisted over the entire time period. Another five miRNAs showed 6-10-fold differences and the remaining miRNAs showed 2-3-fold differences. All of this suggests large post-transcriptional miRNA changes that, to our knowledge, have not been previously reported in other radiation-models either in vitro or in vivo. ROC areas at one or close to one indicate a comparable response among the 17 radiation exposed baboons and with the accompanying low p-values reduce the likelihood of chance findings.
Again, these results indicate a marked radiation-related modification of the post-transcriptome likely occurring in irradiated cells (7 days after exposure) and certainly in the following cell generations up to 75–106 days after exposure. The cellular progeny represent cells that were not directly exposed to radiation. Because our measurements were performed on peripheral whole blood, presumably our results reflect either altered blood stem cells or indicate a selection for less radiosensitive stem cells that reconstitute the peripheral blood after radiation exposure. We wondered about the possible clinical implication of our findings which were not examined within this study and are, therefore, speculative. Interestingly, many of our miRNA species are already known to be linked with radiosensitivity (e.g. miR-22, miR-29c, miR-195, miR-212) or chemotherapy resistance (e.g. miR-331-5p, supplement Table 1). Also, most of these miRNAs are thought to be associated with risk of cancers to the colon, thyroid and lung or acute lymphoblastic or chronic myeloid leukemia (supplement Table 1). Tumorigenesis represents a multistep process. We speculate that this shift in the composition of cells might make the exposed individuals more prone for developing a tumor during their follow-up and as that might represent the first step towards tumorigenesis. It is by chance which of the animals might get the next hit and finally develop a tumor, but, certainly, radiation exposed individuals are under higher risk for developing secondary tumors and the changes observed might represent the first step for tumor initialization. Still, whether these persistent gene expression alterations are associated with risk of primary or secondary tumors is a hypothesis for future research. Further bioinformatic analysis on our over time persistently deregulated miRNAs using the DIANA-miRPath 3.0 tool (search for categories intersection employing FDR adjusted p-values and unbiased empirical distribution as enrichment analysis method) did not result in a further significant KEGG pathways or Gene Ontology processes enrichment of either predicted (TargetScan) or known (Tarbase) target mRNAs (data not shown24). Our findings align with previous observations indicating that miRNA changes perform better than mRNA changes in the classification/diagnosis of tumors10. So, the concept of persistent radiation-related gene expression changes may provide an avenue for early prediction and diagnosis of cancer which, again, has to be proven in future studies.
Long term sequelae of radiation-related or cancer-associated symptoms such as fatigue, depression and sleep disturbances after radiation treatment or chemotherapy have been well documented by others15,17. Our study supports the notion that if irradiation of healthy animals produces persistent molecular changes then this might represent the molecular basis for symptoms persisting over months and years after exposure5,16. Several of our candidate genes are known to impact immunological and inflammatory mechanisms (e.g. miR-130b, miR-29c, miR-720, miR-151a-5p, miR-195, miR-212). This observation is in line with a recent publication where another group showed a persistent altered state of the immune system (based on gene expression measurements) up to 30 days after whole thorax irradiation in non-human primates25. Noteworthy, immunological alterations have been implicated as a mechanism for persistent fatigue after treatment18,19, (supplement Table 1). In our experiment we saw no dramatic changes in animal behavior or activity which could be caused by fatigue. However, a transient body weight loss was observed in three baboons. Clearly, this endpoint (fatigue) was not the main focus of this experiment so that we cannot exclude an unobserved milder grade fatigue.
Interestingly miR-212, which was persistently 48-78-fold downregulated over the entire time period, is already known for its impact on radiosensitivity of glioma cells by targeting BRCA1, its immune modulating effect on primary human natural killer cells through IL-12 and its association with breast and prostate cancer, making it a particularly interesting target for further clinical investigation (supplement Table 1).
Some limitations of our study should be kept in mind. We performed gene expression measurements on microarrays and qRT-PCR using human genomic sequences because the baboon transcriptome was not publically available. Given the high homology of both genomes (93%) and previously reports by other groups26,27,28, we proceeded as described above. Since we used TaqMan chemistry with human primer and probe sequences (high sensitivity and specificity), it is more likely that we lost some of the detectable human RNA species due to a mismatch with the baboon genome, rather than producing false positives. Also, in validation studies using human samples from irradiated patients, we were able to reproduce candidate genes from our previous baboon studies implying that differences in genomic sequences represent a minor problem29.
The small sample size of our study represents a weakness and surely reduced the number of successfully validated candidate genes. Future work should consider larger sample sizes and results should be validated on additional species.
As a better study design it would be preferable performing longitudinal studies in patients and taking biosamples at several time points during weeks and months of follow-up. However, examining this mechanism of radiation-related persistent gene alterations in cancer patients is not possible, because some therapeutic regimens (e.g. bone marrow transplant) will obscure the radiation-related gene expression changes we detected in our healthy baboons. This on the other hand emphasizes the significance of our results.
Finally, we are very much aware about the speculative nature of the clinical implications of our findings, since we neither measured secondary tumors nor sequela such as fatigue in our baboons. What we are trying here is to put our results into a context of clinical implications and to provide possible hints on underlying molecular mechanisms to be examined in future studies.
In summary, weeks and months after radiation exposure we identified substantial and persistent changes in the post-transcriptional level of 21 miRNAs with known associations to tumorigenesis, radiosensitivity and immune response. We speculate whether this might provide the molecular basis for the development of secondary tumors and a radiation-related genesis of persisting behavioral symptoms such as fatigue observed over months and years after cancer treatment.
Data Availability Statement
The datasets generated during or analyzed during the current study are available from the corresponding author on reasonable request.
References
Gilbert, E. S. et al. Stomach Cancer Following Hodgkin Lymphoma, Testicular Cancer and Cervical Cancer: A Pooled Analysis of Three International Studies with a Focus on Radiation Effects. Radiat. Res. 187, 186–195 (2017).
Morton, L. & Gilbert, E. Risk of esophageal cancer following radiotherapy for Hodgkin lymphoma. Haematologica 193–196 https://doi.org/10.3324/haematol.2014.108258 (2014).
Black, A. et al. Pooling prospective studies to investigate the etiology of second cancers. Cancer Epidemiol. Biomarkers Prev. 23, 1598–1608 (2014).
Morton, L. M. et al. Evolving risk of therapy-related acute myeloid leukemia following cancer chemotherapy among adults in the United States, 1975-2008. Blood 121, 2996–3004 (2013).
Reddy, K. B. MicroRNA (miRNA) in cancer. Cancer Cell Int. 15, 4–9 (2015).
Lawrie, C. H. et al. Detection of elevated levels of tumour-associated microRNAs in serum of patients with diffuse large B-cell lymphoma. Br. J. Haematol. 141, 672–675 (2008).
Bianchi, F. et al. A serum circulating miRNA diagnostic test to identify asymptomatic high-risk individuals with early stage lung cancer. EMBO Mol. Med. 3, 495–503 (2011).
Cummins, J. M. & Velculescu, V. E. Implications of micro-RNA profiling for cancer diagnosis. Oncogene 25, 6220–6227 (2006).
Iorio, M. V. & Croce, C. M. MicroRNA dysregulation in cancer: Diagnostics, monitoring and therapeutics. A comprehensive review. EMBO Mol. Med. 4, 143–159 (2012).
Lu, J. et al. MicroRNA expression profiles classify human cancers. Nature 435, 834–838 (2005).
Bower, J. E. et al. Inflammation and behavioral symptoms after breast cancer treatment: Do fatigue, depression, and sleep disturbance share a common underlying mechanism? J. Clin. Oncol. 29, 3517–3522 (2011).
Donovan, K. A. et al. Course of fatigue in women receiving chemotherapy and/or radiotherapy for early stage breast cancer. J. Pain Symptom Manage. 28, 373–380 (2004).
Taunk, N. K. et al. Comparison of radiation-induced fatigue across 3 different radiotherapeutic methods for early stage breast cancer. Cancer 117, 4116–4124 (2011).
Bower, J. E. et al. Inflammatory biomarkers and fatigue during radiation therapy for breast and prostate cancer. Clin. Cancer Res. 15, 5534–5540 (2009).
Wang, X. S. et al. Longitudinal study of the relationship between chemoradiation therapy for non-small-cell lung cancer and patient symptoms. J. Clin. Oncol. 24, 4485–4491 (2006).
Wang, X. S. & Woodruff, J. F. Cancer-related and treatment-related fatigue. Gynecol. Oncol. 136, 446–452 (2015).
Kolak, A. et al. The problem of fatigue in patients suffering from neoplastic disease. Współczesna Onkol. 2, 131–135 (2017).
De Sanctis, V. et al. Cytokines, fatigue, and cutaneous erythema in early stage breast cancer patients receiving adjuvant radiation therapy. Biomed Res. Int. 2014 (2014).
Petty, R. D., McCarthy, N. E., Dieu, R. L. & Kerr, J. R. MicroRNAs hsa-miR-99b, hsa-miR-330, hsa-miR-126 and hsa-miR-30c: Potential diagnostic biomarkers in natural killer (NK) cells of patients with Chronic Fatigue Syndrome (CFS)/myalgic encephalomyelitis (ME). PLoS One 11, 1–19 (2016).
Valente, M. et al. Revisiting Biomarkers of Total-Body and Partial-Body Exposure in a Baboon Model of Irradiation. PLoS One 10, e0132194 (2015).
Port, M. et al. Pre-exposure gene expression in baboons with and without pancytopenia after radiation exposure. Int. J. Mol. Sci. 18, (2017).
Abend, M. et al. Iodine-131 Dose Dependent Gene Expression in Thyroid Cancers and Corresponding Normal Tissues Following the Chernobyl Accident. PLoS One 7, e39103 (2012).
Cui, Y., Cai, M. & Stanley, H. E. Comparative Analysis and Classification of Cassette Exons and Constitutive Exons. Biomed Res. Int. 2017, 1–8 (2017).
Vlachos, I. S. et al. DIANA-miRPathv3.0: Deciphering microRNA function with experimental support. Nucleic Acids Res. 43, W460–W466 (2015).
Ghandhi, S. A. et al. Whole thorax irradiation of non-human primates induces persistent nuclear damage and gene expression changes in peripheral blood cells. PLoS One 13 (2018).
Sherwin, J. R. A. et al. The Endometrial Response to Chorionic Gonadotropin Is Blunted in a Baboon Model of Endometriosis. Endocrinology 151, 4982–4993 (2010).
Shi, Q. et al. Molecular pathways mediating differential responses to lipopolysaccharide between human and baboon arterial endothelial cells. Clin. Exp. Pharmacol. Physiol. 37, 178–184 (2010).
Aavik, E. et al. Correlation between gene expression and morphological alterations in baboon carotid after balloon dilatation injury. FASEB J. 19, 130–2 (2005).
Port, M. et al. Validating Baboon Ex Vivo and In Vivo Radiation-Related Gene Expression with Corresponding HumanData. Radiat. Res. RR14958.1 https://doi.org/10.1667/RR14958.1 (2018).
Acknowledgements
We are very thankful for Reinhard Ullmann’s support regarding bioinformatics. We appreciate the sophisticated and carefully performed technical assistance of Sven Doucha-Senf and Eva Grumpelt. This work was supported by the French and the German Ministry of Defense.
Author information
Authors and Affiliations
Contributions
The in vivo study on baboons was conducted by F. Herodin, M. Valente and M. Drouet. Whole blood samples were provided for further transcriptome and post-transcriptome analysis. This work was conducted by M. Majewski and P. Ostheim under the guidance of M. Abend and M. Port. Results were analyzed and figures and tables were developed by M. Majewski and P. Ostheim. The manuscript was written by M. Port and M. Abend. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Port, M., Hérodin, F., Valente, M. et al. Persistent mRNA and miRNA expression changes in irradiated baboons. Sci Rep 8, 15353 (2018). https://doi.org/10.1038/s41598-018-33544-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-33544-2
Keywords
This article is cited by
-
Serum microRNA profile of rhesus macaques following ionizing radiation exposure and treatment with a medical countermeasure, Ex-Rad
Scientific Reports (2024)
-
MicroRNA: a novel implication for damage and protection against ionizing radiation
Environmental Science and Pollution Research (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
