
Abstract
Bestinopathies are a spectrum of retinal disorders associated with mutations in BEST1 including autosomal recessive bestrophinopathy (ARB) and autosomal dominant Best vitelliform macular dystrophy (BVMD). We applied whole-exome sequencing on four unrelated Indian families comprising eight affected and twelve unaffected individuals. We identified five mutations in BEST1, including p.Tyr131Cys in family A, p.Arg150Pro in family B, p.Arg47His and p.Val216Ile in family C and p.Thr91Ile in family D. Among these, p.Tyr131Cys, p.Arg150Pro and p.Val216Ile have not been previously reported. Further, the inheritance pattern of BEST1 mutations in the families confirmed the diagnosis of ARB in probands in families A, B and C, while the inheritance of heterozygous BEST1 mutation in family D (p.Thr91Ile) was suggestive of BVMD. Interestingly, the ARB families A and B carry homozygous mutations while family C was a compound heterozygote with a mutation in an alternate BEST1 transcript isoform, highlighting a role for alternate BEST1 transcripts in bestrophinopathy. In the BVMD family D, the heterozygous BEST1 mutation found in the proband was also found in the asymptomatic parent, suggesting an incomplete penetrance and/or the presence of additional genetic modifiers. Our report expands the list of pathogenic BEST1 genotypes and the associated clinical diagnosis.
Similar content being viewed by others

Introduction
Mutations in BEST1 gene have been described in a variety of ocular disease phenotypes including autosomal recessive bestrophinopathy (ARB, MIM 611809)1,2, Best vitelliform macular dystrophy (MIM 153700)3,4, autosomal dominant vitreoretinochoroidopathy (MIM 193220)5,6, autosomal dominant microcornea, rod-cone dystrophy, early-onset cataract posterior staphyloma syndrome and retinitis pigmentosa (MIM 613194)7,8. The BEST1 (bestrophin-1) protein is expressed in the basolateral plasma membrane of the retinal pigment epithelium (RPE) where it regulates multiple functions essential for normal vision7,9. It primarily functions as a calcium-activated chloride channel7,9,10. A role for bestrophin-1 in normal ocular development has been proposed, though its precise role in this context is not fully understood7.
Best vitelliform macular dystrophy (BVMD) is an autosomal dominant syndrome associated with BEST1 mutations10. BVMD primarily affects the macula and is characterized by yellowish, vitelliform or egg-yolk-like lesions with considerable morphologic variations based on the stage of the disease11,12. Electrophysiological characteristics in BVMD patients include normal full-field electroretinography (ERG), with a marked decrease or absence of light rise in electro-oculography (EOG)13. Early onset choroidal neovascular membranes in patients with BVMD14, often occur before vitelliruption and it responds to intravitreal therapies. Autosomal recessive bestrophinopathy (ARB) (MIM 611809) is another condition associated with mutations in the BEST1 gene15. ARB usually manifests in the first two decades of life, but may become symptomatic as late as the fifth decade2,15,16. Clinical presentation of ARB is distinct from BVMD17 and is characterized by central visual loss with typical hyperopic conditions, sub-retinal lipofuscin deposits that are predominantly outside the macula, absence of light rise in EOG, reduced ERG, accumulation of fluid within and/or beneath the neurosensory retina and development of angle-closure glaucoma17,18. ARB may manifest as the result of a total absence (null phenotype) of functional bestrophin-1 protein in the RPE15,19, improper localization to the cell membrane with intact anion channel activity20 or lack of channel activity specifically2. Among the roughly 270 mutations21 reported in BEST1 thus far, only about 40 compound heterozygous and homozygous mutations are associated with ARB15,17,18,22. In this study, we analyzed four Indian families with clinically diagnosed bestrophinopathy using whole exome sequencing and identified novel BEST1 mutations.
Materials and Methods
Study cohort
The prospective study was approved by the Narayana Nethralaya Institutional Review Board and was performed as per institutional ethics guidelines and in accordance with the tenets of the Declaration of Helsinki. Subjects were recruited for the study after obtaining informed written consent either from the patient or the guardian and family members. A total of eight patients with Bestrophinopathy from four unrelated families from southern India were investigated. Age at the time presentation ranged between 11–26 years with mean age of 20 years.
Clinical examination
Detailed medical history was obtained, followed by clinical examination including best-corrected Snellen visual acuity (BCVA), slit-lamp examination, gonioscopy, indirect ophthalmoscopy and fundus photography. Fundus autofluorescence (FAF) imaging with a confocal scanning laser ophthalmoscope (Spectralis, Heidelberg Engineering, Heidelberg, Germany) in all eight patients and selected family members was performed. Spectral domain optical coherence tomography (SD OCT; Spectralis, Heidelberg Engineering, Heidelberg, Germany) was also performed simultaneously in these patients. Two of the eight patients underwent fundus fluorescein angiography (FFA). Electrophysiologic examinations were conducted according to the standards given by the International Society of Clinical Electrophysiology in Vision23,24. Viking 5.0 Ganzfeld dome (Nicolet Biomedical Instruments, Madison, Wisconsin, USA) with a light-emitting diode for light stimulation was used for both electro-oculography and full-field electroretinography.
DNA isolation, exome library preparation and sequencing
DNA was isolated from whole blood using QIAamp DNA Blood Mini Kit (Qiagen, CA, US; cat no. 51104). Exome library was performed using Agilent SureSelect (Santa Clara, CA, US) Human All Exome kit v5 (50 Mb). The library was sequenced on Illumina HiSeq. 4000 (Illumina, CA, US) to obtain 2 × 75 bp paired end reads. An average exome coverage of 71x (range 45–94x) with 81% of the bases over 20x (range 76–91) was obtained.
RNA isolation, sequencing and analysis
RNA was isolated from a healthy donor retina sample using RNeasy Mini Kit (Qiagen; catalogue no 74104). About 0.5 μg of total RNA was used to generate RNA-seq library using TruSeq RNA Sample Preparation kit (Illumina). The library was sequenced on HiSeq2500 to obtain 166,513,102 paired-end (2 × 75 bp) reads. RNA-seq reads were mapped to the human reference genome version GRCh37 using GSNAP25. Analysis of BEST1 transcript isoforms was performed using the SGSeq software package (version 1.4.0)26 with gene annotation from Ensembl (release 75).
Variant calling and annotation
Whole exome sequencing data was processed using GATK’s best practices workflow for DNA-Seq27. Briefly, the raw sequencing reads were aligned to the human reference genome version GRCh37 using BWA-MEM (version 0.7.10)28. Picard tools (version 1.126) were used for removing duplicate reads. Indel realignment and base quality score recalibration were performed using GATK (version 3) to improve alignment accuracy and recalibrate base quality29. HaplotypeCaller (in reference confidence model) was applied for each sample to generate gVCF files. The resulting gVCF files from all samples were used for joint variant calling using GenotypeGVCFs walker. Variant Quality Score Recalibration (VQSR) was carried out to estimate the confidence of called variants. Variant annotation was carried out using SnpEff program (version 4.2)30.
Variant filtering and analysis
To identity candidate nucleotide variants, we applied a filtering strategy. Those variants which were present at >0.2% in either the 1000 Genome Project31 or NHLBI-EVS Exome Sequencing Project32 or ExAC database33 were filtered out. Protein-altering or potentially protein-altering variants that follow recessive or dominant modes of inheritance were selected for further analysis. Given that BEST1 is a well-established Bestrophinopathy gene, all nucleotide variants present in BEST1 were additionally reviewed. The genes and corresponding mutations that qualified these filtering criteria were investigated to determine their significance and relevance in Bestrophinopathy. Genes that were associated with an ocular disease phenotype, as reported on RetNet (https://sph.uth.edu/retnet/), were used for further investigation. Read evidence of the final list of nucleotide variants was manually inspected using Integrative Genomics Viewer (IGV)34 to detect sequencing artifacts.
Ethics approval and consent to participate
Written informed consent was obtained from all the study participants. Consent from the parents or legal guardian was obtained wherever samples from minors were included in the study. The study was approved by the Narayana Nethralaya Institutional Review Board and was performed as per institutional ethics guidelines and in accordance with the tenets of the Declaration of Helsinki.
Availability of data and materials
Filtered variant data is available as a supplementary table. Genotype data for patients consenting to share it for research use shall be made available upon request.
Results
Patients and Clinical Characteristics
Twenty individuals, including eight affected and twelve unaffected members from four families, were selected for the study (Figs 1–4; Table 1).

Family A. (a) Pedigree of the family with two affected members. The proband is marked with an arrow. (b,c) Colored fundus photographs of right and left eye of proband (II.1) and affected sibling (II.2) respectively, showing focal areas of subretinal fibrosis (black arrow) with very sparse vitelliform deposit marked by white arrow. (d,e) Fundus autofluorescence images of proband (II.1) and affected sibling (II.2) respectively, showing focal dot like areas of increased autofluorescence (white arrow) corresponding to the areas of vitelliform deposit seen in fundus photography. (f) Optical coherence tomography (OCT) images of the proband showing intra retinal cystoid and schitic changes (asterisk) with minimal sub retinal fluid (white arrow). The sub retinal area is marked with the retinal layers as RPE = Retinal Pigment Epithelium, IZ = Interdigitation Zone, EZ = Ellipsoid Zone and ELM = External Limiting Membrane.
Family A
In family A, two members were affected (Fig. 1a; Table 1). The proband (II.1) was a twenty one year old male with a BCVA of 20/60 in both eyes with hypermetropic refraction. No anterior segment abnormalities were observed in the proband. Fundus image revealed very sparse sub-retinal fibrosis with deposits (Fig. 1b). The affected sibling (II.2) also presented with a normal anterior segment with the corresponding fundus image displaying less conspicuous changes (Fig. 1c). Fundus autofluorescence (FAF) images for both proband and sibling indicated few areas of hyper autoflourescence (Fig. 1d,e). Optical coherence tomography (OCT) images of the proband revealed cystoid and schitic changes in inner and outer nuclear layers of retina (Fig. 1f). Similar OCT features were observed in the affected sibling (Supplementary Fig. S1a). Electrophysiological tests indicated rod cone dysfunction in ERG (Supplementary Fig. S1b–e) with absent light peak in EOG for both proband and the sibling (Supplementary Fig. S1f–i). The electrophysiologic characteristics were similar to the earlier reported cases of ARB35. No ocular abnormalities were observed in the unaffected parents. Although the proband and affected sibling were clinically managed with topical and oral carbonic anhydrase inhibitors, the macular fluid persisted.
Family B
The proband was a twenty four year old male (from non-consanguineous parents) (Fig. 2a) with a BCVA of 20/30 OD and 20/40 OS. Proband’s fundus exhibited sub-retinal fibrosis with yellowish white, vitelliform deposits located primarily outside the arcade (Fig. 2b) and peripapillary area. These deposits were clearly evident on the FAF image (Fig. 2c). Corresponding OCT images exhibit intra-retinal and sub-retinal fluid collection (Fig. 2d). The proband’s ERG indicated rod-cone dysfunction and the corresponding EOG was abnormal (Supplementary Fig. S2b–d). No ocular abnormalities were observed in unaffected parents and sibling, which is consistent with ARB.

Family B. (a) Pedigree of the family with one affected subject (II.1; indicated by arrow). (b) Colored fundus photographs of the right and left eyes of proband demonstrating focal areas of sub-retinal fibrosis (black arrow) and extensive areas of sub-retinal vitelliform deposits across the arcade and nasal to the disk (white arrow). (c) Corresponding fundus autofluorescence (FAF) images of right and left eyes of the proband highlight the vitelliform deposits (white arrows). (d) OCT images of right and left eyes of proband showing sub retinal (white arrow) and intra retinal (white asterisk) changes with fluid accumulation.
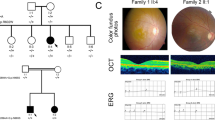
Family C
In this family, four members were affected (Fig. 3a) and the pedigree analysis indicated an autosomal recessive inheritance pattern. The proband (II.1) presented with a history of decreased central vision and a BCVA of 20/125 in both eyes. This subject was diagnosed with angle closure glaucoma along with juvenile retinoschisis and had previously undergone peripheral iridotomy. Fundus examination (Fig. 3b) revealed yellowish white sub-retinal precipitates with corresponding hyper autoflourescence on FAF (Fig. 3c) that was primarily concentrated around the posterior pole. Two affected brothers (patient II.1 and II.4) presented with similar clinical features but the yellowish lesions were more confluent, whereas, the affected sister demonstrated less confluent lesions(Fig. 3d–i). OCT analysis (Fig. 3j) of the proband revealed sub-retinal yellowish lesions and scars which corresponded with the hyper-reflective accumulations on or within the RPE cell layer with serous sub-retinal fluid. The OCT features were essentially similar across all the affected members of family C and correlated with the clinical features (Supplementary Fig. S3a–c) exhibiting predominant subretinal fluid with some intraretinal involvement, which was much lower than that observed in families A and B. ERG performed on the proband was normal (Supplementary Fig. S3f,g). However, the corresponding EOG was abnormal with absent light peak (Fig. 3i). The proband underwent multiple surgeries for her glaucoma including bilateral trabeculaectomy and lensectomy. The affected elder brother had glaucoma that was managed medically.

Family C. (a) Pedigree of the family with four affected members. The proband is marked with an arrow. Colored fundus photographs (b, d, f and h) of the proband (II.3) (b, c), affected siblings (brother II.1 (d,e), brother II.4 (f,g), sister (II.2 (h,i) show yellowish white sub-retinal deposits (white arrows). Fundus autofluoresence (c, e, g and i) of the proband (II.3) (b,c), affected siblings (brother II.1 (d,e), brother II.4 (f,g), sister (II.2 (h,i) show hyper autoflourescence (white arrows) mainly concentrated around posterior pole. A localised area of subretinal fibrosis (black arrow; c) is observed in affected sibling II.1. (j) OCT of right and left eyes of proband show outer segment elongation (black circle) with sub retinal changes and subretinal fluid (white arrow).
Family D
The proband in this family was an eleven year old male (Fig. 4a), who presented with a history of blurred vision and BCVA of 20/40 in both eyes. Fundus examination revealed yellowish, vitelliform deposits in the macula exhibiting a pseudohypopyon appearance (Fig. 4b) that is appreciated better in the FAF image (Fig. 4c). The proband’s father was asymptomatic with visual acuity of 20/20 OU but displaying vitelliform lesion in the left eye (Fig. 4d,e and Supplementary Fig. S4a). The younger sibling of the proband did not present any changes in the retina at the time of examination (Fig. 4f,g and Supplementary Fig. S4b). OCT of the proband indicated sub-retinal deposits with sub-retinal fluid in both eyes (Fig. 4h). The ERG was normal while EOG was abnormal in the proband (Supplementary Fig. S4c–f).

Family D. (a) Pedigree chart of family with one affected subject indicated by black arrow. Colored fundus photographs of right and left eyes of proband (b), father (d) and sister (f). Corresponding FAF images of right and left eyes of proband (c), father (e) and sister (g). Proband (b), shows prominent yellowish vitelliform deposits in macula giving pseudohypopyon appearance, better highlighted in FAF (c). Proband’s father (d) shows a normal fundus in the right eye but left eye had focal vitelliform lesion (white arrow) better highlighted in FAF (white arrow) (e). Sister (f,g) did not show abnormal retinal features. (h) OCT of the proband shows sub-retinal deposits with sub-retinal fluid in both eyes (white arrows).
Exome sequencing and causal variant identification
We performed exome sequencing on twenty individuals with at least 45x coverage (Supplementary Fig. S5) from four unrelated families (Supplementary Fig. S6). Joint-variant calling using data from twenty samples from the four families in our study resulted in 2,630,722 nucleotide variants (Fig. 5a, Methods). Of these, 317,443 (~12%) were rare variants (MAF <= 1%). We predicted 8,788 of the rare variants to have an impact on protein function and/or have disease relevance. The pathogenic variants that segregated through affected and unaffected family members were filtered after overlaying the disease inheritance pattern observed in each pedigree. Additional filters were applied for genes previously implicated in retinopathy as reported on RetNet (https://sph.uth.edu/retnet/). We found BEST1 mutations in all four families - p.Tyr131Cys in Family A, p.Arg150Pro in Family B, p.Arg47His and p.Val216Ile in Family C, and p.Thr91Ile in Family D. (Fig. 5a,b, Table 1 and Supplementary Table S1).

Identification of casual variants in bestrophinopathy families. (a) Flowchart depicting the analysis of exome data from bestrophinopathy patients and their relatives. (b) Pedigrees of four bestrophinopathy families analyzed in this study. Candidate variants identified in each family is shown. White circles - unaffected females; white squares - unaffected male; filled circles - affected females; filled squares - affected males. Number within circle/square indicates that the sample was sequenced.
In Family A, a homozygous transition c.392 A > G in BEST1 was found in both affected children (proband II.1 and his brother II.2) resulting in a substitution of tyrosine at codon 131 with cysteine (p.Tyr131Cys) (Fig. 5b, Supplementary Fig. S7). Both SIFT36 and Polyphen-237 predicted this mutation to be deleterious (Supplementary Table S1). Consistent with the clinical observation, both unaffected parents (I.1 and I.2) were found to be heterozygous for this BEST1 mutation confirming an autosomal recessive mode of inheritance. Interestingly, in this family OCT showed predominant intraretinal schitic and cystoid changes with negligible subretinal fluid.
In family B, we found a missense mutation, p.Arg150Pro, in the BEST1 gene that followed a recessive mode of inheritance (Fig. 5b, Supplementary Fig. S8). Consistent with the clinical findings, BEST1 p.Arg150Pro was observed to be homozygous in the affected proband II.1, heterozygous in both the unaffected parents (I.1 and I.2) and was absent in the unaffected brother (II.2). This alteration was not reported in the 1000 Genome Project31 or the ExAC database33 and was predicted to be deleterious by both SIFT36 and Polyphen-237.
Analysis of family C identified two mutations, p.Arg47His and p.Val216Ile, in the BEST1 gene that showed a recessive segregation pattern within the family (Fig. 5b, Supplementary Figs S9–10). The p.Arg47His mutation resulted from a transition c.140 G > A in exon 2 of BEST1 transcript ENST00000378043. However, the p.Val216Ile mutation was a result of a transition c.646 G > A at exon 6 of another BEST1 transcript ENST00000526988. ENST00000378043 (RefSeq - NM_0041830), encoding a 585 amino acid protein isoform, is the best characterized mRNA transcribed from BEST1. ENST00000526988 is a poorly characterized transcript (RefSeq - XM_017018230), encoding a shorter protein isoform (329aa) and its exon 6 is unique among all 7 protein-coding transcripts of BEST1 (Fig. 6). Manual review of all BEST1 variants found this compound heterozygous mutation, p.Arg47His and p.Val216Ile, to follow a recessive inheritance mode in this family.

RNA-seq analysis of BEST1 transcript isoforms. Top panel shows structure of seven human protein-coding BEST1 transcript isoforms annotated by Ensembl (release 75), and the location of two mutations p.Arg47His and p.Val216Ile. Gray boxes represent exons. Green vertical line and red vertical line indicate ORF start and stop, respectively. Bottom panel shows average per-base read coverage and junction read counts (middle).
We confirmed the expression of both ENST00000378043 and ENST00000526988 isoforms in retina by analyzing RNA-seq data obtained from the retinal tissue of a healthy donor (see Materials & Methods). RNA-seq split-reads (n = 11) supporting the intron junction between exon 6 and exon 7 of ENST00000526988 confirmed that this alternate transcript is expressed in the retina and hence likely to be a relevant isoform of BEST1 in the eye (Fig. 6).
All four affected members in family C (including proband) carried both the p.Arg47His and p.Val216Ile mutations. The unaffected members (parents and children of proband) were carriers for either p.Arg47His or p.Val216Ile. The heterozygous p.Arg47His mutation was reported in a German patient with adult-onset vitelliform macular dystrophy (AVMD)38 and, more recently, in a Chinese patient with Best Vitelliform Macular Dystrophy (BVMD)39. The p.Val216Ile mutation was not present in the 1000 Genome Project31 or the ExAC database33 and was predicted to be damaging by SIFT36 and benign by Polyphen-237.
In family D, we did not observe any homozygous pathogenic mutations through the segregation analysis. However, upon manual review, the proband (II.1) was found to have a heterozygous p.Thr91Ile mutation in the BEST1 gene which was predicted to be deleterious by both SIFT36 and Polyphen-237 (Fig. 5b, Supplementary Fig. S11, Supplementary Table S1). The same mutation was also observed in the father and sister but was absent in the mother. The father (I.1) who did not have any clinical symptoms, demonstrated vitelliform lesions in his left eye, indicating that the mutation may show an incomplete penetrance.
Discussion
The BEST1 gene has been associated with a wide range of ocular phenotypes. Although clinical presentation of the autosomal recessive form of bestrophinopathy is distinct from the autosomal dominant form, both these conditions are known to arise from BEST1 (VMD2) gene alterations. Mutations in BEST1 gene have pleiotropic effects and the phenotypes observed are influenced by age, gender, environment, epigenetic factors and presence of modifier genes7,40.
Our study included four families where probands from three were classified as ARB (families A, B and C) and the proband from the fourth (Family D) was suspected to have BVMD (family D) based on clinical observations. The BEST1 mutations and the pattern of inheritance confirmed the ARB clinical diagnosis in family A and B. Interestingly, the sequencing results established the family C proband to carry a compound heterozygous BEST1 mutation that likely led to ARB. Finally, in family D we identified a potential autosomal dominant BEST1 mutation that exhibited an incomplete penetrance leading to BVMD in the affected proband.
In Family A, we observed a homozygous p.Tyr131Cys BEST1 mutation. The Y131 is a conserved residue (Fig. 7d) and substitution at this position likely affects BEST1 function. The p.Tyr131Cys mutation has not been previously reported. Further, in family B we found a novel homozygous c.449 G > C transversion that led to a replacement of a highly conserved arginine to proline at codon 150. Family A and Family B correlated both genotypically and phenotypically to ARB.

BEST1 mutations depicted on (a) a cartoon of BEST1 with the conserved pfam domain, (b) the predicted topology of BEST156, (c) the structure of BEST1 (ProteinModelPortal, ID O76090), and (d) a multiple sequence alignment of BEST1 protein sequences from multiple species. ARB - Autosomal Recessive Bestrophinopathy.
In family C, the flecks on fundus imaging and autofluorescence tests showed widespread posterior pole distribution with very minimal but definitive focal schitic changes with predominant subretinal fluid, suggestive of ARB. However, phenotypically, the affected members of family C were dissimilar to those of families A and B. In particular, the intraretinal involvement observed in OCT images from family C was relatively lesser compared to families A and B. Interestingly, in family C we observed two missense substitutions that map to two different isoforms of BEST1. One mutation led to arginine to histidine change at codon 47 (p.Arg47His; ENST00000378043) while the other mutation was a valine to isoleucine substitution at codon 216 (p.Val216Ile; ENST00000526988) of a previously known alternate BEST1 transcript. We confirmed the expression of this alternate transcript in normal donor retinal tissue by RNA sequencing. The p.Arg47His mutation was identified previously in a BVMD39 and an AVMD case38 although, the inheritance patterns were not reported. BVMD and AVMD caused by BEST1 mutations are known to show incomplete inheritance and penetrance41. A recent study functionally assessed the p.Arg47His mutation and suggested that it might be a mildly impaired mutant42. Given these, it is possible that in the unaffected carriers of family C, the disease was not penetrant in the absence of the additional BEST1 mutation observed in the proband and other affected family members.
A BEST1 compound heterozygous mutation, p.Trp29X/p.Arg141His, was previously described in a Swedish ARB family43. Similarly, in another ARB family the affected members were reported to carry heterozygous p.Ala243Glu/p.Arg200X BEST1 mutations44. To the best of our knowledge, our study is the first report of a compound heterozygous mutations involving two BEST1 isoforms. However, the molecular function of the alternate transcript ENST00000526988 and the impact of compound heterozygous mutations (p.Arg47His and p.Val216Ile) on the function of BEST1 in associated disease phenotypes require additional studies.
Our analysis of family D found a heterozygous BEST1 p.Thr91Ile mutation in the affected proband. Remarkably, the proband’s father also carried this mutation but was only mildly affected with no loss of vision; only a small foveal lesion was observed by OCT and FAF. Incomplete penetrance in BVMD has been reported45 and it should be noted that variable presentation of the disease as well as lack of clinical symptoms are also associated with mutations in certain families46. While the BEST1 p.Thr91Ile mutation identified in family D is suggestive of a dominant mutation, the mild phenotype in the father is indicative of the variable penetrance of this mutation that might be modulated by other background genomic variations. Consistent with this, incomplete penetrance of an autosomal dominant BEST1 mutation has been reported in multiple studies35,47,48. Interestingly, in 23 patients with heterozygous BEST1 mutations, six were found to have no clinically detectable disease related phenotype49. While the effect of BEST1 mutations on its function remains to be fully elucidated, a dominant negative role for some mutations50,51 and a functional role for compound heterozygous mutations in BEST1 disease phenotypes has been proposed based on in vitro functional studies15,52.
The 585 amino acid long BEST1 protein is characterized by a highly conserved N-terminal region followed by four to six transmembrane domains. BEST1 functions as a calcium-activated chloride channel (CaCC) which regulates the flow of chloride and other monovalent anions across cellular membranes in response to intracellular calcium levels53. The N-terminus of this protein (amino acids 1–390) contains all of the putative membrane-spanning domains and is sufficient for its CaCC activity while the carboxy-terminal region (amino acids 391–585 of BEST1) is predicted to be unstructured54. Structural models of BEST1 have been proposed, which describe topological positions of the N-terminal region, the transmembrane domains and the C-terminus55,56. Such models propose the N- and C-termini as being cytosolic with the presence of four transmembrane domains (domain 1,2 and 5, 6) while domain 3 and 4 are cytoplasmic55,56 (Fig. 7a–c). The crystal structure of chicken BEST1, which is 74% identical to human BEST1, has been solved. This structure is distinct from other channel proteins and is comprised of a pentamer of five BEST1 subunits symmetrically arranged around a central axis which creates a pore for chloride ions to pass through the protein complex57. Although the detailed mechanisms that lead to the disease are not fully understood, most of the characterized BEST1 mutations alter electrophysiological properties of the channel51,54,57. Crystallographic studies of wild-type and mutant proteins suggest that BEST1 mutations altering the cytoplasmic pore structure affects the permeability of anions or anion-cation selectivity58. Interestingly, the mutations discovered in this study are localized to the N-terminal region (Fig. 7a–c). The mutations identified in family A (p.Tyr131Cys) and family B (p.Arg150Pro) lie in the third cytoplasmic domain. Mutations observed in family C (p.Arg47His) localize to the first transmembrane domain, while the mutation in family D (p.Thr91Ile) alters an amino acid proximal to the second transmembrane domain. The amino acids at these positions are conserved among mammals (Fig. 7d).
References
Burgess, R. et al. Biallelic mutation of BEST1 causes a distinct retinopathy in humans. American journal of human genetics 82, 19–31 (2008).
Lee, C. S. et al. A Novel BEST1 Mutation in Autosomal Recessive Bestrophinopathy. Invest Ophthalmol Vis Sci 56, 8141–8150 (2015).
Marquardt, A. et al. Mutations in a novel gene, VMD2, encoding a protein of unknown properties cause juvenile-onset vitelliform macular dystrophy (Best’s disease). Hum Mol Genet 7, 1517–1525 (1998).
Petrukhin, K. et al. Identification of the gene responsible for Best macular dystrophy. Nature genetics 19, 241–247 (1998).
Yardley, J. et al. Mutations of VMD2 splicing regulators cause nanophthalmos and autosomal dominant vitreoretinochoroidopathy (ADVIRC). Invest Ophthalmol Vis Sci 45, 3683–3689 (2004).
Burgess, R. et al. ADVIRC is caused by distinct mutations in BEST1 that alter pre-mRNA splicing. J Med Genet 46, 620–625 (2009).
Boon, C. J. et al. The spectrum of ocular phenotypes caused by mutations in the BEST1 gene. Progress in retinal and eye research 28, 187–205 (2009).
Davidson, A. E. et al. Missense mutations in a retinal pigment epithelium protein, bestrophin-1, cause retinitis pigmentosa. The American Journal of Human Genetics 85, 581–592 (2009).
Marmorstein, A. D., Cross, H. E. & Peachey, N. S. Functional roles of bestrophins in ocular epithelia. Progress in retinal and eye research 28, 206–226 (2009).
Hartzell, H. C., Qu, Z., Yu, K., Xiao, Q. & Chien, L.-T. Molecular physiology of bestrophins: multifunctional membrane proteins linked to best disease and other retinopathies. Physiological reviews 88, 639–672 (2008).
Boon, C. J. et al. Clinical and genetic heterogeneity in multifocal vitelliform dystrophy. Archives of ophthalmology 125, 1100–1106 (2007).
Boon, C. J. et al. Clinical and molecular genetic analysis of best vitelliform macular dystrophy. Retina 29, 835–847 (2009).
Deutman, A. Electro-oculography in families with vitelliform dystrophy of the fovea: detection of the carrier state. Archives of ophthalmology 81, 305–316 (1969).
Khan KN, et al. Functional and Anatomical Outcomes of Choroidal Neovascularization Complicating Best1-Related Retinopathy. Retina, (2016).
Burgess, R. et al. Biallelic mutation of BEST1 causes a distinct retinopathy in humans. The American Journal of Human Genetics 82, 19–31 (2008).
Borman, A. D. et al. Childhood-onset autosomal recessive bestrophinopathy. Archives of Ophthalmology 129, 1088–1093 (2011).
Crowley, C. et al. Autosomal recessive bestrophinopathy associated with angle-closure glaucoma. Documenta Ophthalmologica 129, 57–63 (2014).
Kinnick, T. R. et al. Autosomal recessive vitelliform macular dystrophy in a large cohort of vitelliform macular dystrophy patients. Retina 31, 581–595 (2011).
Davidson, A. E. et al. Functional characterization of bestrophin-1 missense mutations associated with autosomal recessive bestrophinopathy. Investigative ophthalmology & visual science 52, 3730–3736 (2011).
Johnson, A. A. et al. Autosomal Recessive Bestrophinopathy Is Not Associated With the Loss of Bestrophin-1 Anion Channel Function in a Patient With a Novel BEST1 Mutation. Invest Ophthalmol Vis Sci 56, 4619–4630 (2015).
Leiden Open Variation Database (LOVD). LOVD gene homepage: Bestrophin 1 (BEST1) database [database online]. Available at: https://databases.lovd.nl/shared/genes/BEST1. Accessed January 10, 2018.
Sodi, A. et al. BEST1 sequence variants in Italian patients with vitelliform macular dystrophy. Molecular vision 18, 2736–2748 (2011).
Bach, M. et al. Standard for pattern electroretinography. Documenta ophthalmologica 101, 11–18 (2000).
Marmor, M. F. An international standard for electroretinography. Documenta Ophthalmologica 73, 299–302 (1989).
Wu, T. D. & Nacu, S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26, 873–881 (2010).
Goldstein, L. D. et al. Prediction and Quantification of Splice Events from RNA-Seq Data. PloS one 11, e0156132 (2016).
Van der Auwera, G. A. et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Current protocols in bioinformatics/editoral board, Andreas D Baxevanis [et al.] 11, 11.10.11-11.10.33 (2013).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature genetics 43, 491–498 (2011).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strainw1118; iso-2; iso-3. Fly (Austin) 6, 80–92 (2012).
Genomes Project, C. et al. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012).
Fu, W. et al. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature 493, 216–220 (2013).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
Robinson, J. T. et al. Integrative genomics viewer. Nat Biotechnol 29, 24–26 (2011).
Sharon, D. et al. Ocular phenotype analysis of a family with biallelic mutations in the BEST1 gene. American journal of ophthalmology 157, 697–709. e692 (2014).
Ng, P. C. & Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res 11, 863–874 (2001).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat Methods 7, 248–249 (2010).
Kramer, F. et al. Mutations in the VMD2 gene are associated with juvenile-onset vitelliform macular dystrophy (Best disease) and adult vitelliform macular dystrophy but not age-related macular degeneration. European journal of human genetics: EJHG 8, 286–292 (2000).
Liu, J., Zhang, Y., Xuan, Y., Liu, W. & Wang, M. Novel BEST1 Mutations and Special Clinical Features of Best Vitelliform Macular Dystrophy. Ophthalmic Res 56, 178–185 (2016).
Guziewicz, K. E., Slavik, J., Lindauer, S. J., Aguirre, G. D. & Zangerl, B. Molecular consequences of BEST1 gene mutations in canine multifocal retinopathy predict functional implications for human bestrophinopathies. Investigative ophthalmology & visual science 52, 4497–4505 (2011).
Johnson, A. A. et al. Bestrophin 1 and retinal disease. Prog Retin Eye Res 58, 45–69 (2017).
Johnson, A. A. et al. Disease-causing mutations associated with four bestrophinopathies exhibit disparate effects on the localization, but not the oligomerization, of Bestrophin-1. Exp Eye Res 121, 74–85 (2014).
Schatz, P., Klar, J., Andréasson, S., Ponjavic, V. & Dahl, N. Variant phenotype of Best vitelliform macular dystrophy associated with compound heterozygous mutations in VMD2. Ophthalmic genetics 27, 51–56 (2006).
Fung, A. T. et al. New Best1 mutations in autosomal recessive bestrophinopathy. Retina (Philadelphia, Pa) 35, 773 (2015).
Boon, C. J. et al. Clinical and molecular genetic analysis of best vitelliform macular dystrophy. Retina 29, 835–847 (2009).
Wabbels, B., Preising, M. N., Kretschmann, U., Demmler, A. & Lorenz, B. Genotype-phenotype correlation and longitudinal course in ten families with Best vitelliform macular dystrophy. Graefe’s archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie 244, 1453–1466 (2006).
Weber, B., Walker, D. & Müller, B. Molecular evidence for non-penetrance in Best’s disease. Journal of medical genetics 31, 388–392 (1994).
Piñeiro-Gallego T, et al. Clinical evaluation of two consanguineous families with homozygous mutations in BEST1 (2011).
Querques, G. et al. The spectrum of subclinical Best vitelliform macular dystrophy in subjects with mutations in BEST1 gene. Investigative ophthalmology & visual science 52, 4678–4684 (2011).
Qu, Z., Wei, R. W., Mann, W. & Hartzell, H. C. Two bestrophins cloned from Xenopus laevis oocytes express Ca2+ -activated Cl− currents. Journal of Biological Chemistry 278, 49563–49572 (2003).
Sun, H., Tsunenari, T., Yau, K.-W. & Nathans, J. The vitelliform macular dystrophy protein defines a new family of chloride channels. Proceedings of the National Academy of Sciences 99, 4008–4013 (2002).
Lee, C. S. et al. A Novel BEST1 Mutation in Autosomal Recessive BestrophinopathyA Novel BEST1 Mutation in ARB. Investigative ophthalmology & visual science 56, 8141–8150 (2015).
Tsunenari, T. et al. Structure-function analysis of the bestrophin family of anion channels. Journal of Biological Chemistry 278, 41114–41125 (2003).
Xiao, Q., Prussia, A., Yu, K., Cui, Y.-Y. & Hartzell, H. C. Regulation of bestrophin Cl channels by calcium: role of the C terminus. The Journal of general physiology 132, 681–692 (2008).
Moskova-Doumanova, V., Pankov, R., Lalchev, Z. & Doumanov, J. Best1 shot through the eye—structure, functions and clinical implications of bestrophin-1 protein. Biotechnology & Biotechnological Equipment 27, 3457–3464 (2013).
Milenkovic, V. M., Rivera, A., Horling, F. & Weber, B. H. Insertion and topology of normal and mutant bestrophin-1 in the endoplasmic reticulum membrane. Journal of biological chemistry 282, 1313–1321 (2007).
Dickson, V. K., Pedi, L. & Long, S. B. Structure and insights into the function of a Ca2+ -activated Cl-channel. Nature 516, 213–218 (2014).
Vaisey, G., Miller, A. N. & Long, S. B. Distinct regions that control ion selectivity and calcium-dependent activation in the bestrophin ion channel. Proceedings of the National Academy of Sciences of the United States of America 113, E7399–E7408 (2016).
Acknowledgements
The authors would like to acknowledge Genentech DNA Sequencing, Oligo and Bioinformatics groups for their help with the project and members of the Departments of Molecular Biology and Human Genetics for helpful discussions and review of the manuscript. Work in AG’s lab was supported by Narayana Nethralaya Foundation.
Author information
Authors and Affiliations
Contributions
S.S., A.G., A.S.P. designed the study. R.B. and P.B. identified the families, performed clinical assessment of the patients and provided clinical care. A.V. and R.A.M. coordinated patient data collection and sample preparation. S.P. and V.L.R. managed the sequence data collection. T.T.N., S.S. managed and analyzed exome and RNA-seq data. T.T.N., S.S., A.G. and A.S.P. co-wrote the manuscript with the assistance and contributions from all the co-authors.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nguyen, T.T., Poornachandra, B., Verma, A. et al. Next generation sequencing identifies novel disease-associated BEST1 mutations in Bestrophinopathy patients. Sci Rep 8, 10176 (2018). https://doi.org/10.1038/s41598-018-27951-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-27951-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
