
Abstract
All Plasmodium species express variant antigens which may mediate immune escape in the vertebrate host. In Plasmodium falciparum, the rif gene family encodes variant antigens which are partly exposed on the infected red blood cell surface and may function as virulence factors. Not all rif genes are expressed at the same time and it is unclear what controls rif gene expression. In this work, we addressed global rif transcription using plasmid vectors with two drug resistance markers, one controlled by a rif 5′ upstream region and the second by a constitutively active promoter. After spontaneous integration into the genome of one construct, we observed that the resistance marker controlled by the rif 5′ upstream region was expressed dependent on the applied drug pressure. Then, the global transcription of rif genes in these transfectants was compared in the presence or absence of drugs. The relative transcript quantities of all rif loci did not change profoundly between strains grown with or without drug. We conclude that either there is no crosstalk between rif loci or that the elusive system of allelic exclusion of rif gene transcription is not controlled by their 5′ upstream region alone.
Similar content being viewed by others


Introduction
The infection with one of the five Plasmodium species which cause malaria in humans still is a challenge for the public health predominantly in underdeveloped countries. Plasmodium falciparum alone is still responsible for 445000 deaths per year, mostly children under five years or pregnant women1. Part of the virulence exerted by Plasmodium is caused by the presence of variant antigens expressed on the surface of the host’s infected red blood cells. Members of the best-characterized family of variant antigens, the P. falciparum erythrocyte membrane protein 1 (PfEMP1), play a central role in immune evasion. PfEMP1 are encoded by approximately 50–60 different var genes2 which are highly recombinogenic3,4, possibly through specific three-dimensional DNA conformations near breakpoints5, and possess a modular structure6. In order to successfully evade immune mechanisms exerted by the human host, var gene transcription is tightly controlled in a way that normally only one or two var genes are expressed. A number of factors are involved in this control and these include not only sequences in the 5′ upstream regions of var genes7,8, including untranslated ORFs9,10, the pairing of var promoters and var introns11, but also specific DNA/chromatin binding factors12 and the involvement of several chromatin modifiers (reviewed in13). Recently, the participation of non-coding GC-rich RNAs in var transcription control was shown14. The current model of var transcription regulation also suggests a specific subnuclear site in which var transcription occurs and to which var loci translocate in order to be transcribed. However, the exact factors and dynamics which orchestrate this process and license one var locus for transcription while excluding all other var loci are largely unknown. Still more elusive is what determines that a var locus and its associated histone modifications switch from an active to a silent state or vice versa.
Another major variant gene family which is not only found in human or primate Plasmodium species but also in murine species is the pir (Plasmodium interspersed repeat) gene family15, and the biological function of gene products from this family is not well understood. Recent results indicate that their encoded proteins may function at different points of the parasite-host interface16. If results from Plasmodium chabaudi can be extrapolated to all Plasmodium species, pir transcription seems to be reset during mosquito passage and reinfection17 and earlier evidence pointed to transcriptional diversity18 and quick switching in several models19,20. A recent study implied a specific PIR protein of P. falciparum (termed RIFINs, repetitive interspersed family21) as a factor involved in the pathogenic process of erythrocyte rosetting22. Another study revealed that a specific motif in RIFINs promoted binding to leucocyte immunoglobulin-like receptor B1 (LILRB1) or leucocyte-associated immunoglobulin-like receptor 1, thereby inhibiting activation of B-cells and natural killer cells which express the LILRB1 receptor23. This turns evident that at least some RIFINs can be understood as virulence factors. Importantly, VIR proteins of Plasmodium vivax also appear to mediate cytoadherence and participate in pathogenic processes and probably immune evasion24,25. In P. falciparum, the vast majority of rif genes are localized adjacent to var genes, often in a tandem organization. In version 36 of PlasmoDB, there are 221 genes in the 3D7 strain genome which encode PIR proteins. Of these, 158 are full-length RIFINs, 27 are truncated or defect RIFINs, and the remaining are STEVOR (subtelomeric variant open reading frame) or truncated or defect STEVOR. RIFINs can be categorized into two major groups of rif genes and RIFINs: A and B. These groups differ by a short conserved 25mer peptide sequence in the first half of the protein which is present only in the 97 A-group RIFINs in the P. falciparum strain 3D7 genome26. While the A-group RIFINs seem to be exported to the infected red blood cell (IRBC) surface, B-group RIFINs are believed to remain associated with the vesicular network in the IRBC (Maurer’s clefts). It is still unclear what controls rif transcription, a recent study pointed to the transcription factor AP2-SP27 which somehow seems to influence transcription in blood stage P. falciparum28. While earlier studies indicated that rif and var gene transcription may be controlled by similar factors29, no clear-cut allelic exclusion mechanism could be detected in other studies which specifically addressed rif transcription or switching20,30. A drawback in the study from Howitt and colleagues29 was that the tested rif promoter controlled the unique drug resistance marker blasticidin deaminase in their construct, turning a considerable baseline activity of the promoter essential in order to obtain transfectant parasite lines. This did not permit a genuinely “switched-off” state of the rif 5′ ups-controlled transgene. In another study, the activity profile of a 5′-rif upstream region appeared more related to var genes31 and no significantly different regulation could be discerned upon activation. Also, no “crosstalk” – understood as the influence of the activity of one promoter on the activity of remaining promoters - between 5′ upstream regions such as occurs in var gene regulation was detectable. In contrast, in the study by Goel and colleagues, phenotypic selection procedures pointed to the expression of a single rif gene in parasites with PfEMP1-independent rosetting22, supporting the view that allelic exclusion and crosstalk may occur. In order to settle the question if there are allelic exclusion and crosstalk between rif 5′ ups regions, we used three different rif 5′ upstream regions in bicistronic transfection plasmids. Previously, this approach was successfully applied on var promoters7,32. After transfection, we divided transfected parasite lines and in one culture we selected the growth of parasites which actively transcribed a drug resistance marker (human dihydrofolate reductase). Afterwards, we compared rif transcripts between pyrimethamine-derivate sensitive and resistant lines by RNAseq.
Results
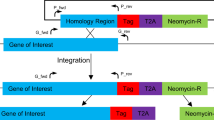
The expression mode of rif genes and the proteins they encode, RIFINs, is unclear. In order to test if rif genes are expressed in a similarly coordinated way as var genes33, we created bicistronic plasmids similar to those used by Voss7 or Witmer31 and colleagues. In these, one resistance marker gene was controlled by the constitutive plasmodial heat shock protein 86/90 promoter and the other by potentially inducible rif 5′ upstream sequences. We chose three rif upstream sequences (ups) based on previous observations using wild-type parasites of the 3D7 lineage where rif transcripts from these 5′ upstream regions were detected20. This assured that functional promoters were being used. The size (~1500 nt) of the inserted putative promoter sequence was chosen observing the distance to adjacent ORFs (Supplemental Fig. 1). In Fig. 1, an outline of the plasmid constructs is shown. Plasmids were transfected and blasticidin-resistant parasite lines were readily established. It is possible that transcription of episomal 5′ upstream sequences is different from genomic loci, due to an increase in the number of circulating episomes (for example31). When testing the copy number of the hDHFR gene, we observed that the transfectant line containing the PF3D7_0200700 5′ rif ups showed only one copy. In contrast, the hDHFR gene in a freshly transfected parasite line containing the Photinus luciferase encoding plasmid pCLH (a derivate of pDC1034 where the chloramphenicol acetyltransferase was changed for a luciferase coding sequence from pGL2 (Promega)) appeared in approximately three copies per cell (selection using 2.5 nM WR99210, Fig. 1). Also, the transfectant lines containing the PF3D7_0900500 or the PF3D7_1300400 5′ rif ups showed five or two hDHFR copies per parasite genome, respectively (Fig. 1). The appearance of only one copy hints to the integration of the bicistronic plasmid. To verify this, we used two PCR amplifications to specifically detect integrated or episomal forms of plasmids in all transfectant lines containing rif 5′ ups. As shown in Fig. 1, no amplicon was detected using an oligo pair which amplifies over the putative breakpoint upon single crossover integration of the construct carrying PF3D7_0200700 5′ rif ups. On the other hand, an amplicon representing the plasmid backbone both in integrated or episomal forms was detected in both transfectants genomic DNA and purified plasmid controls. The transfectants with the PF3D7_0900500 or the PF3D7_1300400 5′ rif ups showed each several amplicons, one of which consistent with episomal forms of the plasmids (Supplementary Fig. 2). Since the transfection plasmid backbone contains four sequence regions which may provoke single crossover recombination into the genome (the hrp3 terminator, the hsp86/90 5′ ups and the hsp86/90 3′ region plus the rif 5′ ups region), we conducted a Southern blot analysis using a digoxigenin-labeled bsd-gene fragment as a probe. The observed pattern indicated that recombination occurred at the hsp86/90 locus and not at the PF3D7_0200700 rif 5′ locus (Fig. 1).

Only the PF3D7_0200700 construct shows single integration in the hsp90 locus. In (A) the outline of the transfection plasmid constructs is shown. rif 5′ ups regions were interchanged in this plasmid by ligation of XhoI/BamHI fragments representing the rif 5′ ups sequence. See Material and Methods for oligonucleotide sequences used for amplification of rif 5′ ups. Due to identity with P. falciparum genomic sequences, Integration may occur in 4 possible sites: hsp90 5′ ups and 3′ downstream regions, in the hrp3 3′ downstream region and in rif 5′ ups regions. Due to the size of identical stretches, we focused on the possible integration events in hsp90 5′ ups and rif 5′ ups. Bold arrows indicate the expected direction of transcription in the 5′-ups regions. (B) Copy number comparison of different transfected parasite lines with gDNA from the integrated NF54::RESA-GFP strain54, an episomal pCLH NF54 strain, and the parasite lines after transfection with plasmids containing different 5′ rif ups. The primer performance of hDHFR primers and for the t-seryl RNA synthetase were tested and judged identical (less than 0.5 Ct difference on the same substrate in qPCR using gDNA from the NF54::RESA-GFP strain). Then, copy numbers of the hDHFR locus in relation to the genomic t-seryl RNA synthetase were calculated. In (C) results from PCRs using long-range polymerases provide indirect evidence that the PF3D7_0200700 5′ rif ups was integrated in the genome. On the left, PCR results showing amplification with forward hDHFR and forward bsd primers (in red) over the backbone of the transfection plasmid (positive for the untransfected plasmid (2) as well as the gDNA from the PF3D7_0200700 5′ rif ups transfected strain). On the right, amplification products from PCRs using reverse hDHFR and reverse bsd oligos. This amplification is only possible when the plasmid is in the episomal form (see scheme for episomal construct and integrated locus in A). No amplification product is seen for the gDNA from the strain with the PF3D7_0200700 5′ rif ups. See Supplementary Figure 2 for results with the other rif ups constructs. In (D), digestion with SpHI of NF54 genomic DNA, transfectant line genomic DNA and transfected plasmid DNA and subsequent Southern blot analysis with a probe consisting of a digoxigenin-labeled bsd fragment. Note that only in the case of integration in the hsp90 locus a 3097 bp fragment is formed, while linearized or concatemerized plasmids will result in 8045 bp fragments.
We then tested if transfectant parasites were able to survive in the presence of the second drug WR99210. A prerequisite for survival is the sufficient transcription from the cloned 5′ rif ups. To test this, the cultures of the three parasite lines were split and one half of each culture was cultivated in the presence of 2.5 µg/ml blasticidin and 2.5 nM WR99210, while the other half was solely grown in the presence of 2.5 µg/ml blasticidin. The parasite line with PF3D7_0200700 5′ rif ups grew slowly for the first few cycles and then proliferated normally, in accordance with a selection of parasites which showed upregulation of the hDHFR controlling PF3D7_0200700 5′ rif ups (Fig. 2). In contrast to this, the parasite line containing the PF3D7_0900500 5′ rif ups was unable to grow in the presence of WR99210 meaning that no parasites were present which had activated this locus. Three experiments over ten growth cycles were tried with this parasite line and parasites never became resistant to WR99210. This means that the cloned PF3D7_0900500 5′ rif ups is either not sufficiently functional or completely silenced. The third construct containing episomes with the PF3D7_1300400 5′ rif ups controlling hDHFR readily grew in the presence of WR99210 and no striking difference could be discerned between blasticidin/WR99210 and blasticidin treated parasites (Fig. 2). Taken together, from the three constructs, only the integrated construct with PF3D7_0200700 5′ rif ups showed a dynamic that was expected for a transcriptionally variable member of the multigene family which is functional in blood stage parasites.

Differential growth of strains containing different 5′ rif ups controlling hDHFR mediated resistance. In (A–C), transfected NF54 parasite strains with plasmids containing the indicated 5′ rif ups controlling hDFHR transcription, established using blasticidin at 2.5 µM as described in methods, were synchronized with plasmagel flotation and sorbitol treatment and submitted to additional WR99210 treatment (2.5 nM) or not. Parasitemias were observed every 48 h hours briefly after reinvasion. In (D) the strain with the PF3D7_0200700 5′ rif ups construct was analyzed for hDHFR transcript abundance dependent on the blood stage form. This experiment was done in three biological replicates and data are shown. In (E) the same trophozoite stage parasites as in (D) were submitted to higher concentrations of WR99210 and the hDHFR transcript was measured by RT-qPCR. Error bars in all graphs show standard deviation. Statistical differences between relative transcript quantity values were calculated using the two-way ANOVA test (*p < 0.05).
We then quantified the relative transcript quantity in the lineage with PF3D7_0200700 5′ rif ups in parasites “on” and “off” WR99210 drug pressure during the intraerythrocytic cycle. As shown in Fig. 2D and E, the relative transcript quantity difference between parasites under WR drug treatment was higher by a factor ranging from 1:4 to 1:40 using reverse transcription-qPCR in three independent experiments. The highest and significant differences were observed in trophozoite stage parasites. In order to monitor if the rif promotor activity could be further increased, we analyzed the steady-state transcript quantities in this parasite lineage grown under 5 nM WR99210 instead of 2.5 nM. As shown in Fig. 2E, the relative transcript quantities strongly increased under these conditions, indicating that the promoter activity may be modulated in a wide range. Importantly, the cultures that were grown in 5 nM WR99210 also did not increase the relative copy number of the artificial hDHFR, reinforcing the view that the 5′ rif ups PF3D7_0200700 had integrated into the genome (Supplementary Fig. 3).
The transcriptional activity of 5′ ups regions of var genes is strictly regulated resulting normally in the expression of one unique gene33 and silencing of all other loci, meaning that var loci are in crosstalk. Accordingly, it was observed that genomic var loci can be silenced by artificial activation of an episomal var 5′ upstream region7,32. To detect if the artificially activated rif locus had any influence on rif transcripts from other genomic loci, RNAseq was performed using a paired sample from the PF3D7_0200700 5′ rif ups-construct containing trophozoites which were submitted or not to WR99210 treatment. As shown in Fig. 3, parasites of this transgenic lineage showed a number of transcripts from genomic rif loci that appeared in higher RPKM numbers (>20 RPKM). In parasites that were not grown in the presence of WR99210, few hDHFR transcripts from the modified PF3D7_0200700 locus were detected (RPKM ~3), corroborating previous qPCR results. In contrast, higher RPKM values for hDHFR as the most abundantly detected rif transcripts were observed in parasites grown in the presence of WR99210. This indicates that the hDHFR-controlling rif promoter can be either active or inactive, pre-requisites of a variant gene promoter. Also, the RPKM values of the modified PF3D7_0200700 were in the same range as other simultaneously active rif promoters (e.g., PF3D7_1372600). When the relative rif transcript quantities from other loci were compared between parasites grown under WR99210 pressure or not, no profound changes were found. This can be interpreted that the activity of the modified PF3D7_0200700 rif promoter did not influence transcription from other loci. Such a result is in contrast with similar experiments using var promoters where the activity of a var 5′ ups led to a substantial silencing of the remaining genomic var loci7. When analyzing the RPKM values of all transcripts (Supplementary Table 1), a number of genes appeared differentially expressed between the two samples, and the observation that invasion-related genes were detected with higher RPKM values indicates that the sample cultivated without WR99210 was slightly advanced in the erythrocytic cycle (Supplementary Fig. 4).

RNAseq shows no substantial differences between rif transcription profiles upon presence or absence of activity from a specific 5′ rif ups (PF3D7_0200700). A: RNAseq was conducted as described with RNA from parasites grown for four reinvasions in the presence of 2.5 nM WR99210 or in the absence of this drug. In red, the RPKM values for all rif loci with the presence of transcripts in parasites grown without WR99210 and in green in the presence of 2.5 nM WR99210. The arrow depicts the strongly different RPKM values for the hDHFR transcript in these cultures. Note that the difference in relative hDHFR transcript quantity is higher than measured by RT-qPCR (see Fig. 2D,E). For individual RPKM data see Supplementary Table 1.
It is possible that subgroups of rif upstream regions are regulated independently, meaning that RIFIN A and/or RIFIN B are not subject to allelic exclusion. When focusing only on rif transcripts which were detected in larger quantities in RNAseq (cutoff RPKMs > 20), we observed that only one rif B type transcript was detected in slightly elevated levels in both treated or untreated parasites, while several rif A-type transcripts were present (Fig. 4). This reinforces that “A”-grouped rif 5′ ups, encoding antigens which are potentially associated with the infected red blood cell surface, are not influenced by an artificially activated “A” type rif 5′-ups.

Rif loci with higher (any value above >20) RPKM values were mostly from loci with A-type rif genes. Results from Fig. 3 were filtered for higher RPKM values and are shown. Note that the locus with an almost identical 5′ rif ups (asterisk) on the same chromosome as the transfected 5′ rif ups controlling hDHFR (bars on the left) is also transcriptionally active independently of activation of the transgene 5′ rif ups PF3D7_0200700. The arrow indicates the only significantly transcribed type B rif.
Discussion
The expression control of variant gene families is an intriguing phenomenon in many human parasites ranging from Trypanosoma brucei (reviewed in35) and Giardia lamblia36 to Plasmodium. Each parasite seems to have developed different molecular mechanisms to ensure that antigenic repertoires are not prematurely exhausted. In Giardia trophozoites, an RNAi-based mechanism enables the translation of a single variant surface antigen per parasite37. In Trypanosoma brucei, selective variant surface antigen (VSG) production is controlled at the transcriptional level and also through genetic recombination. Single vsg genes are transcribed by RNA Polymerase 1 at a specific subnuclear expression site38 and the activation of vsg expression sites is associated with specific chromatin modifications (reviewed in39). Var gene expression in P. falciparum is tightly controlled at the transcriptional level in ring-stage parasites40 and all but one or two sites remain silenced. Silencing of sites is associated with a number of specific chromatin modifications, mainly at histone H3 which - when trimethylated at lysine 9 - recruits heterochromatin protein 112, initially perceived as a landmark of silenced chromatin in different cell types41. A still not answered question is why not all var sites are silenced and the reason for this possibly lies in the concentration of ncRNAs transcribed from a number of GC rich regions present in the nucleus which somehow seem to coordinate gene counting14. Overproduction of members of these ncRNAs lead to the simultaneous transcription of var genes – an effect which is also observed when histone deacetylases SIR2A and SIR2B are repressed or deleted42. Here, we tried to interfere with the transcription of members of the rif gene family using an approach that was successfully applied to monitor var gene transcription. Interestingly, from three different constructs, only one presented a variable transcription activity, which may be considered as a pre-requisite of the corresponding RIFIN to function in antigenic variation. For the other two constructs, which also did not show integration at genomic sites, we found either a permanently activated or silenced status. Notably, all tested 5′-upstream regions have several, almost completely identical regions in the genome but only one of the rif genes controlled by these identical regions seemed transcriptionally active to the same degree as the transgene (not considering the 5′-ups rif construct with PF3D7_0900500 which was always silenced (Table 1)). Interestingly, the only identical locus with considerable amounts of transcripts from the genomic locus appeared for the differentially controlled PF3D7_0200700 rif 5′-ups construct. Intriguingly, the original rif locus PF3D7_0200700 from which the 5′-ups region was cloned had no detectable transcripts at all (Supplementary Table 1), while the rif locus lying on the opposite end of chromosome 2 showed transcripts in the RPKM value range of 91 and 51 depending on the presence or absence of transcripts from the artificially integrated PF3D7_0200700 rif locus (Table 1).
This may be interpreted that absolute sequence may not be associated with promoter activity or silencing. It seems that epigenetic marking of chromatin surrounding rif promoters plus factors associating to them have a decisive role in 5′-ups activation and silencing, similar to what is found for var genes. How this regulation is achieved at the molecular level is still elusive. In the study of Howitt and colleagues, a still unknown - but limited in number- factor which provides transcriptional activation of rif and var transcripts was suggested29. In the present RNAseq analysis only one var locus showed significantly altered transcript levels (data not shown). This var locus – PF3D7_0400200 – is annotated as a pseudogene and consists only of exon 2. PlasmoDB predicts also the presence of the var intron, which itself is also a promoter. It is unclear if RNA from this site is involved in rif transcription regulation. Our data do not support co-regulation of rif and var genes. Of note, the material analyzed in our study may also not permit any conclusion about rif-var co-regulation, since RNAs from middle/late trophozoite stage parasites were employed. Normally, var transcription has ceased in trophozoites 20 h post reinvasion43. It must also be reinforced that the shown RNAseq results are unique for this specific experiment. It may be expected that the rif genes which appear transcribed in the current samples change over time due to switching and that in another experiment different, dominant rif transcripts are detected. Additionally, it is unclear if the observed rif transcripts occurred in the same parasites or if they originated from different parasites, concomitantly with the activated PF3D7_0200700 locus in all parasites. Also, it appears that the untreated sample was slightly advanced in the erythrocytic cycle since a number of schizont related genes such as MSPs showed more transcripts in the sample not treated with WR99210 compared to the treated sample (Supplementary Table 1, spreadsheet “2”, and “3”, Supplementary Fig. 4). Notwithstanding, the main result is still valid and other rif gene transcripts, besides the artificially activated PF3D7_0200700 locus, are not influenced.
Based on our data, it may be postulated the chromosomal context surrounding rif 5′ups-regions may play a pivotal role in rif expression. All of the three 5′-ups regions were designed in the same way and contained almost 1500 nt upstream of the corresponding rif ATG, and all three upstream regions contained either terminator regions or head-to-head promoter regions of the adjacent gene locus possibly including heterochromatin boundary regions (Supplementary Fig. 1). Nonetheless, only two 5′-ups were functional in providing a sufficient number of transcripts and only the apparently integrated allele showed a tunable behavior. In the study by Witmer and colleagues31, the 5′-ups region of rif PF3D7_1300400 was tested. This region is somehow special in that it lies in a head-to-head position to a var upsA promoter region. In their study, in one construct the rif promoter portion could be activated while the adjacent var ups was completely silenced. In a second, slightly modified construct, both promoters could be selected for active transcription. The degree of induction of the rif 5′-ups in their study was not significantly different, indicating that transcription was possibly partly induced even without drug selection for transcription from this promoter. In our study, we used 1465 nt of the same 5′ rif ups and also no differential regulation was observed (construct 3 with PF3D7_1300400 5′ rif ups). Given the ambiguous results for both rif 5′ ups PF3D7_0900500 and PF3D7_1300400 which are either not integrated or may have been rearranged during the transfection procedure, no further conclusions can be drawn why constructs containing these 5′ ups led to the observed results of permanent silencing or activity, respectively.
Here, the copy number of plasmids encountered in parasite lines bearing most probably episomal forms of rif promoted hDHFR was partially lower than in the related study from Howitt29 or Witmer31 and colleagues. This may be due to the fact that we used low concentrations of blasticidin for selection. Howitt and colleagues used 2 or 10 µg/ml blasticidin and copy numbers at 2 µg/ml were in the same range or slightly higher than in our episomal constructs. Witmer and colleagues also used 2.5 µg/ml blasticidin but their plasmid backbones contained the TARE/rep20 element, known to improve plasmid segregation44. When we compared results of the hDHFR marker transcript in the transgenic PF3D7_0200700 parasite line growing or not in the presence of WR99210, we found smaller discrepancies between the “on” and “off” in qPCR than in RNAseq. The reason for this may lie in the differing range of linearity of both techniques, although this was not specifically tested for.
As a central result, we showed that transcription of the rif gene family, specifically of the RIFIN A subset, is most probably not controlled by a mechanism related to allelic exclusion as is valid for var genes and that almost 100% sequence identity in rif 5′ ups regions is not sufficient to predict promoter co-activation or silencing. However, not all rif genes are transcribed meaning that there still must be some kind of controlled activation and repression. Considering the results from Guizetti and colleagues14, it may be postulated that ncRNAs are also involved in the activation of rif loci, with the difference that these ncRNAs allow for activity from multiple loci – similar to what is seen for var genes when certain ncRNA from GC rich regions are overexpressed14. From the study of Guizetti and colleagues, it is not clear what influence the var relevant ncRNAs had on rif gene transcription although these authors speculated that a co-regulation could happen. Importantly, there are ncRNAs for most rif loci with yet elusive function45. A specific transcription machinery in the periphery of the nucleus was postulated for var genes46,47,48. To date, it is unclear if rif genes are also transcribed from the same machinery. This could be elucidated by RNA FISH or other adequate methods to monitor nuclear substructures, and the created parasite line may be useful for this kind of experiment. It also still remains elusive if the B subfamily of rif genes is differentially regulated, given the fact that we found only one B-type rif gene activated in our cultures.
Material and Methods
Parasite culture and transfection
Parasites (strain NF54) were cultured under biological level 2 conditions at 5% hematocrit in human B+ blood supplemented with 0.5% Albumax 1 (Invitrogen) or 10% human B+ plasma in RPMI and 0.23% sodium bicarbonate under a 90% N2, 5% CO2, 5% O2 atmosphere or in candle jars as described earlier49. Human blood and plasma were obtained from the local blood bank and ethical clearance for using this blood for this research was granted by the Ethics Committee of the Institute of Biomedical Sciences at the University of São Paulo (No. 842/2016). The medium was changed daily or every two days when parasitemias were low (<0.5%). Parasitemias were checked by Giemsa-stained thin blood smears. For transfection, the protocol suggested by Hasenkamp et al.50 was used. Essentially, 150 µl of cytomix-washed fresh red blood cells were electroporated in a BioRad Gene Pulser at 310 V, 960 µF with 40 µg purified plasmid and later mixed with 2*107 plasmagel-purified51 schizont stage parasites. On day 2 after transfection, Blasticidin (Sigma) was added at 2.5 µg/ml to the medium. At day 6 after addition of the drug, no more viable parasites were visible and the medium was changed every two days until the appearance of parasites on days 17–25 post-transfection.
Plasmid constructs
The bi-cistronic plasmid used here is based on the pTZ57 (Thermo/Fermentas) backbone, where a cassette containing the hsp86 promoter fragment (BglII-NcoI) from pPF8652 was inserted. The blasticidin-deaminase coding sequence was excised from pBMNL106P-PpLuciBlast53 (NcoI-SalI) and inserted into the vector. Then, the hsp86 terminator sequence was inserted from pPF86 via SalI and BamHI. In this plasmid, the hDHFR resistance cassette from pRESA-GFP-HA54 was inserted via EcoRI/EcoRV inserting in EcoRI/SmaI. This plasmid was used to exchange the Calmodulin promoter for a 5′ upstream region from three rif genes which were previously shown to be transcribed in wild-type 3D7 cultures20. The putative rif promoter regions were amplified using oligonucleotides (forward/reverse) ctcgagatataaatttgtaaaaaccatgtg/ggatccttaattgtgatacgtatattatttaatg (precedes PF3D7_0200700, rif A type), ctcgagtattatatttttatatataattattcgtg/ggatccatttaatgtgatacttatattattttatg (precedes PF3D7_0900500, rif B type), and ctcgagatgtaatatattattatgttaatattc/ggatcctattgtgatacgtatattattttatg (precedes either PF3D7_1300400, PF3D7_0600500, PF3D7_0937500, PF3D7_1150300, all rif A type). The amplified fragments were 1400 to 1500 nt and the identity of the sequences was confirmed by semiautomatic sequencing in an Applied Biosystems 7550 sequencer. An outline of the different rif 5′ upstream region containing plasmid is given in Fig. 1.
Selection of WR99210 resistant parasite lines and total RNA preparation
After outgrowth of parasites resistant to 2.5 µg/ml Blasticidin, parasites were split into two parallel cultures and cultivated in RPMI/Albumax supplemented with either 2.5 µg/ml Blasticidin/2.5 nM or 5 nM WR99210 or solely 2.5 µg/ml Blasticidin. After 4 reinvasion cycles, parasites were synchronized by plasmagel floatation51 and subsequent Sorbitol lysis55 and then harvested after another reinvasion. harvested in ring, trophozoite or schizont stage forms. Harvested IRBC were treated with 0.1% Saponin for 10 min at RT and then pelleted at 12000 g/4 °C for 5 min, washed once in 1 ml PBS and then resuspended in a volume of 100 µl using TE. Afterwards, 1 ml Trizol (Invitrogen) was added and the sample was stored at −80 °C until use. Total RNA was prepared following the Trizol protocol provided by the manufacturer. Final total RNA was dissolved in 20 µl RNAse free water and stored at −80 °C until use.
Real-time PCR with parasite-derived cDNA and PCR test for integration
Total RNA was converted to cDNA using the previously published protocol56 using 5× Hotfire Pol SYBR Mix (Solis Biodyne Inc.) on an Eppendorf realplex2 thermocycler. In order to monitor the hDHFR transcription controlled by rif upstream regions, the following oligonucleotides were employed: forward 5′-ctggttctccattcctgagaag/reverse 5′-ttgtggaggttccttgagttct. These oligos were predicted using Primer3 online software57 using the same settings as used for the design of var oligos and the internal control oligonucleotide pair for the amplification of seryl tRNA ligase (PF3D7_0717700)58. Relative transcript quantities were then calculated by the 2−ΔCt method59 using the seryl tRNA ligase transcript as endogenous control. The performance of the hDHFR oligonucleotide pair does not differ from seryl t-RNA ligase specific oligos (data not shown). To test for integration of plasmids, genomic DNAs (gDNA) were prepared from parasite cultures using the Promega genomic DNA preparation kit and aliquots of the gDNAs were tested combining forward and reverse real-time PCR oligos for hDHFR (see above) and blasticidin deaminase (forward: 5′-tgcagtttcgaatggacaaa, reverse: 5′-aacacaaaacaatctggtgcat). PCRs were performed using Thermo/Invitrogen Elongase with the following thermocycling program: 94 °C, 40 s; 54 °C, 40 s; 65 °C, 4 min, over 30 cycles.
RNAseq with parasite-derived cDNA
Total RNA was isolated from the harvested trophozoite-stage parasites with Trizol reagent following the manufacturer’s instruction. The concentration of the isolated RNA was quantified using a Nanodrop device (Thermo Scientific, USA) and the integrity of the RNA was measured by a 2100 Bioanalyzer (Agilent Technologies, CA). Paired-end sequencing cDNA libraries were constructed from two samples (grown with or without 2.5 nM WR99210 for 6 reinvasions) using a TruSeq RNA Sample Preparation Kit v2 low sample (LS) protocol (Illumina Inc., CA), based on the manufacturer’s instructions. RNAseq was conducted in an Illumina NextSeq500 sequencer following the recommendations of the provider using mid output flow cells and a total of 10 million reads per sample. CLC Genomics Workbench 7.01 platform was used to remove the adapter and assess reads quality from the raw reads. The same platform was used to subject reads to the reference P. falciparum genome available in the Ensemble Genome database (Release 26) and to generate gene reads count table used in the following pipelines. Two packages built in Bioconductor60 were used for further analysis: edgeR61 and limma62. The first was used to filter and normalize the data set, while linear modeling and empirical Bayes to assess differential expression were performed with the limma package. Finally, the RPKM values of trophozoite-derived cDNA from cultures which were treated with WR99210 or not were loaded in MS Access and filtered for product ID containing “rif”, and the corresponding RPKM values for each rif gene were plotted against their ID.
Southern Analysis
Genomic DNA was isolated from the WT (NF54) and NF54::pTZ57PF3D7_0200700 parasites, using the Wizard® Genomic DNA Purification Kit (Promega). 5 μg of each gDNA and 25ηg of plasmid DNA (pDNA) from the pTZ57PF3D7_0200700 construct, were digested using SpHI (Thermo Fisher Scientific). The probe was amplified using standard PCR conditions with digoxigenin-dUTP from DIG High Prime DNA Labeling and Detection Starter Kit I (Roche Diagnostics), using the Blasticidin deaminase cassette from the pTZ57PF3D7_0200700 construct as a template. The oligonucleotide primers used were 5′- atgggaaaaacatttaacatttc-3′ and 5′-aacacaaaacaatctggtgcat-3′. The Southern procedure was performed following the protocol provided by the manufacturer of the labeling kit (Roche), using Hybond N membranes (Amersham/GE Healthcare) and a hybridization temperature of 44 °C. All washings steps were performed at room temperature.
References
WHO: World Malaria Report. 94 (www.who.int/malaria/publications/world-malaria-report-2017/en/) (2017).
Su, X. Z. et al. The large diverse gene family var encodes proteins involved in cytoadherence and antigenic variation of Plasmodium falciparum-infected erythrocytes. Cell 82, 89–100 (1995).
Freitas-Junior, L. H. et al. Frequent ectopic recombination of virulence factor genes in telomeric chromosome clusters of P. falciparum. Nature 407, 1018–1022 (2000).
Claessens, A. et al. Generation of Antigenic Diversity in Plasmodium falciparum by Structured Rearrangement of Var Genes During Mitosis. PLoS Genet. https://doi.org/10.1371/journal.pgen.1004812 (2014).
Sander, A. F. et al. DNA secondary structures are associated with recombination in major Plasmodium falciparum variable surface antigen gene families. Nucleic Acids Res. 42, 2270–2281 (2014).
Rask, T. S., Hansen, D. A., Theander, T. G., Gorm Pedersen, A. & Lavstsen, T. Plasmodium falciparum erythrocyte membrane protein 1 diversity in seven genomes–divide and conquer. PLoS Comput. Biol. 6, (2010).
Voss, T. S. et al. A var gene promoter controls allelic exclusion of virulence genes in Plasmodium falciparum malaria. Nature 439, 1004–8 (2006).
Brancucci, N. M. B., Witmer, K., Schmid, C. D., Flueck, C. & Voss, T. S. Identification of a cis-acting DNA-protein interaction implicated in singular var gene choice in Plasmodium falciparum. Cell. Microbiol. 14, 1836–1848 (2012).
Amulic, B., Salanti, A., Lavstsen, T., Nielsen, M. A. & Deitsch, K. W. An upstream open reading frame controls translation of var2csa, a gene implicated in placental malaria. PLoS Pathog. 5, e1000256 (2009).
Brancucci, N. M. B., Witmer, K., Schmid, C. & Voss, T. S. A var gene upstream element controls protein synthesis at the level of translation initiation in Plasmodium falciparum. PLoS One 9, e100183 (2014).
Frank, M. et al. Strict pairing of var promoters and introns is required for var gene silencing in the malaria parasite Plasmodium falciparum. J Biol Chem 281, 9942–9952 (2006).
Pérez-Toledo, K. et al. Plasmodium falciparum heterochromatin protein 1 binds to tri-methylated histone 3 lysine 9 and is linked to mutually exclusive expression of var genes. Nucleic Acids Res. 37, 2596–606 (2009).
Guizetti, J. & Scherf, A. Silence, activate, poise and switch! Mechanisms of antigenic variation in Plasmodium falciparum. Cell. Microbiol. 15, 718–26 (2013).
Guizetti, J., Barcons-Simon, A. & Scherf, A. Trans-acting GC-rich non-coding RNA at var expression site modulates gene counting in malaria parasite. Nucleic Acids Res. gkw664 https://doi.org/10.1093/nar/gkw664 (2016).
Janssen, C. S., Phillips, R. S., Turner, C. M. R. & Barrett, M. P. Plasmodium interspersed repeats: the major multigene superfamily of malaria parasites. Nucleic Acids Res. 32, 5712–20 (2004).
Yam, X. Y. et al. Characterization of the Plasmodium Interspersed Repeats (PIR) proteins of Plasmodium chabaudi indicates functional diversity. Sci. Rep. 6, 23449 (2016).
Spence, P. J. et al. Vector transmission regulates immune control of Plasmodium virulence. Nature 498, 228–231 (2013).
Ebbinghaus, P. & Krucken, J. Characterization and tissue-specific expression patterns of the Plasmodium chabaudi cir multigene family. Malar J 10, 272 (2011).
Cunningham, D. et al. Rapid changes in transcription profiles of the Plasmodium yoelii yir multigene family in clonal populations: lack of epigenetic memory? PLoS One 4, e4285 (2009).
Cabral, F. J. J. & Wunderlich, G. Transcriptional memory and switching in the Plasmodium falciparum rif gene family. Mol Biochem Parasitol 168, 186–190 (2009).
Cheng, Q. et al. stevor and rif are Plasmodium falciparum multicopy gene families which potentially encode variant antigens. Mol Biochem Parasitol 97, 161–176 (1998).
Goel, S. et al. RIFINs are adhesins implicated in severe Plasmodium falciparum malaria. Nat. Med. 21, 314–17 (2015).
Saito, F. et al. Immune evasion of Plasmodium falciparum by RIFIN via inhibitory receptors. Nature 552, 1–49 (2017).
Bernabeu, M. et al. Functional analysis of Plasmodium vivax VIR proteins reveals different subcellular localizations and cytoadherence to the ICAM-1 endothelial receptor. Cell Microbiol 14, 386–400 (2012).
Carvalho, B. O. et al. On the cytoadhesion of Plasmodium vivax-infected erythrocytes. J. Infect. Dis. 202, 638–647 (2010).
Joannin, N., Abhiman, S., Sonnhammer, E. L. & Wahlgren, M. Sub-grouping and sub-functionalization of the RIFIN multi-copy protein family. BMC Genomics 9, 19 (2008).
Yuda, M., Iwanaga, S., Shigenobu, S., Kato, T. & Kaneko, I. Transcription factor AP2-Sp and its target genes in malarial sporozoites. Mol. Microbiol. 75, 854–63 (2010).
Martins, R. M. et al. An ApiAP2 member regulates expression of clonally variant genes of the human malaria parasite Plasmodium falciparum. Sci. Rep. 7, 14042 (2017).
Howitt, C. A. et al. Clonally variant gene families in Plasmodium falciparum share a common activation factor. Mol Microbiol 73, 1171–1185 (2009).
Wang, C. W., Magistrado, P. A., Nielsen, M. A., Theander, T. G. & Lavstsen, T. Preferential transcription of conserved rif genes in two phenotypically distinct Plasmodium falciparum parasite lines. Int J Parasitol 39, 655–664 (2009).
Witmer, K. et al. Analysis of subtelomeric virulence gene families in Plasmodium falciparum by comparative transcriptional profiling. Mol Microbiol 84, 243–259 (2012).
Dzikowski, R., Frank, M. & Deitsch, K. Mutually exclusive expression of virulence genes by malaria parasites is regulated independently of antigen production. PLoS Pathog 2, e22 (2006).
Scherf, A. et al. Antigenic variation in malaria: in situ switching, relaxed and mutually exclusive transcription of var genes during intra-erythrocytic development in Plasmodium falciparum. EMBO J 17, 5418–5426 (1998).
Fidock, D. A., Nomura, T. & Wellems, T. E. Cycloguanil and its parent compound proguanil demonstrate distinct activities against Plasmodium falciparum malaria parasites transformed with human dihydrofolate reductase. Mol. Pharmacol. 54, 1140–7 (1998).
Horn, D. & McCulloch, R. Molecular mechanisms underlying the control of antigenic variation in African trypanosomes. Curr Opin Microbiol 13, 700–705 (2010).
Prucca, C. G. & Lujan, H. D. Antigenic variation in Giardia lamblia. Cell. Microbiol. 11, 1706–15 (2009).
Prucca, C. G. et al. Antigenic variation in Giardia lamblia is regulated by RNA interference. Nature 456, 750–4 (2008).
Navarro, M. & Gull, K. A pol I transcriptional body associated with VSG mono-allelic expression in Trypanosoma brucei. Nature 414, 759–763 (2001).
Figueiredo, L. M., Cross, G. A. & Janzen, C. J. Epigenetic regulation in African trypanosomes: a new kid on the block. Nat Rev Microbiol 7, 504–513 (2009).
Kyes, S. et al. Plasmodium falciparum var gene expression is developmentally controlled at the level of RNA polymerase II-mediated transcription initiation. Mol. Microbiol. 63, 1237–47 (2007).
Zeng, W., Ball, A. R. & Yokomori, K. HP1: heterochromatin binding proteins working the genome. Epigenetics 5, 287–92 (2010).
Tonkin, C. J. et al. Sir2 paralogues cooperate to regulate virulence genes and antigenic variation in Plasmodium falciparum. PLoS Biol. 7, e84 (2009).
Schieck, E., Pfahler, J. M., Sanchez, C. P. & Lanzer, M. Nuclear run-on analysis of var gene expression in Plasmodium falciparum. Mol. Biochem. Parasitol. 153, 207–12 (2007).
O’Donnell, R. A. et al. A genetic screen for improved plasmid segregation reveals a role for Rep20 in the interaction of Plasmodium falciparum chromosomes. EMBO J. 21, 1231–9 (2002).
Raabe, C. A. et al. A global view of the nonprotein-coding transcriptome in Plasmodium falciparum. Nucleic Acids Res. 38, 608–17 (2010).
Guizetti, J., Martins, R. M., Guadagnini, S., Claes, A. & Scherf, A. Nuclear Pores and Perinuclear Expression Sites of var and Ribosomal DNA Genes Correspond to Physically Distinct Regions in Plasmodium falciparum. Eukaryot. Cell 12, 697–702 (2013).
Ralph, S. A., Scheidig-Benatar, C. & Scherf, A. Antigenic variation in Plasmodium falciparum is associated with movement of var loci between subnuclear locations. in. Proc Natl Acad Sci USA 102, 5414–5419 (2005).
Issar, N., Ralph, S. A., Mancio-Silva, L., Keeling, C. & Scherf, A. Differential sub-nuclear localisation of repressive and activating histone methyl modifications in P. falciparum. Microbes Infect. 11, 403–407 (2009).
Trager, W. & Jensen, J. B. Human malaria parasites in continuous culture. Science (80-.). 193, 673–675 (1976).
Hasenkamp, S., Russell, K. T. & Horrocks, P. Comparison of the absolute and relative efficiencies of electroporation-based transfection protocols for Plasmodium falciparum. Malar. J. 11, 210 (2012).
Lelievre, J., Berry, A. & Benoit-Vical, F. An alternative method for Plasmodium culture synchronization. Exp Parasitol 109, 195–197 (2005).
Militello, K. T. & Wirth, D. F. A new reporter gene for transient transfection of Plasmodium falciparum. Parasitol. Res. 89, 154–7 (2003).
Banaszynski, L. A., Chen, L.-C., Maynard-Smith, L. A., Ooi, A. G. L. & Wandless, T. J. A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell 126, 995–1004 (2006).
de Azevedo, M. F. et al. Systematic Analysis of FKBP Inducible Degradation Domain Tagging Strategies for the Human Malaria Parasite Plasmodium falciparum. PLoS One 7, e40981 (2012).
Lambros, C. & Vanderberg, J. P. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol 65, 418–420 (1979).
Golnitz, U., Albrecht, L. & Wunderlich, G. Var transcription profiling of Plasmodium falciparum 3D7: assignment of cytoadherent phenotypes to dominant transcripts. Malar J 7, 14 (2008).
Untergasser, A. et al. Primer3–new capabilities and interfaces. Nucleic Acids Res. 40, e115 (2012).
Salanti, A. et al. Selective upregulation of a single distinctly structured var gene in chondroitin sulphate A-adhering Plasmodium falciparum involved in pregnancy-associated malaria. Mol. Microbiol. 49, 179–91 (2003).
Livak, K. J. & Schmittgen, T. D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 25, 402–408 (2001).
Huber, W. et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 12, 115–121 (2015).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47–e47 (2015).
Acknowledgements
R.B.D.A., T.M.S. and G.F.L. are/were supported by fellowships from CNPq. G.W. and P.M.R. are CNPq research fellows. G.W. was funded by FAPESP grants (2012/23306-5 and 2015/17174-7).
Author information
Authors and Affiliations
Contributions
R.B.D.A., T.M.S., C.S.K. produced qPCR data, G.F.L. and G.W. constructed transfection plasmids and produced transfectants, D.A. and P.M.R. performed RNAseq and its analysis, G.W. conceived the project and wrote the paper. All authors read and approved the manuscript and its data.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Araujo, R.B.D., Silva, T.M., Kaiser, C.S. et al. Independent regulation of Plasmodium falciparum rif gene promoters. Sci Rep 8, 9332 (2018). https://doi.org/10.1038/s41598-018-27646-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-27646-0
This article is cited by
-
Rapid activation of distinct members of multigene families in Plasmodium spp
Communications Biology (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
