
Abstract
Cystine-stabilized peptides have great utility as they naturally block ion channels, inhibit acetylcholine receptors, or inactivate microbes. However, only a tiny fraction of these peptides has been characterized. Exploration for novel peptides most efficiently starts with the identification of candidates from genome sequence data. Unfortunately, though cystine-stabilized peptides have shared structures, they have low DNA sequence similarity, restricting the utility of BLAST and even more powerful sequence alignment-based annotation algorithms, such as PSI-BLAST and HMMER. In contrast, a supervised machine learning approach may improve discovery and function assignment of these peptides. To this end, we employed our previously described m-NGSG algorithm, which utilizes hidden signatures embedded in peptide primary sequences that define and categorize structural or functional classes of peptides. From the generalized m-NGSG framework, we derived five specific models that categorize cystine-stabilized peptide sequences into specific functional classes. When compared with PSI-BLAST, HMMER and existing function-specific models, our novel approach (named CSPred) consistently demonstrates superior performance in discovery and function-assignment. We also report an interactive version of CSPred, available through download (https://bitbucket.org/sm_islam/cystine-stabilized-proteins/src) or web interface (watson.ecs.baylor.edu/cspred), for the discovery of cystine-stabilized peptides of specific function from genomic datasets and for genome annotation. We fully describe, in the Availability section following the Discussion, the quick and simple usage of the CsPred website to automatically deliver function assignments for batch submissions of peptide sequences.
Similar content being viewed by others


Introduction
Cystine-stabilized peptides are impressively abundant and widespread across the taxa. They form the neurotoxic venom fraction of spiders1, snakes2, scorpions3, sea anemones4, jellyfish, corals and conch5 and may be specific for insects, mammals, or reptiles. Other cystine-stabilized peptides serve as antimicrobials6 and defensins in humans, insects, fungi, plants and most other taxa. Functionally, the venom peptides include sodium7, calcium8 and potassium9 ion channel blockers, acetylcholine receptor inhibitors10, or protease inhibitors11. Antimicrobial peptides generally act as membrane disrupters specifically against bacterial or fungal cells, but, due to their ability to penetrate cell membranes, they can also enter eukaryotic cells to act on host DNA directly and to modulate immune responses6. The stability of these peptides and their specific and powerful functions make them strong candidates for a variety of medical and agricultural applications, including pain relief, disruption of cancer development, and environmentally friendly insecticides, fungicides and bactericides, delivered either directly or via transgenes.
Cystine-stabilized peptides are also achieving commercial success. Clinically, alpha-bungarotoxin has a long history of use in isolating and identifying specific acetylchloline receptors and in the diagnosis of myasthenia gravis10. Aprotinin has been shown clinically effective against flu infection by inhibiting protease cleavage of HA0 to HA1 and HA212, and Linaclotide is licensed for clinical use orally against irritable bowel syndrome13. The calcium channel blocker from conch, ziconotide (Prialt), is used clinically as a pain reliever8, and the chloride channel blocker from scorpion, chlorotoxin, reached Phase III trials as a treatment for glioblastoma cancer14. However, only a tiny fraction of cystine-stabilized peptides has been characterized experimentally15,16,17. To sort through the huge number of remaining cystine-stabilized peptides present in such a wide range of genomes for the purpose of classifying each of these peptides into one of the disparate functional groups, an efficient automated approach is warranted.
Sequence identity of the cystine-stabilized peptides varies broadly and can be distributed into different structural/motif and family-based (the native source of a peptide) classes18. The scorpion toxin-like superfamily17,19,20, agatoxins21, and conotoxins22 are examples of family-based classes, while STPs23, NTPs23, cyclotides24 and knottins25 are examples of structure or motif-based classes. Because of the high degree of heterogeneity in their primary sequences, several sequence alignment independent models have been reported to classify the structure of the cystine-stabilized /disulfide-rich family. For instance, Cypred26 predicts cyclic peptides including cyclotides; Knotter 1D predicts peptides with ICK motifs27; iCTX-Type structures predict types of Conotoxins targeting Ion Channels28; PredCSF predicts conotoxin superfamily from the primary protein sequences29; and PredSTP predicts sequential tri-disulfide motifs in cysteine rich peptide23. In addition, a specific functional group of cystine-stabilized peptides often come from different family or structural classes. Thus, family or structure/motif-based classification will may reveal the functional characteristic of a peptide. Under this context, it is necessary to develop a sequence alignment independent model to discover the functional characteristics in a family of origin or structure agnostic fashion.
Machine learning-based supervised models are widely used to predict the functional and structural class of proteins which are difficult to predict using sequence alignment-based algorithms. However, it is imperative to extract the relevant feature vectors (descriptors) and to implement an optimized classification algorithm to get expected performance from a model. Several classification algorithms have already been exploited to predict protein characteristics from the primary sequences30,31,32, but, extracting proper descriptors from protein sequences remains a challenging task. A number of descriptors, such as amino acid composition33, autocorrelation34, CTD (composition, transition, and distribution)35, conjoint triads36 and pseudo amino acid compositions37 are routinely used to build machine learning-based models. Recently, we demonstrated a complete pipeline of a classifier constructor where the feature generation model is integrated with a logistic regression algorithm38. This training set pipeline is denoted as m-NGSG (modified n-gram and skip-gram) where a modified n-grams39and skip-grams-based40 framework is used to generate descriptors from the protein sequences and utilize the hidden signatures from the descriptors for the supervised classification41. The m-NGSG framework has proven highly accurate for constructing reliable supervised prediction models38.
In this study, we applied m-NGSG to build five individual models to predict ion channel blockers, antimicrobial peptides, acetylcholine receptor inhibitors, serine protease inhibitors, and hemolytic proteins from disulfide stabilized proteins. Identification of hemolytic characteristics will allow the researcher to eliminate from consideration proteins cytotoxic to humans. The results demonstrate superiority of m-NGSG-based models to PSI-BLAST42, HMMER43 and other available models. Finally, we propose the CSPred model which combines the results of the five different models and gives a probability score for the five important functional characteristics of cystine-stabilized proteins. We also present three classifiers that assign ion channel blockers into three subclasses, sodium, potassium and calcium channel blockers.
Material and Methods
Data acquisition and preparation
The positive and negative datasets for ion channel blockers (ICB), antimicrobial peptides (AMP), acetylcholine receptor inhibitors (ACRI), serine protease inhibitors (SPI), and hemolytic proteins (HLP) are generated by obtaining protein sequences from UniprotKB (knowledgebase)44 using the search keys mentioned in Supplement Table 1. All the protein sequences, including positive and negative classes, contain a minimum of one disulfide bond and a chain size of less than 150 amino acid residues. Thereafter, the protein sequences are curated manually based on the functional attribute for each entry. A portion of the HLP positive dataset is collected from the HemoPI server45. Here, only the sequences containing a minimum of one pair of cysteines are selected from the dataset. The CD-HIT software46 is used to organize sequences based on identity thresh-holds to generate final datasets for each functional group of the cysteine stabilized proteins (See Supplement Table 1). From the positive and negative datasets of each selected functional group, 90% of the chains are retained for training sets, while 10% of the chains are reserved for out-of-sample test sets using a random shuffle-split process. The numbers of chains in each training and test sets are mentioned in Supplement Table 1. Further, to construct a separate compound model to classify the ICB into three different subclasses, we made three separate models using six different training sets (Supplement Table 2). The ICB classifier was constructed to classify the ICBs into sodium, potassium and calcium channel blockers. In order of make the process reproducible, the sequences of the training and test sets are provided in a separate supplementary folder (Supplementary Folder 1).
Model construction using m-NGSG
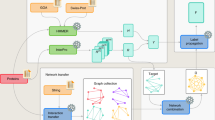
Five different binary classifiers are constructed to predict each of the five selected functional classes using the m-NGSG algorithm38. The m-NGSG algorithm (avialable at https://bitbucket.org/sm_islam/mngsg/src) offers an integrated and fully automated feature generation method followed by a logistic regression-based model construction, feature generation, and parameter optimization as described previously38. Parameter optimizations employed five-fold cross-validation using appropriate training sets. Supplement Table 3 illustrates the parameters selected by the m-NGSG optimizer for each functional group specific model. A combined model CSPred is further derived from the result aggregation of the five-individual function-based models. A diagram of the CSPred model construction is illustrated in Fig. 1.

Work flow of CSPred construction and Implementation.
Model evaluation
The performances of all five models were evaluated using a five-fold cross-validation. Precision (eq. 1), recall (eq. 2), F1-score (eq. 3), accuracy (eq. 4), and Mathews Correlation Coefficient (MCC) (eq. 5) values are calculated for each model as the evaluation matrices. For calculation of these evaluation matrices, the confusion matrices were constructed to calculate the True Positives (TP), False Positives (FP), True Negatives (TN) and False Negatives (FN). TP and TN are correctly predicted positive and negative data points, respectively. Similarly, FP and FN are incorrectly predicted positive and negative data points, respectively. From TP, TN, FP, and FN, the evaluation matrices were calculated using the following equations:
Comparison with PSI-BLAST and HMMER
The performance of each model except the subclasses of ICB is compared with PSI-BLAST42 and HMMER43. The ncbi-blast-2.5.0+ standalone software was downloaded to run PSI-BLAST locally. Similarly, HMMER 3.1b2 was installed in a Linux operating system, and the PHMMER function was used to run HMMER with the default parameters. Evaluation matrices were calculated for the identical training sets with PSI-BLAST using five-fold cross-validation. During cross-validation with PSI-BLAST and PHMMER, the training set was employed to populate the database, while the test set operated as the query. The class of each query sequence was predicted using the highest matching score with the sequences in the database. For PSI-BLAST, cross-validations were conducted using the threshold E-values of 0.01, 0.05, 0.1, 0.5, 1.0 and 5.0 with five iterations. All other parameters were kept as default. Afterwards, the sequence of the out-of-sample test sets from each functional group were predicted keeping the sequence of the corresponding training sets as databases.
Comparison with other available models
Several other models exist to predict subsets of functional groups. iAMP-2L47 and CAMPR348 are available to predict antimicrobial peptides, but are not scoped or optimized to predict cysteine stabilized peptides. We also compare the performance of our AMP model with iAMP-2L and CAMPR3. CAMPR3 offers four different classifiers to predict AMPs: Support Vector Machine (SVM), Random Forest (RF), Artificial Neural Network (ANN), and Discriminant Analysis (DA). We compared our AMP model with all classifiers offered by CAMPR3 using the out-of-sample test set and calculated precision, recall, F1-score, accuracy and MCC values. Similarly, the HemoPI45 model is dedicated to predicting hemolytic peptides, and was used to compare with our HLP model using the out-of-sample test set of HLP and calculated evaluation matrices.
Results
Evaluation of the m-NGSG-based models
ICB, AMP, ACRI, SPI and HLP represent five different functional class-based models constructed using the m-NGSG algorithm38. Each model was evaluated using precision, recall, F1-score, accuracy and MCC scores based on a five-fold cross-validation against a training set. The evaluation matrices are reported in Table 1. The training set accuracies of the five models range from 86.33% to 95.23% where AMP and ACRI rendered the lowest and highest accuracy, respectively. The models also generated F1-Scores ranging from 0.81 to 0.89 and MCC scores ranging from 0.74 to 0.90. In addition, we observed consistent performances from all three ICB subclassifiers. The training set of the NaB, KB and CaB classifiers produced 0.83, 0.82 and 0.73 MCC scores, respectively, from five-fold cross-validations. These classifiers also generated MCC scores of 0.94, 0.87, 0.69, respectively which indicates trivial overfit/underfits of the models. Figure 2 illustrates the other evaluation matrices of the ICB subcalssification models.

Performance of Ion Channel Blocker (ICB) sub-classifiers. NaB, KB and CaB represent the sodium, potassium and calcium channel blocker classifiers, respectively. CV AUC indicates the area under curve (AUC) using five-fold cross-validation; CV ACCURACY indicates the accuracy using five-fold cross-validation; CV MCC indicates the Mathews Correlation Coefficient (MCC) values using five-fold cross-validation; TEST SET ACCURACY indicates the accuracy using the out of sample test set; TEST SET MCC indicates the MCC values using the out of sample test set. These values indicate the robust performance of each classifier.
To judge the robustness of our approach, it is imperative to compare the performance of the model against established reliable available methods. PSI-BLAST and HMMER are used for generalized comparison, while other comparison groups are more specific. iAMP-2L and CAMPR3 are used to evaluate performance against AMPs, and HemoPI for HLP.
Comparison of the evaluation matrices and area under curve (AUC) with PSI-BLAST and HMMER
PSI-BLAST is a dependable and widely used algorithm to discover distantly related protein sequence using PSSM matrices42. HMMER is a Hidden Markov Model-based algorithm designed to detect remote homologs with a high sensitivity43. We compared the performance of each constructed model with PSI-BLAST and HMMER for the corresponding training sets using a five-fold cross-validation. Supplement Fig. 1 illustrates an extensive comparison among the m-NGSG based models, HMMER, and PSI-BLAST models made with different E-values. Precision, recall, F1-score, accuracy and MCC values are used to evaluate the models against PSI-BLAST. Figure 3A and Supplement Fig. 3 specifically show the comparison of the MCC values of each training set with PSI-BLAST and HMMER. Figure 3B and Supplement Fig. 3 illustrates the standard deviation of the MCC values generated from different folds using different models. The area under curve (AUC) for the five-different m-NGSG-based models were also compared with PSI-BLAST and HMMER using the corresponding training sets. The E-values yielding the best MCC values for each function-based training sets were used to run a PSI-BLAST for the comparison. Figure 4 shows the receiver operating characteristic (ROC) curves for m-NGSG-based models with a side by side area under curve (AUC) comparison among each m-NGSG-based model and the corresponding PSI-BLAST and HMMER-based models. For the five training sets, m-NGSG based models generated better AUCs compared to the corresponding PSI BLAST and HMMER based models.

The depth of performance-consistency for each model. (A) (upper panel) illustrates the comparison of MCC (Mathews Correlation Coefficients) among PSI-BLAST (E-value 0.1 and 1), m-NGSG, and HMMER. The Y-axis indicates different function-based models; the X-axis indicates the MCC values with their standard errors. Each bar plot depicts the method used to build the models. (B) (lower panel) illustrates the comparison of standard deviations of MCC (Mathews Correlation Coefficients) scores among PSI-BLAST (E-value 0.1 and 1), m-NGSG, and HMMER. The Y-axis indicates different function-based models; the X-axis indicates the standard deviations of the MCC values with their standard errors. Each bar plot depicts the method used to build the models. Here, the higher the standard deviation, the lower the performance-consistency. The m-NGSG-based models show standard deviations of MCC values lower than 0.05 for each model while HMMER and PSI-BLAST return high standard deviations on some models. Please see Supplement Figs S2 and S3 for more details.

AUC among m-NGSG, HMMER and PSI-BLAST with best scoring MCC value. The left panel indicates the receiver operating characteristics of m-NGSG-based models. The right panel indicates the comparison of AUC among m-NGSG, PSI-BLAST, and HMMER for the corresponding function-based model. The height of each bar represents the AUC for each method. m-NGSG-based models demonstrates better AUC than PSI-BLAST and HMMER.
Comparison of the evaluation matrices with PSI-BLAST and HMMER on the out-of-sample test set
Versatility of the five m-NGSG based models were tested by comparing their performance with PSI-BLAST and HMMER on the corresponding out-of-sample test set. We imported the same E-values from the ROC curve comparison to run the PSI-BLAST on the test sets. The MCC values were measured for each model and the corresponding PSI-BLAST and HMMER to achieve an appropriate comparison. Figure 5 displays comparative bar plots which illustrate the MCC values on the out-of-sample test set produces five different models and PSI-BLAST. According to Fig. 5, each of the five models shows better MCC values compared to their equivalent PSI-BLAST results while four models show better MCC value than HMMER. In the case of AMP, both the m-NGSG-based model and HMMER returns the same MCC value (0.74).

Comparison of MCC values on the out-of-sample test set with function-based model using m-NGSG, PSI-BLAST, or HMMER. While the MCC scores of HMMER are comparable for the AMP and ACRI test tests, the MCC score for ICB, SPI and HLP are noticeably lower compared to the m-NGSG-based models.
Comparison of AMP and Hemolytic peptide prediction models with other currently available models
Along with PSI-BLAST and HMMER, we used the iAMP-2L47 and CAMPR348 models to predict antimicrobial peptides (AMP), and the HemoPI45 algorithm to predict hemolytic peptides. While it is important to note that none of these models are dedicated to the identification of only cystine- stabilized peptides, their performance parameters should generalize to their prediction. We compared performances of iAMP-L2 and CAMPR3 with our m-NGSG-based AMP model and HemoPI with the m-NGSG-based HLP model using the corresponding out-of-sample test sets. Figure 6 shows the comparative precision, recall, accuracy and MCC values among different models. Among the other available models, CAMP-ANN showed the highest precision score 0.43 or 43% while the precision score produced by m-NGSG-based AMP model was 0.85. CAMP-SVM showed a slightly better recall score than m-NGSG, 0.83 and 0.81, respectively. Overall, the best accuracy score was generated by iAMP-2L (0.54), but it was far less than the accuracy score produced by m-NGSG, which was 0.75. Finally, the highest MCC score was generated by CAMP-ANN (0.13) which was also well below the MCC score of m-NGSG (0.74) (see Fig. 6). Similarly, Supplement Fig. 4 illustrates the comparison on the out-of-sample HLP test set among Hemo PI, PSI-BLAST and m-NGSG. Here, precision, recall, accuracy and MCC scores of HemoPI are 0.55 (55%), 0.67 (66%), 0.66 (66%) and 0.31, respectively. These are lower than corresponding scores of m-NGSG-based HLP.

Precision, Recall, Accuracy and MCC. The precision, recall, accuracy and MCC values obtained applying iAMP-2L, CAMP SVM, CAMP RF, CAMP ANN, CAMP DA, PSI-BLAST E-value 0.1 and AMP (m-NGSG-based AMP model) on the out-of-sample AMP test set. (A) illustrates that precision values of iAMP-2L and CAMP models are considerably lower than the m-NGSG based model. (B) illustrates that the recall values of the CAMP models are comparable to the m-NGSG-based model while iAMP-2L demonstrates a noticeably lower recall value. (C,D) shows considerably low MCC and accuracy values displayed by iAMP-2L and CAMP models compared to PSI-BLAST and m-NGSG.
Discussion
In this study, we constructed five different functional classifiers of cystine -stabilized peptides and combined them to build the CSPred model which predicts the probability of the selected five functional characteristics of query peptide sequences. After building a model, the most important step is to assess its performance using a k-fold cross-validation and out-of-sample test sets. We have performed this step carefully using a five-fold cross-validation and an out-of-sample test set for each of the five models. Table 1 shows a comprehensive comparison among evaluation matrices for each model. The cross-validation accuracy ranged from ~86% to ~95% for the models. The accuracies on the out-of-sample test sets were concordant to the cross-validation accuracies. No big difference was detected between the accuracies for the models except the ICB model where the difference between the test and training set was ~5%. However, the increase of accuracy on the test set indicates versatility of the ICB model. The other evaluation matrices such as F1-score and MCC were also quite consistent between the training and test set (see Table 1). The comparative analysis between the evaluation matrices explains the adaptability of each different function-based model.
The protein sequences collected from the Uniprot Knowledgebase were further filtered using the CD-hit46 software to reduce the homoscedasticity of the model by reducing the redundancy of the similar sequences. Although this operation reduced the amount of available training set, particularly of the ICB subclasses, that helped models to be kept optimally unbiased. The fasta sequences of each protein of the training and test sets from Uniprot Knowledgebase may contain some other domains along with the functional domains. Despite the presence of these noises (the subsequences from other domains) in the datapoints (here the data points are the primary sequences of the proteins), CSPred showed a robust performance in the cross-validation and out-of-sample test-set classification. This outcome indicates the strength of m-NGSG algorithm to classify data points with noise. From a previous study, we demonstrated that m-NGSG is capable to differentiate noises in datasets during a classification task38.
The ultimate success and novelty of a machine learning-based model depends on its superiority over other concurrent algorithms. PSI-BLAST and HMMER are successful and widely accepted algorithms to discover distantly related protein chains. Therefore, we compared the performance of each five function-based model with PSI-BLAST and HMMER using the corresponding training and out-of-sample test sets. One complexity and disadvantage to working with PSI-BLAST is choosing the optimal E-value; it is challenging to select an E-value that will give the best results. Supplement Fig. 1 shows a clear superiority of m-NGSG-based methods over the equivalent PSI-BLAST with different E-values and HMMER except better recall values obtained by HMMER compared to m-NGSG based models. That explains only a better sensitivity of HMMER than m-NGSG models but not the overall performance. The MCC score is chosen over an accuracy score for further comparison because MCC is more robust and reflects the sensitivity, specificity, precision and false negative rate while accuracy only reflects the average of sensitivity and specificity of a model49. Similar to the training set, m-NGSG-based models showed better MCC values on the out-of-sample test sets compared to the corresponding HMMER and PSI-BLAST with the optimized E-values, Fig. 5. This result demonstrates consistently better performance over HMMER and PSI-BLAST and unbiased behavior of the m-NGSG-based models. In addition to PSI-BLAST and HMMER, we compared the m-NGSG-based model with other available function specific prediction models. There are two available models to predict antimicrobial peptides: iAMP-2Land CAMPR3. CAMPR3 also has four different classifiers to perform the antimicrobial peptide prediction which are SVM, RF, ANN, and DA. We evaluated all the models by computing the four evaluation matrices (described in Fig. 5) on the out-of-sample test sets. The performance of iAMP-2L and CAMPR3 were significantly low compared to PSI-BLAST and m-NGSG-based AMP model. The reason is possibly the training sets are not optimized to predict the cystine-stabilized AMPs. The similar results are found when we compared HemoPI (see Supplement Fig. 4) with the m-NGSG-based HLP model. These results indicated that the m-NGSG-based models are superior to any other concurrent algorithms to classify functions of cystine-stabilized peptides.
Several cystine-stabilized peptides have already been licensed for clinical or agricultural use. This small fraction demonstrates the potential for new applications hidden among the thousands of undiscovered cystine-stabilized peptide sequences in genomes across many taxa. A voltage-gated calcium channel blocker cystine-stabilized peptide (Hv1a) from spider venom50 is now the primary product of Vestaron, Inc., with commercial production in E. coli for broad-scale application on crops plants as an eco-friendly insecticide that degrades within two weeks after application. This same spider peptide has been fused to a targeting moiety by another group to specifically target aphids as a transgene in plants51. In our own lab (CMK), antimicrobial cystine-stabilized peptides have been targeted for specific toxicity towards individual pathogenic bacterial species, with nontarget toxicity greatly reduced (Islam et al., unpublished data). This has implications for antibiotic treatment without the disruption of the native microbiome. Thus, a diverse array of different cystine-stabilized peptides has realized commercial application.
The model approach outlined here has the potential to greatly impact the discovery of functionally active peptides. A typical pipeline might involve finding top candidate peptide sequences using genome databases and one or more of the prediction models, followed by peptide production of the top candidates in an appropriate heterologous expression system and wet lab evaluation of the peptides. Using an antimicrobial peptide screen as an example, our AMP algorithm would be used to screen a dataset of peptides (see the Availability section below). These candidate sequences would then be screened with the HLP (hemolytic protein) algorithm to eliminate peptides that might be toxic to human cells. From the pool of remaining candidates, synthesized DNA sequences would be cloned into an E. coli expression vector alongside a stabilizing fusion partner such as SUMO52 and a purification tag such as 6x His-Tag. The peptides would be expressed and purified, and then confirmed for stability, toxicity against the target microbe, and lack of toxicity against human cells. Top candidates would be available for pharmaceutical production systems or to be used as transgenes in the organism to be protected by the antimicrobial peptide53. An advantage with using peptides over typical small molecule drugs is the relative ease with which an appropriate modifying peptide can be found and genetically fused to the effector peptide sequence, for example an antimicrobial peptide targeted to a specific pathogenic bacterium with a targeting peptide54. It should be noted that the metadata associated with the peptide candidate sequence can also be used to help select peptides with the desired action. For example, ion channel blockers with oral toxicity might be more commonly found in plants and algae, where an ion channel blocker might serve as an oral insecticide, than in spider or snake venom, where the ion channel blocker would most likely have toxicity only by injection.
Availability
CSPred is an open source collaborative initiative available in the bitbucket repository (https://bitbucket.org/sm_islam/cystine-stabilized-proteins/src). It is also publicly available as a free web application at watson.ecs.baylor.edu/cspred. The web server provides an accessibility to the CSPred, and a user does not need computational experience to use the model. Posting the web address (watson.ecs.baylor.edu/cspred) on a web browser will take the user to the CSPred webpage. There, the user needs to upload the fasta file of the unknown protein sequences and click the submit button. That action will trigger the prediction process. The result is divided into six columns. The first column is the protein ID labels of the fasta sequences. The second, third, fourth, fifth, and sixth columns display the probability values of being an ICB, AMP, ACRI, SRI, and HLP, respectively, for each sequence submitted. Thus, the web interface provides a simple avenue to categorize submitted protein sequences according to these five functional characteristics, and may use a high-throughput batch-style input. Supplementary Figs 5–7 demonstrate the pipeline to use the CSPred web application. The sub classifiers of ICB are not included in CSPred. However, all the training and test datasets are provided as a supplement enabling users to make their own models or reproduce the same models using the m-NGSG framework that is available at watson.ecs.baylor.edu/ngsg.
References
King, G. F. & Hardy, M. C. Spider-Venom Peptides: Structure, Pharmacology, and Potential for Control of Insect Pests. Annu. Rev. Entomol. 58, 475–496 (2013).
Chan, Y. S. et al. Snake venom toxins: toxicity and medicinal applications. Appl. Microbiol. Biotechnol. 100, 6165–6181 (2016).
Ortiz, E., Gurrola, G. B., Schwartz, E. F. & Possani, L. D. Scorpion venom components as potential candidates for drug development. Toxicon 93, 125–135 (2015).
Frazão, B., Vasconcelos, V. & Antunes, A. Sea Anemone (Cnidaria, Anthozoa, Actiniaria) Toxins: An Overview. Mar. Drugs 10, 1812–1851 (2012).
Akondi, K. B. et al. Discovery, Synthesis, and Structure–Activity Relationships of Conotoxins. Chem. Rev. 114, 5815–5847 (2014).
Nguyen, L. T., Haney, E. F. & Vogel, H. J. The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol. 29, 464–472 (2011).
Munasinghe, N. & Christie, M. Conotoxins That Could Provide Analgesia through Voltage Gated Sodium Channel Inhibition. Toxins 7, 5386–5407 (2015).
Bourinet, E. & Zamponi, G. W. Block of voltage-gated calcium channels by peptide toxins. Neuropharmacology, https://doi.org/10.1016/j.neuropharm.2016.10.016 (2016).
Norton, R. S. & Chandy, K. G. Venom-derived peptide inhibitors of voltage-gated potassium channels. Neuropharmacology. https://doi.org/10.1016/j.neuropharm.2017.07.002 (2017).
Dutertre, S., Nicke, A. & Tsetlin, V. I. Nicotinic acetylcholine receptor inhibitors derived from snake and snail venoms. Neuropharmacology, https://doi.org/10.1016/j.neuropharm.2017.06.011 (2017).
Mourão, C. & Schwartz, E. Protease Inhibitors from Marine Venomous Animals and Their Counterparts in Terrestrial Venomous Animals. Mar. Drugs 11, 2069–2112 (2013).
Zhirnov, O. P., Klenk, H. D. & Wright, P. F. Aprotinin and similar protease inhibitors as drugs against influenza. Antiviral Res. 92, 27–36 (2011).
Layer, P. & Stanghellini, V. Review article: linaclotide for the management of irritable bowel syndrome with constipation. Aliment. Pharmacol. Ther. 39, 371–384 (2014).
Cohen-Inbar, O. & Zaaroor, M. Glioblastoma multiforme targeted therapy: The Chlorotoxin story. J. Clin. Neurosci. 33, 52–58 (2016).
Mobli, M., Undheim, E. A. B. & Rash, L. D. Modulation of Ion Channels by Cysteine-Rich Peptides: From Sequence to Structure. Adv. Pharmacol. San Diego Calif 79, 199–223 (2017).
Silverstein, K. A. T. et al. Small cysteine-rich peptides resembling antimicrobial peptides have been under-predicted in plants: Under-predicted cysteine-rich peptides in plants. Plant J. 51, 262–280 (2007).
Kuzmenkov, A. I., Grishin, E. V. & Vassilevski, A. A. Diversity of Potassium Channel Ligands: Focus on ScorpionToxins. Biochem. Mosc. 80, 1764–1799 (2015).
Cheek, S., Krishna, S. S. & Grishin, N. V. Structural Classification of Small, Disulfide-rich Protein Domains. J. Mol. Biol. 359, 215–237 (2006).
Santibáñez-López, C. E. & Possani, L. D. Overview of the Knottin scorpion toxin-like peptides in scorpion venoms: Insights on their classification and evolution. Toxicon 107, 317–326 (2015).
Possani, L. D., Becerril, B., Delepierre, M. & Tytgat, J. Scorpion toxins specific for Na+-channels. Eur. J. Biochem. 264, 287–300 (1999).
Adams, M. E. Agatoxins: ion channel specific toxins from the american funnel web spider, Agelenopsis aperta. Toxicon 43, 509–525 (2004).
Olivera, B. M. et al. Peptide neurotoxins from fish-hunting cone snails. Science 230, 1338–1343 (1985).
Islam, S. M. A., Sajed, T., Kearney, C. M. & Baker, E. J. PredSTP: a highly accurate SVM based model to predict sequential cystine stabilized peptides. BMC Bioinformatics 16, 210 (2015).
Craik, D. J., Simonsen, S. & Daly, N. L. The cyclotides: novel macrocyclic peptides as scaffolds in drug design. Curr. Opin. Drug Discov. Devel. 5, 251–260 (2002).
Gracy, J. et al. KNOTTIN: the knottin or inhibitor cystine knot scaffold in 2007. Nucleic Acids Res. 36, D314–D319 (2007).
Kedarisetti, P., Mizianty, M. J., Kaas, Q. & Craik, D. J. & Kurgan, L. Prediction and characterization of cyclic proteins from sequences in three domains of life. Biochim. Biophys. Acta BBA - Proteins Proteomics 1844, 181–190 (2014).
Gelly, J.-C. The KNOTTIN website and database: a new information system dedicated to the knottin scaffold. Nucleic Acids Res. 32, 156D–159 (2004).
Ding, H. et al. iCTX-Type: A Sequence-Based Predictor for Identifying the Types of Conotoxins in TargetingIon Channels. BioMed Res. Int. 2014, 1–10 (2014).
Fan, Y.-X., Song, J., Kong, X. & Shen, H.-B. PredCSF: An Integrated Feature-Based Approach for Predicting Conotoxin Superfamily. Protein Pept. Lett. 18, 261–267 (2011).
Sharma, A. et al. Computational approach for designing tumor homing peptides. Sci. Rep. 3, 1607 (2013).
Simeon, S. et al. osFP: a web server for predicting the oligomeric states of fluorescent proteins. J. Cheminformatics 8, 72 (2016).
Du, P., Cao, S. & Li, Y. SubChlo: Predicting protein subchloroplast locations with pseudo-amino acid composition and the evidence-theoretic K-nearest neighbor (ET-KNN) algorithm. J. Theor. Biol. 261, 330–335 (2009).
Zhang, G.-Y. & Fang, B.-S. Predicting the cofactors of oxidoreductases based on amino acid composition distribution and Chou’s amphiphilic pseudo-amino acid composition. J. Theor. Biol. 253, 310–315 (2008).
Xia, J.-F., Han, K. & Huang, D.-S. Sequence-Based Prediction of Protein-Protein Interactions by Means of Rotation Forest and Autocorrelation Descriptor. Protein Pept. Lett. 17, 137–145 (2010).
Dubchak, I., Muchnik, I., Holbrook, S. R. & Kim, S. H. Prediction of protein folding class using global description of amino acid sequence. Proc. Natl. Acad. Sci. USA 92, 8700–8704 (1995).
Chang, D., Syu, Y.-T. & Lin, P.-C. Predicting the protein-protein interactions using primary structures with predicted protein surface. BMC Bioinformatics 11, S3 (2010).
Shen, H.-B. & Chou, K.-C. PseAAC: A flexible web server for generating various kinds of protein pseudo amino acid composition. Anal. Biochem. 373, 386–388 (2008).
Islam, S. M. A., Heil, B. J., Kearney, C. M. & Baker, E. J. Protein classification using modified n-grams and skip-grams. Bioinformatics https://doi.org/10.1093/bioinformatics/btx823 (2017)
Keˇselj, V., Peng, F., Cercone, N. & Thomas, C. N-gram-based author profiles for authorship attribution. In Pacific Association for Computational Linguistics (2003).
Guthrie, D., Allison, B., Liu, W., Guthrie, L. & Wilks, Y. A closer look at skip-gram modelling. In Proceedings of the 5th international Conference on Language Resources and Evaluation (LREC- 2006 ), 1–4 (2006).
Islam, S. A., Kearney, C. M., Choudhury, A. & Baker, E. J. Protein Classification using Modified N-Gram and Skip-Gram Models: Extended Abstract. In 586–586, https://doi.org/10.1145/3107411.3108193 (ACM Press, 2017).
Altschul, S. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Finn, R. D., Clements, J. & Eddy, S. R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37 (2011).
Boeckmann, B. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31, 365–370 (2003).
Chaudhary, K. et al. A Web Server and Mobile App for Computing Hemolytic Potency of Peptides. Sci. Rep. 6 (2016).
Huang, Y., Niu, B., Gao, Y., Fu, L. & Li, W. CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics 26, 680–682 (2010).
Xiao, X., Wang, P., Lin, W.-Z., Jia, J.-H. & Chou, K.-C. iAMP-2L: A two-level multi-label classifier for identifying antimicrobial peptides and their functional types. Anal. Biochem. 436, 168–177 (2013).
Waghu, F. H., Barai, R. S., Gurung, P. & Idicula-Thomas, S. CAMP R3: a database on sequences, structures and signatures of antimicrobial peptides: Table 1. Nucleic Acids Res. 44, D1094–D1097 (2016).
Powers, D. M. Evaluation: from precision, recall and F-measure to ROC, informedness, markedness and correlation. J. Mach. Learn. Technol. 2, 37–63 (2011).
Herzig, V. & King, G. The Cystine Knot Is Responsible for the Exceptional Stability of the Insecticidal Spider Toxin ω-Hexatoxin-Hv1a. Toxins 7, 4366–4380 (2015).
Bonning, B. C. et al. Toxin delivery by the coat protein of an aphid-vectored plant virus provides plant resistance to aphids. Nat. Biotechnol. 32, 102–105 (2013).
Butt, T. R., Edavettal, S. C., Hall, J. P. & Mattern, M. R. SUMO fusion technology for difficult-to-express proteins. Protein Expr. Purif. 43, 1–9 (2005).
Peschen, D., Li, H.-P., Fischer, R., Kreuzaler, F. & Liao, Y.-C. Fusion proteins comprising a Fusarium-specific antibody linked to antifungal peptides protect plants against a fungal pathogen. Nat. Biotechnol. 22, 732–738 (2004).
Eckert, R. et al. Targeted Killing of Streptococcus mutans by a Pheromone-Guided ‘Smart’ Antimicrobial Peptide. Antimicrob. Agents Chemother. 50, 3651–3657 (2006).
Acknowledgements
We acknowledge Benjamin J. Heil for making our web-application publicly available through http://watson.ecs.baylor.edu/cspred.
Author information
Authors and Affiliations
Contributions
S.M.A.I. and C.M.K. envisioned the proposal while S.M.A.I. and E.J.B. outlined the project plan, S.M.A.I. prepared the data and conducted the analysis. S.M.A.I., E.J.B. and C.M.K. generated the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Islam, S.M.A., Kearney, C.M. & Baker, E.J. Assigning biological function using hidden signatures in cystine-stabilized peptide sequences. Sci Rep 8, 9049 (2018). https://doi.org/10.1038/s41598-018-27177-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-27177-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
