
Abstract
Previously, we introduced a noninvasive prenatal testing (NIPT) protocol for diagnosing compound heterozygous autosomal recessive point mutations via maternal plasma DNA and simulated control genomic DNA sampling based on fetal DNA fraction. In our present study, we have improved our NIPT protocol to make it possible to diagnose homozygous autosomal recessive point mutations without the need to acquire fetal DNA fraction. Moreover, chi-squared test and empirical statistical range based on the proportion of mutant allele reads among the total reads served as the gatekeeping method. If this method yielded inconclusive results, then the Bayesian method was performed; final conclusion was drawn from the results of both methods. This protocol was applied to three families co-segregating congenital sensorineural hearing loss with monogenic homozygous mutations in prevalent deafness genes. This protocol successfully predicted the fetal genotypes from all families without the information about fetal DNA fraction using one-step dPCR reactions at least for these three families. Furthermore, we suspect that confirmatory diagnosis under this protocol is possible, not only by using picodroplet dPCR, but also by using the more readily available chip-based dPCR, making our NIPT protocol more useful in the diagnosis of autosomal recessive point mutations in the future.
Similar content being viewed by others


Introduction
Conventional methods for prenatal diagnosis have been amniocentesis and chorionic villus sampling, which carry a 1% risk of miscarriage1,2,3. Recently, noninvasive prenatal testing (NIPT) has been gaining popularity, as it only requires maternal peripheral blood4,5. NIPT is recommended as a primary trisomy screening test to all pregnant women6,7. To date, several methods of NIPT have been developed, according to various needs of patients5,8,9,10,11,12. With the development of various methods of NIPT, its overall convenience and accuracy have also been improving. In the previous study, we developed a novel protocol of NIPT applicable to autosomal recessive (AR) monogenic disease in predicting the genotype of a fetus (second baby) based on the first baby’s known genotype, using a higher-resolution picodroplet digital PCR (dPCR)10.
Although our previous protocol showed successful prediction, it required calculating the fraction of fetal DNA (the second baby’s fetal DNA) among the maternal plasma DNA (mpDNA) to produce simulated control samples and perform statistical analyses. In compound heterozygous genotypes, a fraction of fetal DNA can, if not always, be calculated by measuring the fraction of paternal mutation in mpDNA. However, a paternal allele-specific single nucleotide polymorphism (SNP), which does not exist in the maternal allele, should additionally be searched in homozygotes. However, this process is a time and effort-intensive task, which may not always be feasible. This process may be particularly difficult in east Asian populations where there are a lot of prevalent founder alleles, and therefore, homozygous genotypes for autosomal recessive disorders.
In our present study recruiting families segregating such homozygous AR deafness variants, we developed a novel, convenient NIPT protocol, which does not require either searching for a paternal allele specific SNP nor reconstruction of haplotypes. This protocol utilized both chi-squared test and Bayesian method, allowing for prenatal diagnosis without the calculation of fetal DNA fraction. This protocol successfully predicted fetal genotypes.
Methods
Subjects and Ethical Considerations
The institutional review boards of both Seoul National University Hospital (IRBY-H-0905–041–281) and Seoul National University Bundang Hospital (IRB-B-1007-105-402 and IRB-B-1508-312-304) approved all procedures used in this study. All subjects provided written informed consent. All methods were performed in accordance with the relevant guidelines and regulations. Three families with the first baby already confirmed to have SNHL due to AR mutations of known deafness genes and an unborn baby (fetus) were included in this study (Supplementary Figure S1). Causative mutation of SNHL from SH197 and SB275 families has previously been documented as GJB2 c.235delC homozygote through bioinformatic analysis as described13,14. The causative mutation of SNHL from SH162 family has previously been documented as SLC26A4 c.IVS7-2A > G. NIPT was performed for genotyping of the causative deafness gene from the unborn baby of each family.
Plasma DNA extraction protocol
Blood samples were collected from all pregnant mothers. At the time of this procedure, the maternal gestational ages of SH197, SB275, and SH162 families were 16, 27 and 16 weeks, respectively. The maternal body weights were 48, 55, and 51 kg, respectively. Plasma DNA was extracted as described10; 1.5 ml plasma was added to a microcentrifuge tube and centrifuged for 10 min at 2000 g using MACHEREY-NAGEL, NucleoSpin Plasma XS (Germany) kit. Circulating DNA was extracted following the manufacturer’s guidelines.
gDNA preparation
gDNA was prepared as described15. The control samples were made with gDNA previously obtained from the mother, and first baby in each family. To make the size of gDNA close to the size of plasma DNA, gDNA was fragmented using Covaris S220 (Covaris, MA, USA). The fragment size was confirmed as 150 base pair length by Bioanalyzer High Sensitivity DNA Chips (Agilent Technologies, CA, USA). DNA concentration was measured using a fluorescence assay of Picogreen (Invitrogen, Grand Island, NY, USA).
Picodroplet digital PCR (dPCR) methods
Picodroplet dPCR was performed using RainDrop Digital PCR System (RainDance Technologies Inc., Billerica, MA, USA), as previously described10. PCR reaction mixes consisted of primers and probes (Supplementary Table S1 and Supplementary Figure S2) along with 12.5 µl TaqMan Genotyping Master Mix (Life Technologies), 1.25 µl Drop Stabilizer (RainDance Technologies), DNase/RNase-free sterile water, and template DNA (either the minimum 2 ng of plasma DNA or 30 ng of the fragmented gDNA), making up a total reaction volume of 25 µl. Each 25 µl PCR mix was emulsified into 5 pl droplet volumes using RainDrop Source instrument (RainDance Technologies). A single molecule of DNA was partitioned into approximately 5 million droplets. Then, PCR mixes were placed in a C1000 with deep-well (Bio-Rad) and amplified, according to the protocol (Supplementary Table S2). The fluorescent intensity of each droplet for two fluorophores (FAM and VIC) was identified using RainDrop Sense instrument (RainDance Technologies). The data from cluster plots were analyzed using RainDrop Analyst data analysis software, as previously described10.
Chip-based dPCR methods
QX200 Droplet digital PCR System (Bio-Rad Laboratories, Inc., Hercules, CA USA) was used to assess nanodroplet dPCR. In a pre–polymerase chain reaction environment, PCR reaction mixes were combined with primers and probes (Supplementary Table S3) along with 10 μl TaqMan Genotyping Master Mix (Life Technologies), DNase/RNase-free sterile water, and template DNA (either the minimum 700 pg of plasma DNA or 30 ng of the fragmented gDNA), which made up a total reaction volume of 20 μl. A probe was validated (Supplementary Figure S2). Droplets were then generated using DG8 droplet generator cartridges by mixing the aqueous phase with 70 mL of droplet generation oil (Bio-Rad Laboratories). Each 20 μl PCR mix was emulsified into about 1 nl droplet volumes, partitioning a few molecules of DNA into approximately 20 thousand droplets. Droplets were transferred to a 96-well PCR plate and then sealed using the PX1 PCR plate sealer (Bio-Rad Laboratories) for 5 seconds at 180 degree before thermal cycling. The PCR plate was placed in a C1000 Touch thermal cycler with deep-well (Bio-Rad Laboratories) to be amplified, following the protocol outlined in Supplementary Table S4. After thermal cycling, the PCR plate that included droplets was loaded onto QX200 Droplet Reader instrument (Bio-Rad laboratories), identifying the fluorescent intensity of each droplet for two fluorophores (FAM and VIC) simultaneously using Multi-pixel photon counter. This detector reads the droplets to determine the ones that contain a target gene (+) and the ones that do not (−), and plots the fluorescence droplet by droplet. The fraction of positive droplets in a sample determines the concentration of target in copies/μl.
Noninvasive prenatal testing (NIPT) protocol
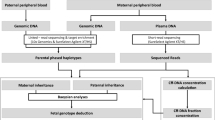
We used both the gatekeeping method and Bayesian method to predict the genotype status of the fetus. The gatekeeping method was composed of chi-squared test and comparison of a proportion of the mutant allele reads among the total reads (θ) against the empirical range of the proportion from a heterozygous genotype, and it facilitated fast decision. The Bayesian method was based on prior information and data distribution, and robust decision was expected (Fig. 1).

Protocol of noninvasive prenatal diagnosis. *The cutoff value of Bayes factor is 0.7 for picodroplet dPCR and 1 for chip-based dPCR. BF, Bayes factor.
In the gatekeeping method, chi-squared test of the plasma sample against the simulated heterozygous control was initially conducted. Maternal gDNA was used as the heterozygous control sample. When dPCR was performed on several samples from one subject, the observed mutant allele proportion was calculated by summing the mutant reads with the total droplet reads in each sample. The heterozygous control was expected to have a wild: mutant allele reads ratio of 50:50; rejecting the null hypothesis of chi-squared test indicated that the plasma sample is more likely to be either wild or mutant homozygote. To rule out any false negative results from chi-squared test due to errors and a small number of reads from dPCR, value of θ less than 48% or greater than 52% was regarded as a wild or mutant homozygous. This empirical range of the heterozygous genotype was obtained from a hypothetical range of the fetus DNA fraction (2.5–11%) and a statistical simulation assuming a normal experimental error rate of 1.0% (standard deviation 0.025%) and 1.5% (standard deviation 0.5%) for picodroplet and chip-based dPCR, respectively. The mean and standard deviation of error were obtained from simulated control with known a fetus DNA fraction. As the effect caused by the difference in the error rate between picodroplet and chip-based dPCR was not significant, the same range was applicable for both methods. Consequently, if a P-value from chi-squared test was less than 0.05 and the value of θ was less than 48% or more than 52%, the result was considered conclusive in the gatekeeping method. If not, the diagnosis was made considering a Bayesian method.
For the Bayesian method, a single droplet read was assumed to be generated from the Bernoulli trial. A mutant allele read was considered as a success. θ was necessarily affected by the amount of fetus DNA in the total plasma DNA (p). We introduced three genotype statuses of the fetus (G); a wildtype (G = 1) with θ = (1-p)/2, a single heterozygote (G = 2) with θ = 1/2, and a homozygous mutant (G = 3) with θ = (1 + p)/2. Prior information of three genotypes denoted as \(\pi (G)\) was 0.25 (G = 1), 0.5 (G = 2), and 0.25 (G = 3), respectively. The priors imply the genotype is determined by the Mendelian inheritance. Prior information of p, \(\pi (p)\), was assumed to follow the Beta distribution with α = 4 and ß = 100. It means that the prior mean of the amount of fetus DNA contained in the total plasma DNA was set as 4%. The observed mutant allele proportion of the ith sample was defined as mutant reads (x i ) out of the total droplet reads (n i ). When dPCR was performed on several samples from one subject, the observed mutant allele proportion was calculated by summing x i and n i in each sample. The posterior distribution of G can be calculated based on the experimental values as follows; \(\pi (p,\,G|x)\propto \,(\begin{array}{c}N\\ x\end{array})\,{(\theta (G,p))}^{x}{(1-\theta (G,p))}^{\{N-x\}}\times \,\pi (G)\times \pi (p)\). We conducted the Bayesian hypothesis test using the Bayes factor, which is a ratio of two competing hypotheses; a higher value indicates the favorability of the alternative hypothesis over the null hypothesis. In our study, the null hypothesis was that the fetus was either a wildtype or a single heterozygote, whereas the alternative hypothesis was that the fetus was a homozygous mutant. If the Bayes factor of a homozygous mutant (G = 3) over a wildtype or a single heterozygote (G = 1 or 2) was greater than the cutoff value, the alternative hypothesis – homozygous mutant – was accepted.
To determine the cutoff value for the Bayes factor, we simulated both numerical and experimental studies. We made three types of biological control samples mimicking the wildtype, homozygous mutant, and heterozygote in each family. The homozygous mutant and wildtype control samples indicated the maternal gDNA with and without a homozygous mutation in a proportion of 5.6%, respectively. We used the first baby’s gDNA as the gDNA components with homozygous mutation. Maternal gDNA was used as the heterozygote control sample. Several datasets were computationally simulated based on the same assumption for the gatekeeping method; the error rate followed normal a distribution with a mean of 1.0% and a standard deviation of 0.025% for picodroplet dPCR and a mean of 1.5% and a standard deviation of 0.5% for chip-based dPCR; the various fetus DNA fraction ratios ranged from 2.5% to 11%. The cutoff value of Bayes factor was determined to be 0.7 for picodroplet dPCR and 1 for chip-based dPCR, since this cutoff provided 100% specificity and 90% sensitivity in our simulated data when the number of read copy was at least 1,000.
We developed a diagnostic protocol using both the gatekeeping method and Bayesian method. The former was first performed to obtain the quick diagnosis, if applicable. If the gatekeeping method yielded inconclusive results, the Bayesian method was conducted. Finally, results from both methods were considered (Fig. 1). Statistical analyses were performed using R 3.4.1.
Confirmation of fetal genotype after birth
In the SH197 family, peripheral blood samples were obtained from the second baby after birth. Allele-specific PCR-based universal array (CapitalBio Corporation, Beijing, China) was applied to targeted the gene11. In SB275 and SH162 families, Sanger sequencing of gDNA from buccal mucosa of the second baby was performed to sequence the target residue after birth. The predicted fetal genotypes were checked against these results.
Results
Prediction of fetal genotypes by our NIPT protocol
In the SH197 and SB275 families, father and mother were single heterozygous carriers of GJB2 c.235delC, and the first babies were homozygotes of c.235delC in GJB2. In the SH162 family, father and mother were single heterozygous carriers of SLC26A4 IVS7-2A > G, and the first babies were homozygotes of IVS7-2A > G in SLC26A4 (Supplementary Figure S1).
In the SH197 family, a chi-squared test was conducted and θ of plasma sample was significantly lower than that of the heterozygous control (P = 0.001). θ of plasma sample (344/786 (43.77%)) was less than 48%. The fetus was diagnosed as a wildtype genotype (Tables 1 and 2 and Fig. 2
).

Two-dimensional histogram of the mutation (GJB2 c.235delC) in wildtype (A), heterozygote (B,C) and homozygote (D,E) controls and maternal plasma DNA (F,G) of SH197 family.
In the SB275 family, a chi-squared test was conducted and θ of plasma sample was significantly higher than that of the heterozygous control (P = 0.018). θ of plasma sample (1,068/2,007 (53.21%)) was more than 52%. The fetus was diagnosed as homozygous genotype (Tables 1 and 2 and Supplementary Figure S3). The prenatal diagnosis was obtained from the gatekeeping method, but the Bayesian method was conducted to verify the results from the gatekeeping method. The two plasma samples were tested, and the sum of mutant reads and total droplet reads were 1,068 and 2,007, respectively, resulting in a Bayes factor of 14.885. The fetus was diagnosed as homozygous genotype consistent with the result of the gatekeeping method (Tables 1 and 2 and Supplementary Figure S3).
In the SH162 family, we conducted several experiments using picodroplet and chip-based dPCR. According to our first result from picodroplet dPCR, θ of plasma sample showed no significant difference when compared with that of the heterozygous control (P = 0.379), although θ of plasma sample (128/242 (52.89%)) was more than 52%. As the number of read copy was less than 1,000, the Bayesian method was not conducted. The fetus was diagnosed as a homozygous genotype, without certainty (Tables 1 and 2 and Supplementary Figure S4). As this result was inconclusive, we conducted dPCR once more using picodroplet dPCR. According to our second result from picodroplet dPCR, θ of plasma sample showed no significant difference when compared with that of the heterozygous control (P = 0.411), although θ of plasma sample (616/1160 (53.10%)) was more than 52%. The Bayesian method was conducted, resulting in a Bayes factor of 2.889. The fetus was diagnosed as a homozygous genotype (Tables 1 and 2 and Supplementary Figure S5).
As for the chip-based dPCR, our first result showed that there was no significant difference of θ between the plasma sample and the heterozygous control (P = 0.389), although θ of plasma sample (135/257 (52.53%)) was more than 52%. As the number of read copy was less than 1,000, the Bayesian method was not conducted. The fetus was diagnosed as likely having a homozygous genotype (Tables 1 and 2 and Supplementary Figure S6). Given the inconclusive result, we conducted dPCR once more using chip-based dPCR. According to our second result from chip-based dPCR, θ of plasma sample was significantly higher than that of the heterozygous control (P = 0.018); θ of plasma sample (139/246 (56.50%)) was more than 52%. The fetus was diagnosed as homozygous genotype (Tables 1 and 2 and Supplementary Figure S7). The diagnosis using chip-based dPCR was consistent with using picodroplet dPCR.
Genetic study for confirmation of fetal genotype
In the SH197 family, allele-specific PCR-based universal array from the second baby confirmed a wildtype of GJB2 c.235delC16. In the SB275 and SH162 families, Sanger sequencing from the second baby confirmed a homozygous mutant of GJB2 c.235delC and SLC26A4 IVS7-2A > G, respectively (Fig. 3). The prenatal diagnosis for the second babies in all families was correct.

Genetic study for confirmation of fetal genotype. (A) Sanger sequencing traces of the second-born baby of SB275 family: GJB2 c.235delC homozygote. (B) Sanger sequencing traces of the second-born baby of SH162 family: SLC26A4 IVS7-2A > G.
Discussion
After discovering the cell-free fetal DNA (cffDNA) in the peripheral blood of pregnant women17, there has been tremendous development in the prenatal diagnosis using cffDNA8,12,18. The first NIPT using cffDNA was performed to determine fetal sex17 and RhD status19,20. The application of NIPT was extended from aneuploidies, such as trisomy 21, to monogenic diseases8,10,18,21,22. The expansion of NIPT applicability has been attributed to the improvement of technology and strategy of NIPT. The representative techniques of NIPT are massive parallel sequencing (MPS)23 and dPCR8,12.
NIPT using targeted MPS technology has also been performed by the reconstruction of fetal haplotypes, which required sequencing numerous single nucleotide polymorphisms (SNPs) around the residue of interest24,25,26,27,28. Recombination of fetal alleles has often made NIPT more complicated. Recently, it has been shown that the reconstruction of only the paired end allelic reads suffice for NIPT4,5. However, it is not possible to make the whole process of MPS technology for NIPT any simpler or shorter, regardless of the frequency of target mutation. The second NIPT technique using dPCR makes it possible to genotype the residue of interest directly. It may relatively be simpler and more straightforward than NIPT using MPS. Once we are well equipped with probes and primers for dPCR, with reaction conditions that have been well calibrated for certain founder mutations, then subsequent NIPT testing that targets a specific mutation may be easier and less time consuming to perform. Therefore, this technology is especially convenient for testing highly prevalent AR variants. Moreover, in our previous study, we showed that the accuracy of NIPT was further improved by utilizing picodroplet dPCR, which guarantees higher resolution10. However, there were procedural difficulties, especially in homozygous genotypes. In our previous protocol, prenatal diagnosis was determined by comparing the mutant fraction of the study sample with those of positive and negative controls10. The production of positive and negative controls was essential for the previous protocol, which can only be obtained via the calculation of the fraction of fetal DNA in mpDNA. In case that the maternal and the paternal mutation are different from each other, the fraction of fetal DNA in mpDNA can be calculated easily by measuring the fraction of the paternal mutant residue in mpDNA, if the fetus inherits the paternal mutation. However, in homozygous genotypes, both paternal and maternal mutations are identical; hence, the fraction of fetal DNA in mpDNA cannot be calculated simply by measuring the fraction of the paternal mutant residue in mpDNA. SNPs (more preferably, homozygous SNPs) that exist exclusively in the paternal gDNA, but not in the maternal gDNA, should be searched. This requires additional cost and time. To circumvent this issue, we developed a novel protocol for diagnosing the fetal genotype in homozygous genotypes without needing any information regarding the fraction of fetal DNA in mpDNA.
Theoretically, in determining genotypes of homozygous AR variants we can predict the genotype of a fetus based on the proportion of mutant allele reads among total reads in mpDNA: a proportion of mutant allele reads of less than 50% is considered a wildtype; a proportion of mutant allele reads that equals 50% is considered a single heterozygote; and a proportion of mutant allele reads of greater than 50% is considered a homozygote. However, since no test is perfectly precise, we must take in to account for any test errors. For example, we may consider two possibilities when a proportion of mutant allele reads is measured to be 50.5%. The first interpretation is that we can regard the proportion to have some experimental error and consider the genotype to be a single heterozygote. The second one is to consider the genotype to be a mutant homozygous, with a fairly small amount of fetus DNA in mpDNA. To distinguish these possibilities statistically, we employed a comprehensive simulation study for both the gatekeeping method and Bayesian method. We assumed the error rate to be within a normal distribution and a fetus DNA fraction to be within a range from 2.5% to 11%. The simulated data was used to determine an empirical range of mutant read proportion. The genotype of a fetus was expected to be wild or mutant homozygous when the proportion of mutant allele reads of mpDNA was smaller than 48% or greater than 52%, respectively, with a P-value from chi-squared test set to less than 0.05. We also generated simulated data for deciding the cutoff value for the Bayesian method. The Bayes factor cutoff value −0.7 for picodroplet dPCR and 1 for chip-based dPCR – was then tested for three types of control samples with known membership information and these proposed values worked successfully for prediction of genotypes of the control samples. Our numerical simulation showed that the proposed cutoff value was applicable to all cases with GJB2 c.235delC and c.SLC26A4 IVS7–2A > G mutation, under the condition that the total droplet read counts in dPCR is greater than or equal to 1,000 for statistical significance. Therefore, in the case of GJB2 c.235delC and SLC26A4 IVS7-2A > G, prenatal diagnosis can be made by examining only mpDNA in a single step, without further simulation. Expanding the scope further, if the cutoff value of Bayes factor is calculated for other prevalent homozygous mutations in this manner, a rapid diagnosis may likely be possible just from obtaining the maternal peripheral blood sample.
Another important implication of this study is that the improved NIPT protocol described in this paper successfully predicted the fetal genotypes using the chip-based dPCR platform. Although picodroplet dPCR has a higher resolution than chip-based dPCR, it is not widely available. If the NIPT protocol is only available using picodroplet dPCR, it may be difficult for most clinics to adopt this protocol, thus limiting the popularization. In this sense, our NIPT protocol coupled with chip-based dPCR and the gatekeeping method is expected to be more widely used and contribute to the popularization of NIPT.
As technology further develops, prenatal diagnosis will become more popular, with increased benefits. Our study shows that NIPT for all monogenic diseases could be easily performed by simply taking peripheral blood samples and performing quick statistical tests using the data generated from readily available chip-based dPCR, if the genotype of the first baby is available.
References
Kong, C. W. et al. Risk factors for procedure-related fetal losses after mid-trimester genetic amniocentesis. Prenatal diagnosis 26, 925–930 (2006).
Lau, K. T. et al. [Outcome of 1,355 consecutive transabdominal chorionic villus samplings in 1,351 patients]. Chinese medical journal 118, 1675–1681 (2005).
Tabor, A. et al. Randomised controlled trial of genetic amniocentesis in 4606 low-risk women. Lancet (London, England) 1, 1287–1293 (1986).
Chen, Y. et al. Development and validation of a fetal genotyping assay with potential for noninvasive prenatal diagnosis of hereditary hearing loss. Prenatal diagnosis 36, 1233–1241 (2016).
Han, M. et al. A quantitative cSMART assay for noninvasive prenatal screening of autosomal recessive nonsyndromic hearing loss caused by GJB2 and SLC26A4 mutations. Genetics in medicine: official journal of the American College of Medical Genetics (2017).
Committee Opinion No. 640: Cell-Free DNA Screening For Fetal Aneuploidy. Obstetrics and gynecology 126, e31–37 (2015).
Norton, M. E. et al. Cell-free DNA analysis for noninvasive examination of trisomy. The New England journal of medicine 372, 1589–1597 (2015).
Barrett, A. N., McDonnell, T. C., Chan, K. C. & Chitty, L. S. Digital PCR analysis of maternal plasma for noninvasive detection of sickle cell anemia. Clinical chemistry 58, 1026–1032 (2012).
Camunas-Soler, J. et al. Noninvasive Prenatal Diagnosis of Single-Gene Disorders by Use of Droplet Digital PCR. Clinical chemistry (2017).
Chang, M. Y. et al. Development of novel noninvasive prenatal testing protocol for whole autosomal recessive disease using picodroplet digital PCR. Scientific reports 6, 37153 (2016).
Li, C. X. et al. Construction of a multiplex allele-specific PCR-based universal array (ASPUA) and its application to hearing loss screening. Human mutation 29, 306–314 (2008).
Tsui, N. B. et al. Noninvasive prenatal diagnosis of hemophilia by microfluidics digital PCR analysis of maternal plasma DNA. Blood 117, 3684–3691 (2011).
Kim, A. R. et al. The Analysis of A Frequent TMPRSS3 Allele Containing P.V116M and P.V291L in A Cis Configuration among Deaf Koreans. International journal of molecular sciences 18 (2017).
Kim, B. J. et al. Discovery of MYH14 as an important and unique deafness gene causing prelingually severe autosomal dominant nonsyndromic hearing loss. The journal of gene medicine 19 (2017).
Han, K. H. et al. ATP1A3 mutations can cause progressive auditory neuropathy: a new gene of auditory synaptopathy. Scientific reports 7, 16504 (2017).
Han, K. H. et al. Establishment of a Flexible Real-Time Polymerase Chain Reaction-Based Platform for Detecting Prevalent Deafness Mutations Associated with Variable Degree of Sensorineural Hearing Loss in Koreans. PloS one 11, e0161756 (2016).
Lo, Y. M. et al. Presence of fetal DNA in maternal plasma and serum. Lancet (London, England) 350, 485–487 (1997).
Debrand, E., Lykoudi, A., Bradshaw, E. & Allen, S. K. A Non-Invasive Droplet Digital PCR (ddPCR) Assay to Detect Paternal CFTR Mutations in the Cell-Free Fetal DNA (cffDNA) of Three Pregnancies at Risk of Cystic Fibrosis via Compound Heterozygosity. PloS one 10, e0142729 (2015).
Faas, B. H., Beuling, E. A., Christiaens, G. C., von dem Borne, A. E. & van der Schoot, C. E. Detection of fetal RHD-specific sequences in maternal plasma. Lancet (London, England) 352, 1196 (1998).
Lo, Y. M. et al. Prenatal diagnosis of fetal RhD status by molecular analysis of maternal plasma. The New England journal of medicine 339, 1734–1738 (1998).
Chitty, L. S. et al. New aids for the non-invasive prenatal diagnosis of achondroplasia: dysmorphic features, charts of fetal size and molecular confirmation using cell-free fetal DNA in maternal plasma. Ultrasound in obstetrics & gynecology: the official journal of the International Society of Ultrasound in Obstetrics and Gynecology 37, 283–289 (2011).
Meaney, C. & Norbury, G. Noninvasive prenatal diagnosis of early onset primary dystonia I in maternal plasma. Prenatal diagnosis 29, 1218–1221 (2009).
Liao, G. J. et al. Targeted massively parallel sequencing of maternal plasma DNA permits efficient and unbiased detection of fetal alleles. Clinical chemistry 57, 92–101 (2011).
Lam, K. W. et al. Noninvasive prenatal diagnosis of monogenic diseases by targeted massively parallel sequencing of maternal plasma: application to beta-thalassemia. Clinical chemistry 58, 1467–1475 (2012).
Lim, B. C. et al. Genetic diagnosis of Duchenne and Becker muscular dystrophy using next-generation sequencing technology: comprehensive mutational search in a single platform. Journal of medical genetics 48, 731–736 (2011).
Meng, M. et al. Noninvasive prenatal testing for autosomal recessive conditions by maternal plasma sequencing in a case of congenital deafness. Genetics in medicine: official journal of the American College of Medical Genetics 16, 972–976 (2014).
New, M. I. et al. Noninvasive prenatal diagnosis of congenital adrenal hyperplasia using cell-free fetal DNA in maternal plasma. The Journal of clinical endocrinology and metabolism 99, E1022–1030 (2014).
Yoo, S. K. et al. Noninvasive prenatal diagnosis of duchenne muscular dystrophy: comprehensive genetic diagnosis in carrier, proband, and fetus. Clinical chemistry 61, 829–837 (2015).
Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2013R1A1A2063237 to B.Y. Choi) and also by the Korean Health Technology R&D project, Ministry of Health & Welfare, Republic of Korea (HI15C1632 and HI14C1867) to B.Y. Choi).
Author information
Authors and Affiliations
Contributions
B.Y.C., M.Y.C. and S.A. designed the study and wrote the paper. M.Y.K., J.H.H., H.R.P., H.K.S., J.Y., S.L. and D.Y.O. performed the experiments. B.Y.C., M.Y.C, C.K. and S.A. analysed the data. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chang, M.Y., Ahn, S., Kim, M.Y. et al. One-step noninvasive prenatal testing (NIPT) for autosomal recessive homozygous point mutations using digital PCR. Sci Rep 8, 2877 (2018). https://doi.org/10.1038/s41598-018-21236-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-21236-w
This article is cited by
-
Droplet-based microfluidics
Nature Reviews Methods Primers (2023)
-
Clinical application of non-invasive prenatal diagnosis of phenylketonuria based on haplotypes via paired-end molecular tags and weighting algorithm
BMC Medical Genomics (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
