
Abstract
Biologically meaningful metabolic pathways are important references in the design of industrial bacterium. At present, constraint-based method is the only way to model and simulate a genome-scale metabolic network under steady-state criteria. Due to the inadequate assumption of the relationship in gene-enzyme-reaction as one-to-one unique association, computational difficulty or ignoring the yield from substrate to product, previous pathway finding approaches can’t be effectively applied to find out the high yield pathways that are mass balanced in stoichiometry. In addition, the shortest pathways may not be the pathways with high yield. At the same time, a pathway, which exists in stoichiometry, may not be feasible in thermodynamics. By using mixed integer programming strategy, we put forward an algorithm to identify all the smallest balanced pathways which convert the source compound to the target compound in large-scale metabolic networks. The resulting pathways by our method can finely satisfy the stoichiometric constraints and non-decomposability condition. Especially, the functions of high yield and thermodynamics feasibility have been considered in our approach. This tool is tailored to direct the metabolic engineering practice to enlarge the metabolic potentials of industrial strains by integrating the extensive metabolic network information built from systems biology dataset.
Similar content being viewed by others
Introduction
Metabolic network, the pseudo-steady state condition (PSSC): Genome-scale metabolic network (directed graph) is used to model the metabolism of biological systems, such as microorganisms. A few of models have been published including E. coli 1, S. aureus 2, H. pylori 3, M. barkeri 4, S. cerevisiae 5, B. subtilis 6, and so on. The pseudo-steady state condition (PSSC) refers to the main assumption that the concentration of internal compounds keeps invariable over time. Thus, internal compounds satisfy dxc/dt = 0 where xc is the concentration of compound C7.
Source and target, external and internal compounds, exchange reactions: For a genome-scale metabolic network, exchange reactions are transport reactions through which cells exchange materials with the environment. External compounds are the compounds in the extracellular environment, but they enter the cell through exchange reactions and then play a rule. Source and target are respectively the start and the end of the pathways we hope to find.
Pathway and path: A metabolic pathway (a subset of the whole metabolic network) is a set of reactions by which a living organism transforms a source compound into a target compound8. Within a graph representation of a metabolic network, there may be multiple pathways. From the source compound to the target compound, there is a directed path with no cycles and in a particular determined metabolic pathway, and we refer to this directed path as metabolic path9. Of course and especially, when the pathway is branched, it may not be unique for this path. The metabolic pathway contains all the compounds and reactions involved in the pathway, all the internal compounds must be mass balanced in PSSC. Non-decomposability condition means that a pathway can’t be separated into two or more independent pathways.
Smallest pathway in large-scale metabolic networks: For a metabolic network, many pathways may have no biological meaning and if we can find experimentally determined pathways, this may provide in-depth knowledge for biomedical or biotechnological applications. So methodologies on metabolic pathway will devote to discover biologically meaningful metabolic pathways in metabolic networks. There may be many pathways between a source and a target in a large-scale network, and it would be computationally impracticable to completely enumerate all these pathways. Thus, pathway finding methods should focus on finding a set of pathways which were defined by the stoichiometric constraints and could be able to span the complete solution space of pathways. The smallest pathway is defined as the pathway with least reactions which convert the source compound to the target compound. Although pathway research should not come down only to the smallest pathway, the smallest pathway is an important aspect of biological meaning10.
Pathway finding and approaches: As we mentioned above, pathway finding approaches aim to find a set of pathways that should satisfy the stoichiometric constraints, so they can be called stoichiometric approaches. 1) Genetically independent pathways (GIP): Seressiotis and Bailey11,12 provided a method to discover a set of genetically independent pathways and their work represented the first stoichiometric methodology for the computation of metabolic pathways. But their algorithm required big computational effort, so could deal with only metabolic networks of relatively small size. In addition, their approach was based on the assumption that the relationship in gene-enzyme-reaction was a one-to-one unique association. 2) Improved genetically independent pathways (IGIP): Mavrovouniotis13,14,15 developed the algorithm of Seressiotis and Bailey, and used it to deal with pathways which comprised multiple targets and sources. His approach can be applied to a moderate size of the metabolic network. 3) Elementary flux modes: Elementary flux modes (EFMs), i.e. non-decomposable pathways at PSSC, were named by Schuster and co-workers16. With the increase in the size of the metabolic network, the number of EFMs entails combinatorial fashion17. In order to overcome this combinatorial explosion, different strategies have been adopted18,19,20. 4) Extreme pathways: Extreme pathways (EPs), a refined set of EFMs, were proposed by Schilling et al.21. Apart from the non-decomposability condition and the PSSC defined above, the systemic independence condition must be satisfied by the set of EPs, i.e. no EP can be written as a non-trivial nonnegative linear combination of other EPs8. As for EFMs, when applied to large-scale networks, computing all the EPs will suffer a combinatorial explosion. But enumerating special EFMs or EPs, such as from a substrate to a product in a given large-scale metabolic network, is computationally feasible. 5) k-shortest EFMs and flux paths: Figueiredo and Planes have presented a method to find the shortest elementary flux modes in genome-scale metabolic networks with integer programming10. By examining carefully the paper and doing computational practice, we found that this method did not consider ATP maintenance and the yield from substrate to product, and at the same time it did not provide the actual flux distribution in the identified pathways.
High yield and thermodynamics feasibility for a pathway: In the area of industrial biotechnology, improving bacterium is an important task and a high yield from substrate to product is the first target. For the construction of microorganism, we should utilize the pathway with high yield. The shortest EFMs may not be pathways with high yield and they are not equivalent to each other. But the two sets of pathways usually were regarded as equivalent, as stated in the literature10. We will show the difference and give a comparison in the result section of this paper. At the same time, although a pathway exists in stoichiometry, it may not always be feasible in thermodynamics. If we regard a pathway as an overall reaction and if we hope it is able to proceed spontaneously, it should satisfy the requirement of free energy change. Moreover, if there are several pathways which satisfy the condition, which one is more probable to occur in the cell?
Motivation and our contribution: At present, the modeling and simulation method for the genome-scale metabolic network is constraint-based method which satisfies steady-state criteria. For the reason of inadequacy assumption (GIP, IGIP), computational difficulty (EFMs, EPs), or ignoring the yield (k-shortest EFMs and flux paths), previous pathway finding approaches as we stated above can’t effectively design optimal pathways to direct the metabolic engineering practice. In this work, by using MIP (Mixed Integer Programming) strategy we put forward an algorithm to identify the smallest balanced pathways (SBPs) which convert the source compound to the target compound in large-scale metabolic networks. Under PSSC, the resulting SBPs of our method can well satisfy the stoichiometric constraints and non-decomposability condition; Multiple pathways which meet the above-mentioned criteria can be found and provided as candidate design; In addition, high yield is a new function; Especially, thermodynamics feasibility has been considered in our approach. The smallest pathways founded by our method can provide good references in the pathway design for the industrial microorganism. Our model can be easily solved by existing optimization software.
Methods
Mathematical description of metabolic network, Flux balance, and FBA
Usually, we can use a stoichiometric matrix, S, to describe genome-scale metabolic network and the elements in S are the coefficients of reactions22. Under steady-state criteria, the time derivatives of metabolite concentrations are zero7, i.e. those internal metabolites should satisfy mass balance, so the equations of mass balance for all the metabolites can be represented as follows
where S is the stoichiometric matrix, and α i and β i define the bounds through each reaction v i , R is the set of reactions.
As for metabolic networks in genome-scale, the fluxes within a cell usually can be computed with flux balance analysis (FBA) that can give optimal growth phenotypes, though not unique. In mathematics, FBA is an equivalent to a large-scale linear programming (LP). In our algorithm, we confine source and target compounds to be external compounds, i.e. there are exchange reactions related to them. For example, for the genome-scale metabolic network of E. coli_iJO13661, there are more than 300 exchange reactions and we can choose any two as source and target.
Mathematical model to find the smallest balanced pathway
In order to find the smallest balanced pathways in large-scale metabolic networks, MIP strategy is used as the mathematic model. We introduce binary variable y of the same number of continuous variable v to indicate the absence or presence of a reaction v i.
If y i = 0 then v i = 0 and If y i = 1 then α i ≤ v i ≤ β i, we can express this idea as a constraint:
The source and target nodes should be external nodes, and there are exchange reactions connected to them. In order to give a connected pathway, two bounds are added.
The reason for the small of v t is to let v i of other reactions in the pathway not be beyond their constraints, and here constant1 and constant2 are positive values. Equation (4) is clearer in describing the input and output of the SBP than those methods of k-shortest EFMs10 and flux paths23.
Now we choose the sum of the number of used reactions as the objective function, i.e.
The strategy to find the smallest balanced pathways in large-scale metabolic networks may be expressed as a MIP model with v i as continuous variable and y i as a binary variable.
The SBPs is different from the null space of the stoichiometric matrix and the null space of the stoichiometric matrix is only the constraints (1). The SBPs is smaller than the null space of the stoichiometric matrix.
Extend to custom-specified conditions
For this model, we can easily preset the metabolic network to meet the requirement of the specific situations. For example, certain reactions must not be appearing, or some genes are to be disrupted, we just preset vi = 0; In other case, certain reactions must be reversible, we can preset vmin = −1000, vmax = 1000. These could be achieved by setting the boundaries of the reactions. Then the solution of smallest balanced pathways is within the scope of the given conditions.
Solve the model and obtain multi solutions
For MIP, some existing software can be used to find its solution and we use Gurobi24 here. With a statistic of the fluxes which are not zero in absolute value (or larger than a given small value 10−5) or which y i is 1 (the two ways are consistent), we can determine those reactions which should appear, and further, we can obtain the smallest balanced pathways.
Sometimes, there exist different states of integer variables but the objective value is the same, i.e. a MIP may have multi integer solutions. Up to date, as we know, there does not exist optimization tool which can give directly multi integer solutions for a MIP. Here we utilize a method proposed by Balas and Jeroslow, named Combinatorial Bender’s cut25. The approach of Bender’s cut is that iteration is used from an existing solution, at the same time in each iteration to exclude an existing solution by adding the following binary cut
All the multi integer solutions will be got by this way.
Smallest balanced pathway with high yield
SBPs have the least number of reactions but may not have the high yield of a chemical which the microorganism produces. High yield means a high amount of desired product and little or no by-product which might make the downstream complicated, costly, and polluted. In another word, high yield means cost-saving. Sometimes, high yield is our interesting aspect, so it is best to find SBPs with high yield. In order to estimate the reachable high yield of the chemical, we can use FBA with the exchange reaction rate vchem of this chemical as the objective and we will get the theoretical ratio Vmax. Then we can use 95% of the value of highest yield Vmax as a constraint in our MIP model. Finally, we will get all SBPs with a high yield which is larger than 95% of the value of highest yield.
Thermodynamics feasibility analysis
Although a pathway exists in stoichiometry, it may not always be feasible in thermodynamics. If we regard a pathway as an overall reaction and if we hope it is able to proceed spontaneously, it should satisfy the requirement that each reaction in the pathway must be thermodynamically feasible individually, i.e. the flux and the free energy change of this reaction must have opposite signs or the reaction is reversible. The data of free energy change for a microbe is not rich in literature and the first one is E. coli 26,27. The thermodynamic data of E. coli model was calculated by Group Contribution Method27,28. There is a range of free energy change for every reaction and it is calculated by min/max delta G. The range of delta G could be used to decide the reversibility of a reaction.
Results
Case 1: The SBPs from glucose to succinic acid in given conditions
The SBPs from glucose to succinic acid
In this example, we hope to know how succinic acid is synthesized by glucose with E. coli. By using our algorithm, we computed out the smallest balanced pathways from glucose (source compound, input exchange reaction is EX_glc[e]) to succinic acid (target compound, output exchange reaction is EX_succ[e]) in the genome-scale metabolic network of E. coli (its SBML model is iJO13661). In the process of computation, we restrict the input rate of glucose (vs = −100 mmol/g(Dw)h) and the output rate of succinic acid (vt ≥ 0.01 mmol/g(Dw)h). The given conditions are that we restrict the input and output of the cell to be only five compounds, i.e. glucose, succinic acid, CO2, H2O, and H. The reason for restricting only these five compounds is that succinic acid can be synthesized by them. There are 12 alternative solutions for this model, seeing Supplementary Material a, and all the solutions have 37 step reactions. Among 12 alternative solutions, 31 step reactions are the same, and they are “ACONTa, ACONTb, ATPM, ATPS4rpp, CO2tex, CO2tpp, CS, ENO, EX_co2(e), EX_glc(e), EX_h2o(e), EX_h(e), EX_succ(e), FBA, FUM, GAPD, GLCt2pp, H2Otex, H2Otpp, Htex, ICL, MALS, MDH, PDH, PFK, PGK, PGM, PPC, SUCCt3pp, SUCCtex, TPI”, while 11 step reactions are different, illustrated in Table 1. The whole names of each reaction in these 12 pathways are provided in the Supplementary Material a.
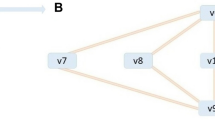
One of these pathways, the first solution, was illustrated in Fig. 1 with red and circle nodes for reactions and with blue and square nodes for compounds. The pathway included 37 reactions and 41 compounds. The number marked beside each line represents the rate of consuming or producing the corresponding compound. For every compound, its mass is balanced, i.e. the sum rate consuming it is equal to the sum rate producing it. At the same time, this pathway includes the least reactions among all the pathways converting glucose to succinic acid.

One of the smallest balanced pathways from glucose to succinic acid in the genome-scale metabolic network of E. coli under given conditions, including 37 reactions and 41 compounds. The number marked beside every line represents consuming or producing rate of compounds.
From this pathway, we can know clearly the pathway to synthesize succinic acid and the balanced proportions among fluxes through every reaction in this pathway. We know also how these enzymes (or reactions) cooperate with each other to synthesize succinic acid. This means these reactions are equally important to fulfill the overall function of succinic acid production. The pathways including the reactions, genes, and fluxes should be regarded as ideal references to guide strain engineering activity. This will greatly reduce the scope of targets to enhance genes in order to accelerate the speed of producing succinic acid.
Thermodynamics feasibility analysis
With the data of free energy change of each reaction for E. coli 26,27, seeing Supplementary Material a, we have made a statistic on the free energy change delta G and the range of delta G of individual reactions for each of the above 12 alternative pathways producing succinic acid, illustrated in Table 2. For each pathway, the fluxes and their corresponding free energy changes of these reactions either have opposite signs or the reactions are reversible, the number of irreversible reactions that the fluxes and their corresponding free energy changes have the same signs is zero, and so all these pathways are feasible in thermodynamics.
Comparison between SBPs with high yield and those without high yield
In the above succinic acid case, the yield 171.43:100 has almost been the theoretical value. In order to show the difference of those with high yield and SBPs without high yield, threonine production with E. coli is selected for the study. Threonine is an important chemical in industry, which can be produced by E. coli with glucose. We have computed all the threonine SBPs with highest yield in E. coli with glucose as substrate, and get 16 SBPs. All the SBPs have 50 step-reactions and the yield is 1.248:1 in a molar ratio which is near the theoretical yield. All the SBPs are in Supplementary Material b. At the same time, we cancel the high yield function of our algorithm and run our algorithm again. Now there are 7 SBPs and all the SBPs have 43 steps of reactions, but the yield is only 0.25:1 in molar ratio. So although the steps are less, the yield is smaller than that of SBPs with high yield. All the SBPs without high yield are in Supplementary Material c. Of course, in the practice of synthesizing threonine, the SBPs with high yield has more significance for commercialization.
Comparison with k-shortest EFMs
In order to make a quantitative comparison with conventional methods, the method of k-shortest EFMs was selected as it is the nearest approach to ours. We use this algorithm to compute out all the shortest EFMs from glucose to succinate in the genome-scale metabolic network of E. coli_iJO1366, illustrated in Supplementary Material d. All the 24 shortest EFMs are 30 step reactions, they are shorter than our above SBPs from glucose to succinate, but the molar yields of these EFMs are 1.0, while the molar yields of our SBPs are 1.71, which is near the theoretical ratio. We checked these EFMs and found the main reason was that they did not consider ATP maintenance and the maximum conversion yield. The SBPs with high yield will be more helpful in the practices of synthesizing chemicals with microbes. ATP maintenance, i.e. ATPM reaction, an artificial reaction, is necessary for the cell to maintain the physiological behaviors of microbes. If we reject ATP maintenance and the requirement of high yield, our SBP algorithm will get a similar result of shortest EFMs. Another aspect is the approach of shortest EFMs did not provide the flux distribution in the computed pathway and all the reaction flux is 1, while our SBP algorithm can give the actual flux distribution in the computed pathway. Flux distribution is a fine reference in pathway design when we want to synthesize chemicals with microbes.
Case 2: The SBPs from glucose to a variety of chemicals which E. coli can produce with maximum productivity
In addition to succinic acid and threonine that we mentioned above, E. coli can produce many other chemicals such as lactic acid, formic acid, fumaric acid and so on. In the model iJO1366, there are 324 exchange reactions. Only 25 reactions have low bounds which less than 0, while all the up bounds equal to 1000. We use glucose (start or source) as the input, at the same time use all these 324 reactions except for glucose as the output (target) respectively, and calculate the SBPs with maximum productivity for every chemical. For a given chemical, if E. coli can’t produce it, i.e. the maximum productivity for it is zero, the algorithm will not return its SBPs.
We have made a statistics for all the SBPs to a variety of chemicals which E. coli can produce and found that in many cases, the number of SBPs is less than 10 and that those cases which are larger than 100 only take a very small proportion, as shown in Fig. 2. Here, we do not provide thermodynamics feasibility analysis (TFA) for each SBP. If we have interest for a certain SBPs in their thermodynamics feasibility, we can do TFA by the method we provided in the section of Methods.

Statistics for all the SBPs to a variety of chemicals which E. coli can produce.
Conclusions
Main idea and difference from previous algorithms
Up to date, modeling a genome-scale metabolic network in dynamics is still beyond the access of most laboratories, so the best way to make use of flooding metabolic network information to direct the metabolic engineering practice is the constraint-based approach which satisfies the pseudo-steady state condition (PSSC). A pathway that converts a given source compound to a given target compound should satisfy the stoichiometric constraints and non-decomposability condition. EFMs and EPs are two pathway finding approaches, but calculating the set of EPs or enumerating all the EFMs will suffer a combinatorial explosion when applied to large networks. Existed approaches of k-shortest EFMs and flux paths are not the methods of considering the yield from substrate to product which is highly biotechnologically relevant.
In this work, by utilizing Mixed Integer Programming (MIP), we present an approach for pathway finding. Our algorithm has a number of good features: 1) It is a method of thorough stoichiometry. It can identify the balanced pathways in the genome-scale metabolic network. The balance here means that the mass of internal compounds is balanced, i.e. stoichiometric balance. The smallest means that the pathway identified has least reactions. 2) The pathways found by this approach are usually short enough, which simplify the metabolic engineering practice and also save the cellular energy consumption for synthesizing proteins for the reactions. It is well-known that protein synthesis is the most energy-intensive process. 3) Our algorithm can return all the alternative solutions, and this can provides more choices in industrial stain design. 4) High yield can be added as required condition, which is important for biotechnology purpose. 5) Thermodynamics data are integrated to allow the thermodynamics feasibility analysis.
We recognized that although the short pathway has the advantages as mentioned above, the shortest pathway may not necessarily be biologically feasible and some biological pathways are not the shortest one in nature. Our algorithm is to break the evolutionary barrier and eventually help to create artificial cell factory. Furthermore, by just simply modifying our code, we can easily found all balanced pathway with the length of shortest plus 1, 2, …, and so on.
Computational complexity
The model of our approach comes down to a MIP and MIP is an essentially combinational problem. Computational complexity will be proportional to the scale of the problem. But for a large scale problem, existing solving software can solve it in not long time. Such as our case study with several thousand of variables, the computation time will take just several minutes by an HPC (high-performance computer) with 48 cores.
Application of industrial stain design
In industrial stain design, high yield from the source substrate to the target product is the first important aspect. To implement this, biologically feasible and high yield pathways should be utilized. The smallest balanced pathway with high yield can provide an ideal reference to guide metabolic engineering practice. In particular, the balanced pathways tells many co-dancing reactions which are beyond the sight of normal biological knowledge. The metabolic bottleneck may not necessarily locate on the traditional biochemical pathway. The accessory reaction which is responsible to recycle the cofactors, intermediate or to supply the precursors is shown to be as important as the reactions in the known biochemical pathway. Meanwhile, the relative strength of the fluxes of different reactions also tells the metabolic engineer how to fine-tune the relative activities of different reactions. Integrating with the experimental determination of intracellular transcriptome, proteome and even metabolome, metabolic engineering should be able to identify the potential rate-limiting reactions which they need to put effort on.
In our first case study, producing succinic acid with E. coli, the input rate of glucose is 100 mmol/g(Dw)h and the output rate of succinic acid is 171 mmol/g(Dw)h, so this pathway almost reaches the theoretical ratio of glucose/succinic acid in E. coli. If we fulfill the pathway in E. coli, it can make a good utilization of glucose in producing succinic acid. SBPs with high yield from glucose to a variety of chemicals, which E. coli can produce, have been calculated by our algorithm. Especially, we can make decisions on the thermodynamics feasibility by integrating the data of free energy change.
References
Orth, J. D. et al. A comprehensive genome-scale reconstruction of Escherichia coli metabolism—2011. Molecular Systems Biology. 7, 535 (2011)
Becker, S. A. & Palsson, B. O. Genome-scale reconstruction of the metabolic network in Staphylococcus aureus N315: an initial draft to the two-dimensional annotation. BMC Microbiology. 5, Art. No. 8 (2005).
Ines, T., Vo Thuy, D., Price, N. D. & Palsson, B. O. Expanded metabolic reconstruction of Helicobacter pylori (iIT341 GSM/GPR): an in silico genome-scale characterization of single- and double-deletion mutants. Journal of Bacteriology. 187, 5818–5830 (2005).
Feist, A. M. et al. Modeling methanogenesis with a genomescale metabolic reconstruction of Methanosarcina barkeri. Molecular Systems Biology. 2, Art. No. 2006.0004 (2006).
Duarte, N. C., Herrgard, M. J. & Palsson, B. O. Reconstruction and validation of Saccharomyces cerevisiae iND750, a fully compartmentalized genome-scale metabolic model. Genome Research. 14, 1298–1309 (2004).
Oh, Y. K., Palsson, B. O., Park, S. M., Schilling, C. H. & Mahadevan, R. Genomescale reconstruction of metabolic network in Bacillus subtilis based on high-throughput phenotyping and gene essentiality data. The Journal of Biological Chemistry. 282, 28791–28799 (2007).
Reed, J. L., Vo, T. D., Schilling, C. H. & Palsson, B. O. An expanded genomescale model of Escherichia coli K-12 (iJR904GSM/GPR). Genome Biology. 4, R54 (2003).
Francisco, J. & Planes, J. E. B. A critical examination of stoichiometric and path-finding approaches to metabolic pathways. Briefings in Bioinformatics. 9(5), 422–436 (2008).
Francisco, J. P. & John, E. B. Path finding approaches and metabolic pathways. Discrete Applied Mathematics. 157, 2244–2256 (2009).
de Figueiredo, L. F. et al. Computing the shortest elementary flux modes in genome-scale metabolic networks. Bioinformatics. 25(23), 3158–3165 (2009).
Seressiotis, A. & Bailey, J. E. MPS. an algorithm and data base for metabolic pathway synthesis. Biotechnology Letters. 8, 837–842 (1986).
Seressiotis, A. & Bailey, J. E. MPS - an artificially intelligent software system for the analysis and synthesis of metabolic pathways. Biotechnology and Bioengineering. 31, 587–602 (1988).
Mavrovouniotis, M. L. Synthesis of reaction mechanisms consisting of reversible and irreversible steps. 2. Formalization and analysis of the synthesis algorithm. Industrial & Engineering Chemistry Research. 31, 1637–1653 (1992).
Mavrovouniotis, M. L. Identification of qualitatively feasible metabolic pathways. In: Hunter L (ed). Artificial Intelligence and Molecular Biology. Menlo Park, CA: AAAI Press/MIT Press. 325–364 (1993).
Mavrovouniotis, M. L. & Stephanopoulos, G. Synthesis of reaction mechanisms consisting of reversible and irreversible steps. 1. A synthesis approach in the context of simple examples. Industrial & Engineering Chemistry Research. 31, 1625–1637 (1992).
Schuster, S. & Hilgetag, C. On elementary flux modes in biochemical reaction systems at steady state. Journal of Biological Systems. 2, 165–182 (1994).
von Kamp, A. & Schuster, S. Metatool 5.0: fast and flexible elementary mode analysis. Bioinformatics. 22, 1930–1 (2006).
Dandekar, T., Moldenhauer, F. & Bulik, S. et al. A method for classifying metabolites in topological pathway analyses based on minimization of pathway number. Biosystems. 70, 255–270 (2003).
Schuster, S., Pfeiffer, T. & Moldenhauer, F. et al. Exploring the pathway structure of metabolism: decomposition into subnetworks and application to Mycoplasma pneumoniae. Bioinformatics. 18, 351–361 (2002).
Teusink, B., Wiersma, A. & Molenaar, D. et al. Analysis of growth of Lactobacillus plantarum WCFS1 on a complex medium using a genome-scale metabolic model. The Journal of Biological Chemistry. 281, 40041–8 (2006).
Schilling, C. H., Letscher, D. & Palsson, B. O. Theory for the systemic definition of metabolic pathways and their use in interpreting metabolic function from a pathway-oriented perspective. Journal of Theoretical Biology. 203, 229–248 (2000).
Xu, Z., Sun, X. & Yu, S. Genome-Scale Analysis to the Impact of Gene deletion on the Metabolism of E. coli: Constraint-Based Simulation Approach. BMC Bioinformatics. 10(Suppl 1), S62 (2009).
Jon, P. et al. Path finding methods accounting for stoichiometry in metabolic networks. Genome Biology. 12, R49 (2011).
Gurobi optimization company, Gurobi Optimizer (http://www.gurobi.com) (Date of access:20/03/2017).
Balas, E. & Jeroslow, R. Canonical cuts on the unit hypercube. SIAM Journal of Applied Mathematics. 23(1), 61–69 (1972).
Xu, Z. et al. Construction and Analysis of the Model of Energy Metabolism in E. coli. PLoS ONE. 8(1), e55137 (2013).
Feist, A. M. et al. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Molecular Systems Biology. 3, Art. No. 121 (2007).
Jankowski Matthew, D. et al. Group contribution method for thermodynamic analysis of complex metabolic networks. Biophysical Journal. 95, 1487–1499 (2008).
Acknowledgements
Support for this work was provided by “National Natural Science Foundation of China (31370829, 31370113)”, “Tianjin Research Program of Application Foundation and Advanced Technology (15JCYBJC23600)”, “Tianjin Science and Technology Committee (11ZCZDSY08600)”. We thank professor Francisco J Planes for providing the Matlab codes of k-shortest EFMS. The authors thank the anonymous reviewers for their valuable suggestions.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: Z.X. Performed the experiments: Z.X. Analyzed the data: Z.X., Q.W., D.Z., J.S. Contributed reagents/materials/analysis tools: Q.W., D.Z., J.S. Wrote the paper: Z.X., D.Z.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, Z., Sun, J., Wu, Q. et al. Find_tfSBP: find thermodynamics-feasible and smallest balanced pathways with high yield from large-scale metabolic networks. Sci Rep 7, 17334 (2017). https://doi.org/10.1038/s41598-017-17552-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-17552-2
This article is cited by
-
Metabolic pathways synthesis based on ant colony optimization
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
