
Abstract
We analysed the ligand-based activation mechanism of the prostate-specific G-protein coupled receptor (PSGR), which is an olfactory receptor that mediates cellular growth in prostate cancer cells. Furthermore, it is an olfactory receptor with a known chemically near identic antagonist/agonist pair, α- and β-ionone. Using a combined theoretical and experimental approach, we propose that this receptor is activated by a ligand-induced rearrangement of a protein-internal hydrogen bond network. Surprisingly, this rearrangement is not induced by interaction of the ligand with the network, but by dynamic van der Waals contacts of the ligand with the involved amino acid side chains, altering their conformations and intraprotein connectivity. Ligand recognition in this GPCR is therefore highly stereo selective, but seemingly lacks any ligand recognition via polar contacts. A putative olfactory receptor-based drug design scheme will have to take this unique mode of protein/ligand action into account.
Similar content being viewed by others


Introduction
Olfactory receptors (ORs), which are part of the G protein-coupled receptor (GPCR) family1, recently turned out to be present not only in the human nose, but in a large set of different human tissues as well2,3. A functional role of these ectopically expressed ORs was shown in human colon tissue4, sperm5, blood cells6, and skin tissue7,8. Furthermore, the expression of some ORs is up regulated in different types of cancer cells9,10,11,12,13, and these ORs have an effect on cell proliferation in at least prostate2, liver14, leukemia15 cancer and melanoma16 cells. One of these cancer-related receptors is OR51E2, known as well as the prostate-specific G protein-coupled receptor (PSGR) as this GPCR was first detected in the prostate before it was classified as an olfactory receptor by sequence homology10,17. Recent studies indicate that PSGR expression is up-regulated in prostate cancer2 and melanoma cells16, and that PSGR activation leads to an inhibition of cell proliferation2,16,18,19. Human melanoma is an aggressive and highly metastatic type of cancer, which is highly resistant to conventional therapies20. Prostate cancer is the 2nd most common cause of cancer, and the 6th leading cause of cancer death in men21. PSGR may therefore be a new target for the diagnosis and therapy of cancer, especially for notoriously difficult to treat melanoma.
To be able to design molecular therapeutics specific for PSGR, the prerequisites of molecular pharmacology for ORs need to be known. It was recently shown that though ORs are classified as rhodopsin-like GPCRs, they do not belong to the common rhodopsin-like small molecule binding GPCRs, but rather form a subclass of their own22. Consequently, OR ligand (odorant) recognition exhibits peculiar activation properties: each single OR can be activated by a large number of ligands, which translates into a cytosolic G protein response level that depends on the respective ligand bound23. Contrary to the seemingly unspecific ligand recognition, ORs can show remarkable ligand specificity: in earlier experimental studies, we established β-ionone as agonist for PSGR, and α-ionone as competitive antagonist2,8,16. PSGR and β-ionone/α-ionone are an OR agonist/antagonist combination with nearly identical ligand chemical scaffolds, i.e., they only differ in the position of a single carbon/hydrogen bond, and thus present a unique case to assess differences in OR/ligand interactions between these pharmacological subclasses. Agonist/antagonist pairs with a lower grade of structural similarity, i.e., they contain similar structural features like ring moieties, polar groups, or hydrophobic tails at comparable positions, are known for several other receptors24,25,26,27,28,29. In addition to ionones, we identified steroid hormones as activators of PSGR2,8. However, as numerous GPCR crystal structures have shown30, cholesterol scaffold-based compounds do not bind to the center of the 7TM helix bundle, but to the protein/membrane interface at helices II and IV. Therefore, we hold these steroids to be allosteric activators of PSGR, and do not investigate them here further. The peculiar ligand recognition properties of olfactory receptors were proposed to be based on recognition by shape components (so-called “odotopes”)31,32,33,34,35, molecular vibrations36, or a combination of both37. In a pilot study on the olfactory receptor hOR2AG1, we established an alternative model, which is a combination of shape recognition and matching protein–ligand dynamics38. According to our “dynamical ligand binding” model, besides ligand affinity39, it is the frequency of receptor and ligand contacts occurrence, which is decisive for receptor activation. Recently, this concept was nicely reproduced for the case of odorant recognition in the mouse olfactory receptors mOR-EG and mOR-EV40, and is not restricted to ORs, but applies to other small-ligand binding GPCRs as well41.
Figure 1a shows a comparison of the two ionones: α- and β-ionone are constitutional isomers, which only differ in the position of one double bond. Structurally, this leads to a different angle at the connection between the ionone ring moiety and the butenoneyl side chain. The two substances thus only differ slightly in their side molecular structure, while being chemically nearly identical. However, this small difference is recognized by PSGR, and leads to the discrimination into agonist and antagonist. α-ionone actually exists as two enantiomers, (R)- and (S)- α-ionone, which will bind to the receptor with different affinities, and potentially will even belong to different pharmacological classes. This substance was applied as racemic mix in earlier experiments2, and separate measurements of the enantiomers are not possible: due to being a β-unsaturated ketone, keto-enol-tautomerism includes the δ-carbon atom, i.e., the asymmetric atom within α-ionones. Enantiomeric pure (R)- or (S)- α-ionone would therefore quickly racemize in aqueous solution. Due to this chemical limitation, it is not clear which of both enantiomers is the actual antagonist.

Structure of α-ionones and β-ionone, and the predicted ligand binding cavity. (R)- α-ionone in cyan, (S)- α-ionone in dark blue, β-ionone in orange. (a) Chemical structures and 3D structures of ionones after overlay of their ring moieties. The main difference between the three isoforms lies in the angle of the butenoneyl side chain to the ring moiety. (b) PSGR structural model with a close-up of the central binding site. Protein backbone in grey, β-ionone in its best docking posture in the inactive protein state in orange sticks, surrounding mutated residues in blue sticks. Helices numbered in roman numbers. The displayed structure is the initial inactive protein model used for ligand docking. Mutation of all displayed residues had an effect on ligand binding and protein activation (see Table 1). We therefore propose this position within the protein model as the β-ionone binding site.
In this work, we investigate the different dynamic binding modes of α- and β-ionone in PSGR by employing a close combination of experimental and theoretical methods. By the creation of a dynamic homology model38,41,42 of PSGR in combination with ligand docking41,43, we propose a model of the binding site for both ligands within our OR of interest, and further analysed the different binding strengths of both enantiomers of α-ionone in comparison to β-ionone within the model. We then assessed their differences in dynamic binding modes of α-ionones in comparison to β-ionone by analysing the frequency of occurrence of their dynamic protein/ligand and protein/protein contacts in the form of dynamic interaction fingerprints (IFPs)38,41,44,45. For this, we build two homology models, based on a rhodopsin crystal structure in its inactive state46, and an opsin crystal structure in an activated state with a bound hydrophobic lipid molecule47 (see Figure S1 for the used sequence alignment). Though olfactory receptors form a separate subclass of GPCRs22, we chose rhodopsin as starting model, as it proved to be a good structural template in our earlier investigation on hOR2AG138, and is the only GPCR that contains a hydrophobic ligand and is at the same time available as structural model in both an inactive and an active-like conformation. We verified our theoretical results by extensive comparison with experimental results on PSGR activation derived from luciferase reporter assays in the heterologous expression system in combination with point mutation analysis of recombinant receptors. Both theoretical and experimental results matched well, and allowed us to propose a dynamic binding mode and a dynamic protein/ligand and protein/protein IFP necessary for activation. To our surprise, this binding mode showed to be highly stereo selective, but not to include any polar contacts: The ionones were recognized by their overall shape, but no significant occurrence of a protein/ligand hydrogen bond via the keto moiety was observed. This matched well with an overall low affinity for ionones observed in experiments. A future development of OR-based drug design will need to take this mode of hydrophobic ligand action into account.
Results
Determination of the β-ionone binding site
To start our investigation, we needed to find a putative ionone-binding pocket within PSGR. For this, we focused on β-ionone, as it is the compound in our investigation, which induces protein activation, and thus results in a clear experimental output signal. As protein model, we used the inactive ligand free rhodopsin-based PSGR homology model, as this would be the structure β-ionone would need to bind to according to the ternary complex model48.
We here have to state that with a 20% sequence identity between PSGR and rhodopsin, the overall homology between target and template is relatively low, and that the cut-off for a “reliable” GPCR homology model is at a minimum of 35% identity, as pointed out by Kufareva et al.49. However, this is a general problem with modelling odorant receptors. The success of generating homology models of ORs based on crystal structures of rhodopsin and other GPCRs (see refs33,38,39,40,50,51,52) justifies homology modelling of ORs to construct molecular models. However, these models per se have predictive character, but cannot be taken as competitors of experimentally resolved structures. Nevertheless, comparison with experiment offers an additional source of data for model refinement: identifying models that integrate most of the experimental data can allow formulating hypotheses and predictions for the actual dynamics of the receptor and receptor activation. We have shown this successfully by reprogramming the selectivity filter of hOR2AG1 by predicting a point mutation that leads to activation of the receptor by a ligand that does not activate wild type protein38.
Figure 1b shows the putative β-ionone binding position appearing during docking runs. The site is positioned within the centre of the 7TM helix bundle between helices III, IV, V, and VI. It therefore is in good agreement with the position of the ionone ring of retinal within rhodopsin46 and earlier works on olfactory receptor binding sites33,38,39,40,50,51,52,53,54. Based on these results, we determined the amino acids constituting this site, and carried out functional analysis of point-mutated receptors in order to verify the binding site. These amino acids (see Fig. 1b; numbering in superscripts are the respective Ballesteros-Weinstein residue numbers55) were His1043.33, Ser1073.36, and Ser1113.40 in helix III; Lys1855.42, Asp1905.47, and Asn1945.51 in helix V; and Tyr2516.48 in helix VI. All mutant receptors were functionally expressed in Hana3A cells as controlled by live-cell immunocytochemical staining (see Supplementary Figure S2). Table 1, and Figs 2a and S3a show the effect of the respective mutations on the experimentally observable threshold concentration and the maximal receptor activation (Emax). We here need to point out that we use threshold concentrations, and not EC50 values, as such EC50 values would have been observable at concentrations >250 µM β-ionone, which showed cytotoxic effects in our luciferase essays. Furthermore, there is a significant difference in β-ionone potencies determined in the study of Neuhaus et al. by single cell calcium imaging of transiently transfected HEK293 cells2 compared to our approach using Hana3a cells and the CRE-mediated luciferase assay, which may result from differential assessment of potencies or from biased signalling of PSGR. We therefore recommend to not to take the respective Emax and response curves as absolute values, but to consider them for a comparison of differences within the framework of this article. Mutation of each investigated residue showed an impact on both thresholds and Emax. Interestingly, we observed not only receptor mutants with decreased thresholds/Emax, but also neutral point mutations as well as mutants with increased thresholds/Emax (“hyperactive” mutants). We provide a detailed analysis of these mutants in the MD simulation section.

Dose-response curves of PSGR variants. (a) Responses of cells transfected either with a plasmid encoding for wild type (WT) and point-mutated PSGR, respectively, to β-ionone as measured by luciferase assay. Receptor activation was normalized to the relative response to β-ionone (250 µM) in WT transfected cells. Dose-response curves were fitted by a Hill equation. Odorant concentrations up to 250 µM were used because higher concentrations of β-ionone exhibited toxic effects on the cells as cells detached during treatment. Error bars indicate the standard error of the mean (SEM) of 4–15 replicas. (b) Quantification of luciferase values of Hana3a cells transiently expressing wild type (WT) or point-mutated PSGR co-stimulated with α-ionone (200 µM) and β-ionone (200 µM) or β-ionone (200 µM) only. The blocking effect of α-ionone was normalized to the corresponding response to β-ionone of the respective receptor. The data are shown as the means ± SEM (n = 3). Significance was calculated by Student’s t-test (*p < 0.05).
As control, we introduced an I2556.52W mutation. This mutation targets a residue close to the proposed binding site, but the respective native side chain does not make a contact with the docked ligand, nor does it interfere with the neighbouring helices (see Supplementary Figure S4). As to be expected, the mutant exhibits a near-native threshold and Emax, which further confirms our proposed protein homology model and the binding position for β-ionone.
As an additional check to correlate individual amino acids and receptor activation, we investigated if the mutants had an effect on the basal activity of the receptor56,57. The results are summarized in Table SI: we observed 2-8fold decreases in basal activities for the mutants K1855.42R, K1855.42Q, and D1905.47L, which further supports the involvement of these residues in receptor activation.
We need to point out that a recent study of Sanz et al.58 found α-ionone to be an agonist of heterlogously in HEK293 cells expressed PSGR. However, this work used a small number of experiments (>3), and the relative fluorescence intensity in non-ratiometric measurments of a small number of cells (>20) as main observable. For the results displayed in Fig. 3a, we rely on ten individual experiments (each with 600 to 1600 cells), and the relative number of cells responding to an odorant stimulus above a certain fluorescence threshold from a large number of individual cells. Furthermore, Sanz et al. do not comment on the statistical significance of their data, which exhibit a large standard deviation58. We therefore believe that the experimental setup and the number of experiments of Sanz et al.58 are too small provide the same granularity of results as we did.

Experimental ligand competition between α-ionones, and β-ionone in comparison with predicted binding affinities from Docking and free energy calculations. (a) Experimental competition assay2. The β-ionone induced Ca2+ responses of HEK293 cells transiently expressing PSGR is inhibited by the co-application of α-ionone in a dose-dependent manner. Co-application of increasing concentrations of α-ionone caused an increased reduction of the number of cells responding to 50 μM β-ionone. Shown are relative numbers of cells responding to 50 μM β-ionone in the presence of varying concentrations of a racemic mix of α-ionones. Odorants were applied for 20 s. The data are the mean of 10 independent experiments for every α-ionone concentration tested, each with 600 to 1600 cells. Significance was calculated by Student’s t-test for each sample group referring to cell responses to β-ionone (50 µM). Error bars represent SEM, (*p < 0.05, **p < 0.01 and ***p < 0.001). α-ionone blocks cellular responses to β-ionone, with an affinity that is seemingly twofold higher than the one of β-ionone. (b) Best docking poses of (R)-, (S)- α-ionone, and β-ionone found in the initial ligand-free inactive PSGR model with respective calculated ∆Gbind values. Protein backbone in grey, β-ionone in orange sticks, (R)- α-ionone as cyan sticks, (S)- α-ionone as blue sticks. Selected surrounding protein residues in grey sticks. Helices numbered in roman numbers. β-ionone and (R)- α-ionone bind in a very similar fashion with the head groups oriented towards Asp1905.47; the detailed binding mode however differs, as (R)- α-ionone presents its keto group oxygen atom to Asp1905.47, while β-ionone presents the terminal methyl group to Asp1905.47. (S)- α-ionone binds with the head group oriented towards Lys1855.42. Both α-ionone enantiomers bind with a slightly higher affinity than β-ionone, which is in line with α-ionone being a competitive antagonist for β-ionone. (c) Results from free energy calculations (see Table SIII). Both α-ionone stereoisomers exhibit a clearly separated affinity for the active and the inactive receptor conformation, with the affinity for the inactive conformation being significantly higher than the one for the active conformation. β-ionone exhibits an affinity for the receptor, which is comparable to the one of α-ionones for the inactive receptor, but within the range of its standard deviation the same for the active and the inactive receptor.
Analysis of α-ionone binding and antagonistic effect
Our previous experimental investigations2 showed α- and β-ionone to be competitive antagonist and agonist, respectively. To understand the molecular basis of this inhibition, we measured the cellular response of HEK293 cells to 50 µM β-ionone at varying concentrations of α-ionone2 via Ca2+-imaging. Figure 3a displays the resulting inhibition curve: a β-ionone/α-ionone concentration ratio of 2:1 led to 50% relative cellular response, a ratio of 1:1 to 36% cellular response, a ratio of 1:2 to 32% response, and a ratio of 1:4 to 20% cellular response. From this observation, we conclude that β-ionone and α-ionone compete for the same binding site, and that α-ionone exhibits the 1-2 – fold affinity of β-ionone for the receptor. However, as Fig. 1 shows, α-ionone exists as two different enantiomers, and the experimental investigations are based on a racemic mixture. To investigate the molecular mechanism of inhibition, we carried out docking runs with (R)- α-ionone, (S)- α-ionone, and β-ionone within the static initial ligand-free inactive PSGR model. As stated above, this is the structure that ionone would need to bind to according to the ternary complex model48. Figure 3b shows the respective best binding modes found. All three tested compounds are found in the same binding cavity, with their ionone ring positioned at the same position. The observed differences in binding result from the differences in stereochemistry at the connection between ring and butenoneyl side chain (in the following called “head group”). β-ionone and (R)- α-ionone bind in a very similar fashion with the head groups oriented towards Asp1905.47, while (S)- α-ionone binds with the head group oriented towards Lys1855.42. Both α-ionone enantiomers bind with a slightly higher affinity (∆Gbind = −6.4 kcal/mol and −6.9 kcal/mol) than β-ionone (∆Gbind = −6.1 kcal/mol). Furthermore, as free energy calculations performed on our MD trajectories (see Fig. 3c and Table SII) show that both α-ionone stereoisomers exhibit comparable and clearly separated affinities for the active and the inactive receptor conformation, with the affinity for the inactive conformation being significantly higher than the one for the active conformation. β-ionone exhibits an affinity for the receptor, which is comparable to the one of α-ionones for the inactive receptor, but is within the range of its standard deviation the same for the active and the inactive receptor. This result is in excellent agreement with the experimental observations: both α-ionones preferably bind to the inactive receptor conformation, and thus inhibit receptor activation. β-ionone exhibits a comparable affinity to both receptor conformations, and thus partially stabilizes the active conformation, leading to receptor activation. Due to the matching experimental and theoretical results, we assume that α-ionones bind to the same position as β-ionone, and act as orthosteric agonists opposed to a possible allosteric binding mode. In addition to this experiment, we investigated if our PSGR mutants exhibit a different response to co-application of rac- α-ionone and β-ionone. As Fig. 2b shows, while most mutants behave like WT receptors, we indeed observe an altered response for S1073.36V and K1855.42R: while S1073.36V activation receives a stronger block from rac- α-ionone than the WT, K1855.42R is less sensitive to the antagonist. This is a further indication that we have correctly identified the location of the odorant othosteric binding site within PSGR. In the following we will analyse the differences in dynamic binding modes of (R)- α-ionone, (S)- α-ionone and β-ionone in MD simulations to determine the molecular basis for ligand agonism/antagonism in PSGR.
Dynamic binding modes define ligand effect on PSGR
For a better sampling of ligand dynamics, we carried out three independent simulations with each ligand bound to the binding site as depicted in Fig. 3b. These ligand postures were generated by Docking as starting positions, but changed significant within the 40 ns equilibration phase of the respective models. Supplementary Figure S5 shows the resulting Cα-RMSD plots of the transmembrane helices for all simulation systems. All models stayed within a range of 2.5–3.5 Å in respect to the initial rhodopsin models. In our 50 ns runs of free MD simulations, we chose the last 10 ns for data assessment, as the protein models seemed structurally equilibrated and sufficiently stable during this period. Figure 4 depicts the ligand residence volumes of (S)- α-ionone, (R)- α-ionone, and β-ionone in both inactive and active model during these last 10 ns of MD simulation. We did not observe any specific binding position of ionones within the protein, as all three ligands were highly dynamic within the residence volumes. Furthermore, the comparison of binding positions of the correct ligand/protein model combinations (α-ionones in inactive protein conformation, β-ionone in active protein conformation) with the artificial residence volumes (α-ionones in active protein conformation, β-ionone in inactive protein conformation) shows that the residence volumes of all three ligands in both states exhibit a considerable overlap. Therefore, (R)- α-ionone, (S)- α-ionone and β-ionone should in theory be able to easily switch positions between active and inactive state – related positions. A difference in binding position alone cannot explain the selective activation of PSGR by β-ionone. In our earlier investigations, we found that ligand action on the protein is determined by distinct dynamic protein/ligand contacts (IFPs)38,41. In the following, we therefore analysed differences in such dynamic IFPs, and derived a model for the protein/protein and protein/ligand interactions relevant for protein activation. We here focus on the residues predicted by Docking to form part of a putative ligand binding site, and at the same time affect receptor activation upon mutation in experiments.

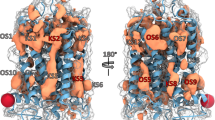
Selected ligand residence volumes during the last 10 ns of free MD simulation. Protein backbone in grey, selected surrounding protein residues in grey sticks. Helices numbered in roman numbers. (a) (S)- α-ionone. The dark blue mesh depicts the residence volume of (S)- α-ionone in the inactive state, and the light blue mesh depicts the residence volume in the active state. (b) (R)- α-ionone. The dark cyan mesh depicts the residence volume of (S)- α-ionone in the inactive state, and the light cyan mesh depicts the residence volume in the active state. (c) β-ionone. The dark orange mesh depicts the residence volume of β-ionone accessed in the inactive state model, and the light orange mesh the residence volume of β-ionone accessed in the active state model. The active and inactive state volumes of all ligands show a significant overlap. All ligands theoretically could transfer from their inactive state to their active state positions. Thus, binding position selectivity is no criterion for protein activation.
Table 2 displays dynamic protein/ligand contacts appearing in MD simulations with residues determined as crucial for protein activation via mutation analysis. Contacts were judged to be significant if they existed in at least two out of three simulation runs during >60% of simulated time. The following significant protein/ligand contacts were observed:
His1043.33: this residue only forms van der Waals-contacts with β-ionone, but not with α-ionones, in both active and inactive protein models. As the H1043.33F mutant is affected in its experimentally measured activity, His1043.33 seems to form part of the ligand cavity wall.
Ser1073.36: β-ionone forms significant contacts with this residue in the inactive state, but not in the active state. α-ionones do not exhibit any contacts with this residue. In experiment, mutation of Ser1073.36 to valine resulted in a hyperactive receptor. Therefore, Ser1073.36 might play a direct role in receptor activation by β-ionone. Concerning the increased efficiency of blockage by rac-α-ionone in S1073.36V, Table 2 shows that Ser1073.36 furthermore exhibits more frequent van der Waals contacts with α-ionones than with β-ionone. A hydrophobic mutation might increase the contact frequency with α-ionones, leading to better binding and thus blockage of β-ionone binding.
Ser1113.40: only a significant contact with (S)- α-ionone in the active state is observable. As (S)- α-ionone cannot activate the receptor, this is considered as an artificial contact. Nevertheless, Ser1113.40 was experimentally shown to be important for ligand-induced protein activation, as S1113.40V is an affected mutant (see Table 1).
Lys1855.42: no significant contacts are present for all ligands in the inactive conformation. In the active conformation, Lys1855.42 forms a significant van der Waals contact with β-ionone, but not with α-ionones. The experimentally observed affected activity/inactivity of the K1855.42L and K1855.42M mutants proves that this side chain is important for ligand-induced activation of PSGR. However, it further implies that the crucial feature of the Lys1855.42 side chain is its positive charge: a mutation retaining the charge distribution (K1855.42R) is still fully active. The K1855.42Q mutant, which does not exhibit a side chain charge, but still exhibits hydrogen bond capacity, is affected in its activity. Last, both K1855.42R and K1855.42Q exhibit a decreased basal activity, which further supports the involvement of the Lys1855.42 side chain in protein activation. Concerning the decreased efficiency of blockage by rac-α-ionone in K1855.42R, it might be possible that an intermediate distance charge interaction between the Lys1855.42 ammonium moiety and the α-ionone carbonyl atom can be formed in WT protein, similar to the situation in the Docking pose of (S)- α-ionone depicted in Fig. 3b, which is not present for β-ionone. The K1855.42R mutation therefore would affect (S)- α-ionone binding more than β-ionone binding.
Asp1905.47: this residue forms van der Waals contacts with β-ionone in the inactive state, but not in the active state, while (S)- α-ionone exhibits a contact pattern, which shows exactly the opposite behavior. (R)- α-ionone forms significant van der Waals contacts with this residue in both active and inactive state. Like in the case of Lys1855.42, the experimental mutant data implies that the side chain charge is the crucial feature for protein activation: while the D1905.47L mutant is affected in both ligand induced and basal activity, the D1905.47N mutant, which retains the possibility to form a hydrogen bond, is completely inactive.
Asn1945.51: as in the case for Ser1113.40, no significant protein/ligand contacts can be observed. However, as the N1945.51L mutant is experimentally inactive, Asn1945.51 has to be important for ligand-induced protein activation.
Tyr2516.48: in its inactive state, this residue forms van der Waals contacts with both α- and β-ionone. In the active state however, only a van der Waals contact with α-ionone is present. Like Ser1073.36, this residue therefore might play a significant role in protein/ligand activation. In agreement with this, in our sequence alignment used for creating the PSGR homology model, Tyr2516.48 is found at the position of Trp2656.48, the “rotameric toggle switch” in rhodopsin-like GPCRs59, which is crucial for the outward movement of helix VI, leading to G-protein binding and activation. A recent investigation found that this substitution of this tryptophane by a tyrosine is a general feature for the whole family of olfactory receptors57. However, the experimental mutant data shows that the hydrogen bonding capability of the tyrosine side chain seems to be the crucial determinant for ligand-induced protein activation: the Y2516.48F mutant is inactive.
From this list, it appears that the ligand does not form any side chain hydrogen bonds at all during simulation. As can be seen in Supplementary Table SII, even hydrogen bonds with the protein backbone are only observed for (S)- α-ionone in the active state, which is an artificial state. Selectivity of PSGR for ionones therefore seems to exclusively result from van der Waals contacts. However, it seems that van der Waals protein/ligand contacts can only explain the effect of the mutants of His1043.33 and Ser1073.36. The remaining mutants are either not easily explainable (Lys1855.42/Asp1905.47/Tyr2516.48) or not even coupled to protein/ligand contacts (Ser1113.40/Asn1945.51). It is possible that in the case of these residues, not only OR/ligand, but protein-internal contacts bonds are crucial for protein activation. In the following, we will therefore monitor the possible presence of protein/protein side chain interactions between these residues.
Table 3 displays such significant protein/protein contacts appearing during simulation. Judged by these data, (R)- α-, (S)- α- and β-ionone differ by the following protein/protein contacts:
Lys1855.42/Asp1905.47: both amino acids exhibit a salt bridge with β-ionone and (R)- α-ionone in the inactive state, but not in the active state. In simulations with (S)- α-ionone, this salt bridge is not significantly present. The presence of the salt bridge is in good agreement with our experimental mutant data: a mutation retaining the charge distribution (K1855.42R) is still fully active. Mutations quenching side chain charges, while still exhibiting hydrogen bond capacity (K1855.42Q, D1905.47N) are either affected or inactive. Lys1855.42 and Asp1905.47 therefore seem to be connected via a salt bridge, which needs to be formed in the inactive state, and to be broken by ligand binding to activate the receptor. This is in agreement with β-ionone exhibiting van der Waals contacts with Lys1855.42 in the active, but not in the inactive state. None of the (S)- α-ionone-bound models exhibits a Lys1855.42/ligand contact or the Lys1855.42/Asp1905.47 salt bridge, indicating a different protein side chain conformation distribution upon binding of this ligand. However, (R)- α-ionone behaves like β-ionone, allowing no clear discrimination of antagonism and agonism solely based on this intraprotein contact. We have to add here that the rupture of this salt bridge by an odorant would be energetically highly unfavourable. However, in simulations, we observe that the salt bridge is actually not broken, but shifted from Lys1855.42/Asp1905.47 to Asp1825.39/Lys1855.42 (see Table 3 and Figure S6), cancelling this enthalpic penalty.
Ser1113.40/Asn1945.51: similar to the case of Lys1855.42/Asp1905.47, Ser1113.40/Asn1945.51 exhibit a hydrogen bond, which exists in the inactive, β-ionone-bound state, while it is not significant in the active states and in simulations of α-ionones. The existence of this hydrogen bond in the inactive state and subsequent breakage of the bond seems therefore to be a prerequisite for receptor activation. Unlike in the case of the Lys1855.42/Asp1905.47 salt bridge, none of the two residues exhibits a significant van der Waals contact with any of the ligands. The alteration of the present hydrogen bond present here therefore seems to be an indirect effect of ligand binding.
Ser1073.36/Tyr2516.48: these two residues exhibit a hydrogen bond in the active, β-ionone bound state, which is not present in any other state. This coincides with both α-ionones and β-ionone forming a van der Waals contact with Tyr2516.48 in the inactive state, while in the active state, only (S)- α-ionone forms this contact. It therefore seems that formation of this hydrogen bond is crucial for protein activation, which is hindered by bound α-ionone molecules. This finding is in agreement with the observation that the “rotameric toggle” tyrosine in helix VI of olfactory receptors forms interactions with amino acid side chains in helix III57.
Furthermore, we checked if our investigated residues are found at evolutionary conserved or variable positions: residues conserved within the OR family are believed to be involved in receptor activation57, while hyper variable residues constitute to ligand selectivity60. We find that the interaction pair Ser1073.36/Tyr2516.48 was found to be a conserved contact motif crucial for receptor activation57, although position 3.36 is mostly occupied by a small hydrophobic residues, and that this contact is mostly a van der Waals contact. All other residues are highly variable throughout the OR family. Therefore, while ligand induced interruption of the Ser1073.36/Tyr2516.48 contact pair observed by us might be a conserved activation feature, the combination of all other residues forming the ligand binding site, and the resulting contact pairs Ser1113.40/Asn1945.51 and Lys1855.42/Asp1905.47 seem to be a specific feature of PSGR.
Summarizing these results, binding of α-ionones and β-ionone to PSGR seems to be coupled to the rearrangement of a protein-internal hydrogen bond network, which consists of the residues Ser1073.36, Ser1113.40, Lys1855.42, Asp1905.47, Asn1945.51, and Tyr2516.48, and is displayed in Fig. 5. This network links helices III (Ser1073.36, Ser1113.40) with V (Lys1855.42, Asp1905.47, Asn1945.51) and VI (Tyr2516.48), which are all known to change their relative positions upon receptor activation61. The modulation of this network is not achieved via hydrogen bond interaction between receptor and ligands, but via van der Waals interactions, which affect amino acid side chain conformations. Such a ligand-mediated modulation further offers an explanation for the weak effect of the K1855.42L and D1905.47L mutants, and the hyperactivity of the S1073.36V mutant: here, van der Waals contacts with the ligand might compensate for the missing crucial hydrogen bond connections listed above. Further support for this model comes from in silico data on changes of ligand binding affinities in a virtual alanine scanning we performed as displayed in Table 1: all investigated mutants, with the exception of I2556.52, exhibit a change in ligand affinity upon mutation to alanine in silico for all investigated ligands, which is in agreement with experiments on β-ionone, and further supported by the data on mutation effect on activation inhibition by α-ionones. The control mutant residue I2556.52 does not exhibit a significant influence on ligand binding. Interestingly, we find again that the best rationale between experiment and our theoretical data is provided by our hypothesis that disruption of intraprotein interaction partner connections by the ligand: The influence of mutation of Ser1113.40 cannot be explained by the impact of this residue on ligand binding affinities in the in silico alanine scanning. However, its interaction partner Asn1945.51 exhibits a significant impact on ligand binding, respectively. More pronounced even is this effect in the case of the Lys1855.42/Asp1905.47 pair: here, while Lys1855.42 only shows a weak influence on ligand binding, its interaction partner Asp1905.47 exhibits a strong influence.

Proposed protein-internal hydrogen bond network in PSGR in the active state. The figure represents a snapshot out of the last 10 ns of free MD simulation (1st β-ionone simulation in the active state listed in Table 2). The protein backbone in yellow, β-ionone in orange sticks, hydrogen bond network forming protein residues in grey sticks. Helices numbered in roman numbers. Upon protein activation, the Ser1113.40/Asn1945.51 hydrogen bond.and the Lys1855.42/Asp1905.47 salt bridge are broken. A new hydrogen bond is formed between Ser1073.36 and Tyr2516.48.
Discussion
Taking together our results so far, we see a good agreement between our theoretically predicted binding site model and the experimental point mutation analysis. Like in the case of our study on the olfactory receptor hOR2AG138, receptor activation is achieved by modulation of the dynamics of a hydrogen bond system. However, in contrast to hOR2AG1, the respective hydrogen bond network in this case does not connect protein and ligand, but is completely protein-internal, connecting helices III, V, and VI. Especially the connection of helices III and VI via a “rotameric toggle” tyrosine is known to be an essential feature in olfactory receptor activation57. The ligands merely alter the connectivity via dynamic van der Waals interactions with amino acid side chains belonging to the hydrogen bond network. This mode of ligand-induced changes of protein-internal connections is in good agreement with the model that ligands alter the overall energetic landscape of possible protein conformations62,63. Interestingly, the two α-ionones do not induce an identical contact pattern: while they have the same effect on the interhelical hydrogen bonds of Ser1073.36/Tyr2516.48 and Ser1113.40/Asn1945.51, they differ in the effect on the hydrogen bond of Lys1855.42/Asp1905.47. We assume that this intrahelical salt bridge is less important for receptor activation than the interhelical hydrogen bonds, which are more likely to influence the packing and dynamics of the transmembrane helix bundle. Ligand recognition is highly stereo selective, as the investigated (R)- α-, (S)- α- and β-ionone majorly differ in the angle connecting the ionone ring and the butenoneyl side chain. To our surprise, we observe almost no hydrogen bonds to the keto moiety, which excludes this group as a possible pharmacophor for ligand recognition. At first glance, the absence of such a hydrogen bond seems energetically unfavourable, as it would require a desolvation of the keto moiety upon binding from the solvent. However, ionones are highly hydrophobic compounds, and therefore most likely do not enter the receptor from the solvent phase, but first enter the hydrophobic volume created by the membrane lipids, and then enter the protein from the membrane/protein interface, as it is the case with retinal in rhodopsin64. This cancels out the desolvation step during binding, removing this enthalpic penalty. It seems that the receptor simply does not form any hydrogen bond to the ligands, and relies on shape recognition via van der Waals interactions. This suggestion fits with the experimentally observed low affinity of ionones and PSGR, and the binding mode of β-ionone bound to rhodopsin in an x-ray structure (PDB ID 3OAX)65. The receptor therefore seems not to recognize the keto moiety as odotope31,32,33,34, but the whole shape of the ligands. While molecular models of OR/ligand interaction so far have mostly taken van der Waals contacts into account as elements of the binding site defining ligand binding affinity33,38,39,50,51,52, we here find them to be actively involved in receptor activation. At first glance, this result is in agreement with the recent findings of Baud et al.40, who used Docking on mOR-EG as major tool to assess receptor activation, and with comparisons of crystal structures of other classes of GPCRs in their active and inactive state66,67. However, we here find in our MD simulations that these van der Waals contacts are highly dynamic, and do not result in a singular stable ligand binding position. The resulting contact pattern seems to be specific enough to cause receptor activation only for β-ionone, but not for α-ionones. We do not expect this result to be an artefact from the modelling procedure, as our model can nicely explain the experimental data on amino acid mutations. We do not assume that the connection patterns found between PSGR and the tested ionones are representative for all ligands that possibly can activate PSGR. However, we believe that the general principle of activation, namely the modulation of protein-internal side chain contact frequencies, is valid for all olfactory GPCRs. We here have to state that in contrast to this low affinity, the detection threshold for β-ionone in the human nose is extremely low (<1ppb, i.e. <44 pM)68. It is well recognized that heterologous expression systems for ligand screening differ in composition of the signalling machinery compared to the native cellular environment. As such, our luciferase assay does not use the native olfactory signal cascade present in olfactory cilia. Consequently, the signalling efficacy might differ between our in vitro system and the human nose. Furthermore, we cannot exclude the missing presence of metal cofactors in the experiments69. However, PSGR signalling seems to depend on the cell type. PSGR in cancer cells does not signal via the native Golf, but hijacks different signalling pathways18,19 as well, which may account for the decreased sensitivity of these cells to β-ionone as well as different response kinetics compared to olfactory sensory neurons. While the receptor exhibits a low overall affinity for ionones and seemingly lacks a stable binding mode, it is highly stereo selective and can discriminate between the subtle structural differences of α-ionones and β-ionone.
In summary, we investigate the molecular interaction of the antagonist/agonist combination (R)- α-/ (S)- α-/ β-ionone on the ectopically expressed olfactory receptor PSGR, which is a potential anti-cancer drug target for prostate cancer, and especially the difficult to treat melanoma cancer. Based on our combined theoretical and experimental investigations, we suggest that this OR is activated by a ligand-induced rearrangement of a protein-internal hydrogen bond network. Like in our earlier investigations on hOR2AG1, this is achieved by the dynamic interaction between receptor and odorants38. Though the investigated ligands only exhibit a small geometric difference, the dynamic protein/ligand interaction pattern differs significantly. The network rearrangement is not performed by competing hydrogen bond interaction of the ligand with the network, but by van der Waals contacts of the ligand with the involved amino acid side chains, altering their conformations. Thus, the odotope recognition in this OR is highly stereo selective, but seemingly lacks any selectivity based on protein/ligand hydrogen bonds. In agreement with this, the experimentally observed affinity for β-ionone is very low, with threshold concentrations in the higher µM range. While the absence of a hydrogen bond between receptor and ionones seems surprising, binding of β-ionone to a GPCR (in this case: rhodopsin) has already been experimentally observed in an x-ray crystal structure65. As mentioned above, a further reason for the observed low affinity and efficacy might be the usage of a luciferase assay, which does not work via the native olfactory signal cascade. However, as this resembles the situation within prostate cancer cells18,19, the luciferase assay might be a valid model for signalling in ectopically expressed ORs. A future OR-based drug design scheme will have to be based on reproducing the correct dynamic protein/ligand IFPs41 instead of simply searching for complementarity of ligand and binding pocket shape. Furthermore, low-affinity ORs will need to be targeted with local high-dose drug applications.
Methods
Computational Methods
PSGR homology models were build by dynamic homology modelling42. The sequence of PSGR (Uniprot70 accession number Q9H255) was aligned to the one of rhodopsin (see Figure S1 for the alignment) with a focus on reproducing positions of the highest conserved helical residues according to Ballesteros-Weinstein numbering55. We chose rhodopsin as structural template, as is has been proven to be a good basis for modelling olfactory receptors38,47, and is the only GPCR that contains a hydrophobic ligand and is at the same time available as structural model in both an inactive and an active-like conformation. The model of PSGR in its inactive state was based on the inactive rhodopsin structure (PDB entry 1U19)46, while the model of active PSGR was based on the octylglycoside-bound active opsin structure (PDB ID 4J4Q)47. Ionone topologies and structures were obtained from the PRODRG server71. Ligand atomic charges were obtained from RESP charges from B3LYP/6-31 G* calculations42 in Gaussian0972. Docking was performed with Autodock Vina43, using a box of 20 × 20 × 20 Å3 with a grid spacing of 1 Å, covering the extracellular half of the protein. For conformational search, we kept the side chain single bonds in the ionones rotatable, and used an exhaustiveness factor of 8041. The most stable binding mode of each ligand (see Fig. 3b) was used as initial structure for simulation system creation. Protonation states were assigned with PROPKA73, with Asp69 and Glu110 being protonated. MD simulations were carried out with GROMACS (v4.0.5)74 and the GROMOS96 force field with Berger lipid parameters75 following the protocol of Wolf et al.42 with the receptor model being embedded in an explicit membrane (POPC)/solvent environment. Membrane insertion was performed with inflategro76. After addition of SPC water molecules, a steepest descent minimization with harmonic restraints on protein and ligand atomic coordinates (1000 kJ mol−1 nm−2) was followed by 5 ns of membrane/solvent equilibration with harmonic restraints on protein and ligand atomic coordinates (1000 kJ mol−1 nm−2). The full system was then energetically minimized using the steepest descent algorithm without any restraints. After velocity assignment for temperature generation (310 K), two consecutive steps of 100 ps MD simulations with stepwise reduced restraints (500 and 200 kJ mol−1 nm−2, respectively) followed, and free MD production runs without any restraints were carried out for 50 ns with 3 independent replica runs each (differing in their initial velocity distribution). We chose 50 ns as a suitable simulation length, as homology models with docked ligands quickly find into suitable binding poses41,42, but tend to diverge from a comparable crystal-structure based dynamic model: Figure S7 displays a short 10 ns MD simulation comparing the 7TM Cα RMSD of a beta(2) adrenergic receptor (B2AR) model build from a rhodopsin structure (PDB ID 1U1946) in comparison to a simulation of a beta(2) adrenergic receptor crystal structure (PDB ID 2RH177). Already within this short time period, the RMSD quickly reaches 3.5 Å, which stands for a not reliable overall structure. Contrary to general belief, a MD simulation with a homology model therefore does not necessarily improve the overall structural model. This result might be discouraging concerning the usability of GPCR homology models in general. However, for the same example of a B2AR homology model, we found that when focussing on the binding site itself (ref.41, Figure S4), the model actually results in the right receptor/ligand contacts within 5 ns, and continues to exhibit this contact pattern for 95 ns. As summary of these two points, we judged that when using GPCR homology models to analyse protein/ligand interactions, it is more important to use repetitions of short trajectories instead of few long simulations. Taking the structural development of our PSGR models into account (see Figure S5), we took 50 ns simulation time as a good compromise. Replica runs were generated by the addition of different velocity distributions to each starting structure after the 2nd minimisation. In each MD simulation period, the simulation system was held at 310 K with a Nose-Hoover thermostat (τT = 0.1 ps), and at a pressure of 1 bar with a Parinello-Rahman barostat (τP = 0.5 ps; semiisotropic coupling of membrane area axes and the transmembrane axis). As the transmembrane helix Cα-RMSD showed to be stable in all simulation systems during the last 10 ns of simulated time (see Figure S5), we used these 10 ns for data assessment. Residue contacts were evaluated with MOBY78. Ligand residence volumes were calculated based on snapshots taken each 0.1 ns during the assessment period of the last 10 ns from all replica runs. Free energy calculations were performed with GMXPBSA2.179,80 As both α- and β-ionone are constitutional isomers with the same number and types of chemical bonds, the rotational and vibrational contribution to the overall entropy can be neglected while calculating relative and not absolute binding affinities. We needed to exclude two trajectories from the inactive state as outliers, as in these cases, the ligands spend a significant amount of time at the protein/membrane interface with parts of their structure, which led to an underestimation of the nonpolar ligand interaction energy (as no membrane is present in the energy evaluation process).
Mutagenesis by Overlap Extension PCR and Cloning
Point mutations in PSGR were introduced at different positions using overlap extension PCR. Full-length rho1D4-tagged PSGR in pCI (Addgene, Cambridge, Massachusetts (MA), USA) served as a template. Initial PCRs provided mutated gene segments, with overlapping complementary 3′ ends carrying the desired point mutation, which were then mixed and used as a template for a subsequent PCR to generate the full-length product using flanking primers. Full-length primers included NotI and MluI restriction sites for further cloning into pCI (Promega, Madison, Wisconsin (WI), USA). The nucleotide sequence of the mutants was verified by sequencing. The primer pairs used for mutations are listed in the Supplementary Materials.
Immunocytochemistry
Live-cell immunocytochemistry to evaluate cell-surface expression of PSGR variants was performed as described by Zhuang and Matsunami81 (see Supplementary Figure S2). Pictures were taken with Zeiss Axioskop 2 fluorescence microscope and Axiovision software (Zeiss, Jena, Germany).
Luciferase assay
The Dual-Glo Luciferase Assay System (Promega, Madison, Wisconsin, USA) was used to measure cellular responses as an indirect measure of receptor activation as previously described. It is the most commonly used method for high-throughput screening of odorant-receptor pairs81,82. This method quantifies cellular responses as an indirect measure of odorant receptor activation Hana3A cells seeded on a 96-well plate (NUNC, Thermo Fisher Scientific, Waltham, Massachusetts (MA), USA) were transfected with Lipofectamine 2000 (Invitrogen, Invitogen, Carlsbad, California (CA), USA) according to the manufacturer’s protocol using 18 µl Lipofectamine, 1 µg of RTP1S plasmid83, 1 µg of pRL-TK-Renilla (Promega), 2 µg of pGL4.29-luciferase (Promega), 1 µg of hM384 and 5 µg of plasmids encoding for olfactory receptors for an entire well plate. 18–24 h after transfection, transfection medium was removed and replaced with the appropriate concentration of odorant, 0.1% DMSO (negative control) or 10 µM forskolin (positive control) in CD293 medium (Gibco; Life Technologies, Carlsbad, CA, USA) with 2 mM L-glutamine. Ultrapure β-ionone was a generous gift of Dr. J. Panten (Symrise, Holzminden, Germany). rac-α-ionone was purchased from Sigma-Aldrich. Odorant stocks were diluted in DMSO (Sigma–Aldrich, Munich, Germany). Four hours after odor stimulation, luminescence was measured using the microplate reader Fusion (Packard BioScience, Meriden, Connecticut (CT), USA). Firefly luminescence values were divided by the Renilla luciferase activity to control for transfection efficiency in a given well. The firefly-Renilla luciferase ratio was normalized against the lowest/highest luciferase ratios obtained for that experiment. Normalized luciferase activity was calculated by the formula (Luc/Ren(N) − Luc/Ren(lowest))/(Luc/Ren(highest) − Luc/Ren(lowest), where Luc/Ren(N) is the luminescence of firefly luciferase divided by luminescence of Renilla luciferase in a certain well; Luc/Renilla(lowest) is the lowest luciferase ratio of PSGR mutants transfected cells to negative control; Luc/Ren(highest) is the maximum luciferase ratio of PSGR mutant- transfected cells to the positive control of a plate. Mock-transfected cells were stimulated to exclude unspecific responses to the tested compounds. The dose response curves were fitted by Hill equation. The threshold concentration (10% signalling output level of 10 µM forskolin) was determined using dose response curve. The luciferase value of receptor mutants after stimulation with 250 µM β-ionone (highest applicable odorant concentration) was normalized to the value of wild type receptor to calculate the Emax value. Data were analysed with Microsoft Excel and SigmaPlot.
Calcium imaging
HEK293 cells were grown in 35-mm cell culture dishes (50% confluence) and transfected with pcDNA3-PSGR (1 µg) using a standard calcium-phosphate precipitation technique. After 48 hours cells were incubated for 30 minutes at 37 °C with Ringer’s solution (140 mM NaCl, 5 mM KCl, 10 mM HEPES, 2 mM CaCl2, 1 mM MgCl2; pH 7.4) and 3 µM Fura-2-AM (Life Technologies, Carlsbad, California, USA). Calcium imaging experiments were performed as described in Spehr et al.5 and Neuhaus et al.2.
References
Buck, L. & Axel, R. A novel multigene family may encode odorant receptors: A molecular basis for odor recognition. Cell 65, 175–187 (1991).
Neuhaus, E. M. et al. Activation of an Olfactory Receptor Inhibits Proliferation of Prostate Cancer Cells. J. Biol. Chem. 284, 16218–16225 (2009).
Flegel, C., Manteniotis, S., Osthold, S., Hatt, H. & Gisselmann, G. Expression Profile of Ectopic Olfactory Receptors Determined by Deep Sequencing. Plos One 8, e55368 (2013).
Braun, T., Voland, P., Kunz, L., Prinz, C. & Gratzl, M. Enterochromaffin Cells of the Human Gut: Sensors for Spices and Odorants. Gastroenterology 132, 1890–1901 (2007).
Spehr, M. et al. Identification of a testicular odorant receptor mediating human sperm chemotaxis. Science 299, 2054–2058 (2003).
Zhao, W. et al. Decreased level of olfactory receptors in blood cells following traumatic brain injury and potential association with tauopathy. J. Alzheimers Dis. 34, 417–429 (2013).
Busse, D. et al. A Synthetic Sandalwood Odorant Induces Wound-Healing Processes in Human Keratinocytes via the Olfactory Receptor OR2AT4. J. Invest. Dermatol. 134, 2823–2832 (2014).
Gelis, L. et al. Functional Characterization of the Odorant Receptor 51E2 in Human Melanocytes. J. Biol. Chem. jbc.M116.734517, https://doi.org/10.1074/jbc.M116.734517 (2016).
Weng, J. S. et al. PSGR2, a novel G-protein coupled receptor, is overexpressed in human prostate cancer. Int. J. Cancer 118, 1471–1480 (2006).
Wang, J. H. et al. The prostate-specific G-protein coupled receptors PSGR and PSGR2 are prostate cancer biomarkers that are complementary to alpha-methylacyl-CoA racemase. Prostate 66, 847–857 (2006).
Fuessel, S. et al. Transcript quantification of Dresden G protein-coupled receptor (D-GPCR) in primary prostate cancer tissue pairs. Cancer Lett. 236, 95–104 (2006).
Cui, T. et al. Olfactory Receptor 51E1 is a Potential Novel Tissue Biomarker for the Diagnosis of Small Intestine Neuroendocrine Tumors. Pancreas 42, 373–373 (2013).
Giandomenico, V. et al. Olfactory receptor 51E1 as a novel target for diagnosis in somatostatin receptor-negative lung carcinoids. J. Mol. Endocrinol. 51, 277–286 (2013).
Maßberg, D. et al. Monoterpene (−)-citronellal affects hepatocarcinoma cell signaling via an olfactory receptor. Arch. Biochem. Biophys. 566, 100–109 (2015).
Manteniotis, S. et al. Functional characterization of the ectopically expressed olfactory receptor 2AT4 in human myelogenous leukemia. Cell Death Discov. 2, 15070 (2016).
Gelis, L., Jovancevic, N., Bechara, F., Neuhaus, E. M. & Hatt, H. Functional expression of olfactory receptors in human primary melanoma and melanoma metastasis. Exp. Dermatol. 26, 569–576 (2017).
Xu, L. L. et al. Quantitative expression profile of PSGR in prostate cancer. Prostate Cancer P. D. 9, 56–61 (2006).
Spehr, J. et al. G Protein-coupled Receptor Signaling via Src Kinase Induces Endogenous Human Transient Receptor Potential Vanilloid Type 6 (TRPV6) Channel Activation. J. Biol. Chem. 286, 13184–13192 (2011).
Wiese, H. et al. Quantitative phosphoproteomics reveals the protein tyrosine kinase Pyk2 as a central effector of olfactory receptor signaling in prostate cancer cells. Biochim. Biophys. Acta 1854, 632–640 (2015).
Schatton, T. & Frank, M. H. Review Article: Cancer stem cells and human malignant melanoma. Pigm. Cell Melanoma Res. 21, 39–55 (2007).
Center, M. M. et al. International variation in prostate cancer incidence and mortality rates. Eur. urol. 61, 1079–1092 (2012).
Wolf, S. & Grünewald, S. Sequence, Structure and Ligand Binding Evolution of Rhodopsin-Like G Protein-Coupled Receptors: A Crystal Structure-Based Phylogenetic Analysis. Plos One 10, e0123533 (2015).
Firestein, S. How the olfactory system makes sense of scents. Nature 413, 211–218 (2001).
Firestein, S., Araneda, R. C. & Kini, A. D. The molecular receptive range of an odorant receptor. Nat Neurosci 3, 1248–1255 (2000).
Oka, Y., Omura, M., Kataoka, H. & Touhara, K. Olfactory receptor antagonism between odorants. EMBO J. 23, 120–126 (2004).
Sanz, G., Schlegel, C., Pernollet, J.-C. & Briand, L. Comparison of odorant specificity of two human olfactory receptors from different phylogenetic classes and evidence for antagonism. Chem. Senses 30, 69–80 (2005).
Shirokova, E. et al. Identification of Specific Ligands for Orphan Olfactory Receptors: G protein-dependent agonism and antagonism of odorants. J. Biol. Chem. 280, 11807–11815 (2005).
Jacquier, V., Pick, H. & Vogel, H. Characterization of an extended receptive ligand repertoire of the human olfactory receptor OR17-40 comprising structurally related compounds. J. Neurochem. 97, 537–544 (2006).
Bavan, S., Sherman, B., Luetje, C. W. & Abaffy, T. Discovery of Novel Ligands for Mouse Olfactory Receptor MOR42-3 Using an In Silico Screening Approach and In Vitro Validation. Plos One 9, e92064 (2017).
Katritch, V., Cherezov, V. & Stevens, R. C. Diversity and modularity of G protein-coupled receptor structures. Trends Pharmacol Sci 33, 17–27 (2012).
Shepherd, G. M. Discrimination of molecular signals by the olfactory receptor neuron. Neuron 13, 771–790 (1994).
Kraft, P., Bajgrowicz, J. A., Denis, C. & Fráter, G. Odds and Trends: Recent Developments in the Chemistry of Odorants. Angew. Chem. Int. Ed. 39, 2980–3010 (2000).
Doszczak, L. et al. Prediction of Perception: Probing the hOR17-4 Olfactory Receptor Model with Silicon Analogues of Bourgeonal and Lilial. Angew. Chem. Int. Ed. 46, 3367–3371 (2007).
Sanz, G. et al. Relationships Between Molecular Structure and Perceived Odor Quality of Ligands for a Human Olfactory Receptor. Chem. Senses 33, 639–653 (2008).
Wolf, S., Gelis, L., Dörrich, S., Hatt, H. & Kraft, P. Evidence for a shape-based recognition of odorants in vivo in the human nose from an analysis of the molecular mechanism of lily-of-the-valley odorants detection in the Lilial and Bourgeonal family using the C/Si/Ge/Sn switch strategy. Plos One 12, e0182147 (2017).
Turin, L. A spectroscopic mechanism for primary olfactory reception. Chem. Senses 21, 773–791 (1996).
Brookes, J., Horsfield, A. & Stoneham, A. The Swipe Card Model of Odorant Recognition. Sensors 12, 15709–15749 (2012).
Gelis, L., Wolf, S., Hatt, H., Neuhaus, E. M. & Gerwert, K. Prediction of a ligand-binding niche within a human olfactory receptor by combining site-directed mutagenesis with dynamic homology modeling. Angew. Chem. Int. Ed. Engl. 51, 1274–1278 (2012).
Topin, J. et al. Discrimination between Olfactory Receptor Agonists and Non-agonists. Chem. Eur. J. 20, 10227–10230 (2014).
Baud, O. et al. Exchanging ligand-binding specificity between a pair of mouse olfactory receptor paralogs reveals odorant recognition principles. Sci Rep 5, 14948 (2015).
Schneider, M., Wolf, S., Schlitter, J. & Gerwert, K. The structure of active opsin as a basis for identification of GPCR agonists by dynamic homology modelling and virtual screening assays. FEBS Lett. 585, 3587–3592 (2011).
Wolf, S., Böckmann, M., Höweler, U., Schlitter, J. & Gerwert, K. Simulations of a G protein-coupled receptor homology model predict dynamic features and a ligand binding site. FEBS Lett. 582, 3335–3342 (2008).
Trott, O. & Olson, A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 (2010).
Deng, Z., Chuaqui, C. & Singh, J. Structural Interaction Fingerprint (SIFt): A Novel Method for Analyzing Three-Dimensional Protein−Ligand Binding Interactions. J. Med. Chem. 47, 337–344 (2004).
Kooistra, A. J., Leurs, R., de Esch, I. J. P. & de Graaf, C. Structure-Based Prediction of G-Protein-Coupled Receptor Ligand Function: A β-Adrenoceptor Case Study. J. chem. inf. Model. 55, 1045–1061 (2015).
Okada, T. et al. The Retinal Conformation and its Environment in Rhodopsin in Light of a New 2.2 Å Crystal Structure. J. Mol. Biol. 342, 571–583 (2004).
Park, J. H. et al. Opsin, a structural model for olfactory receptors? Angew. Chem. Int. Ed. Engl. 52, 11021–11024 (2013).
Kenakin, T. Efficacy at G-protein-coupled receptors. Nat. Rev. Drug Disc. 1, 103–110 (2002).
Kufareva, I., Rueda, M., Katritch, V., Stevens, R. C. & Abagyan, R. Status of GPCR Modeling and Docking as Reflected by Community-wide GPCR Dock 2010 Assessment. Structure 19, 1108–1126 (2011).
Abaffy, T., Malhotra, A. & Luetje, C. W. The molecular basis for ligand specificity in a mouse olfactory receptor: a network of functionally important residues. J. Biol. Chem. 282, 1216–1224 (2007).
Launay, G. et al. Automatic modeling of mammalian olfactory receptors and docking of odorants. Protein Eng. Des. Sel., 25, 377–386 (2012).
Li, Y. et al. Aldehyde recognition and discrimination by mammalian odorant receptors via functional group-specific hydration chemistry. ACS Chem. Biol. 9, 2563–2571 (2014).
Man, O. Prediction of the odorant binding site of olfactory receptor proteins by human-mouse comparisons. Protein Sci. 13, 240–254 (2004).
Sekharan, S. et al. QM/MM Model of the Mouse Olfactory Receptor MOR244-3 Validated by Site-Directed Mutagenesis Experiments. Biophys. J. 107, L5–L8 (2014).
Ballesteros, J. & Weinstein, H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In Methods in Neurosciences (eds Sealfon, S. & Conn, P.) 25, 366–428 (1995).
Yu, Y. et al. Responsiveness of G protein-coupled odorant receptors is partially attributed to the activation mechanism. Proc. Natl. Acad. Sci. USA 112, 14966–14971 (2015).
de March, C. A. et al. Conserved Residues Control Activation of Mammalian G Protein-Coupled Odorant Receptors. J. Am. Chem. Soc. 137, 8611–8616 (2015).
Sanz, G. et al. Structurally related odorant ligands of the olfactory receptor OR51E2 differentially promote metastasis emergence and tumor growth. Oncotarget, 8, 4330–4341 (2017).
Shi, L. beta 2 Adrenergic Receptor Activation. Modulation of the proline kink in transmembrane 6 by a rotamer toggle switch. J. Biol. Chem. 277, 40989–40996 (2002).
Pilpel, Y. & Lancet, D. The variable and conserved interfaces of modeled olfactory receptor proteins. Protein Sci. 8, 969–977 (1999).
Tehan, B. G., Bortolato, A., Blaney, F. E., Weir, M. P. & Mason, J. S. Unifying family A GPCR theories of activation. Pharmacol. Therapeut. 143, 51–60 (2014).
Kim, T. H. et al. The Role of Ligands on the Equilibria Between Functional States of a G Protein-Coupled Receptor. J. Am. Chem. Soc. 135, 9465–9474 (2013).
Lamichhane, R. et al. Single-molecule view of basal activity and activation mechanisms of the G protein-coupled receptor β2AR. Proc. Natl. Acad. Sci. USA 112, 14254–14259 (2015).
Piechnick, R. et al. Effect of channel mutations on the uptake and release of the retinal ligand in opsin. Proc. Natl. Acad. Sci. USA 109, 5247–5252 (2012).
Makino, C. L., Riley, C. K., Looney, J., Crouch, R. K. & Okada, T. Binding of More Than One Retinoid to Visual Opsins. Biophys. J. 99, 2366–2373 (2010).
Kobilka, B. K. Structural insights into adrenergic receptor function and pharmacology. Trends Pharmacol Sci 32, 213–218 (2011).
Congreve, M., Oswald, C. & Marshall, F. H. Applying Structure-Based Drug Design Approaches to Allosteric Modulators of GPCRs. Trends Pharmacol Sci. 38, 837–847 (2017).
Plotto, A., Barnes, K. W. & Goodner, K. L. Specific Anosmia Observed for b-Ionone, but not for a-Ionone: Significance for FlavorResearch. J. Food Sci. 71, S401–S406 (2006).
Wang, J., Luthey-Schulten, Z. A. & Suslick, K. S. Is the olfactory receptor a metalloprotein? Proc. Natl. Acad. Sci. USA 100, 3035–3039 (2003).
The UniProt Consortium. Activities at the Universal Protein Resource (UniProt). Nucleic Acids Res 42, D191–D198 (2013).
Schuttelkopf, A. W. & van Aalten, D. M. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D 60, 1355–1363 (2004).
Frisch, M. J. et al. Gaussian 09. Gaussian Inc (2009).
Olsson, M. H., Søndergaard, C. R., Rostkowski, M. & Jensen, J. H. PROPKA3: consistent treatment of internal and surface residues in empirical p K a predictions. J. Chem. Theory Comput. 7, 525–537 (2011).
Hess, B., Kutzner, C., Van Der Spoel, D. & Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 4, 435–447 (2008).
Berger, O., Edholm, O. & Jähnig, F. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys. J. 72, 2002–2013 (1997).
Schmidt, T. H. & Kandt, C. LAMBADA and InflateGRO2: efficient membrane alignment and insertion of membrane proteins for molecular dynamics simulations. J.chem. inf. Model. 52, 2657–2669 (2012).
Rosenbaum, D. M. et al. GPCR Engineering Yields High-Resolution Structural Insights into 2-Adrenergic Receptor Function. Science 318, 1266–1273 (2007).
Höweler, U. MAXIMOBY 8.1 and MOBY 3.0 (2007).
Spiliotopoulos, D., Spitaleri, A. & Musco, G. Exploring PHD Fingers and H3K4me0 Interactions with Molecular Dynamics Simulations and Binding Free Energy Calculations: AIRE-PHD1, a Comparative Study. Plos One 7, e46902.
Paissoni, C., Spiliotopoulos, D., Musco, G. & Spitaleri, A. GMXPBSA 2.1: A GROMACS tool to perform MM/PBSA and computational alanine scanning. Comput. Phys. Commun. 186, 105–107 (2015).
Zhuang, H. & Matsunami, H. Evaluating cell-surface expression and measuring activation of mammalian odorant receptors in heterologous cells. Nat Protoc 3, 1402–1413 (2008).
Mainland, J. D. et al. The missense of smell: functional variability in the human odorant receptor repertoire. Nat Neurosci 17, 114–120 (2013).
Zhuang, H. & Matsunami, H. Synergism of Accessory Factors in Functional Expression of Mammalian Odorant Receptors. J. Biol. Chem. 282, 15284–15293 (2007).
Li, Y. R. & Matsunami, H. Activation State of the M3 Muscarinic Acetylcholine Receptor Modulates Mammalian Odorant Receptor Signaling. Sci. Signal. 4, ra1–ra1 (2011).
Acknowledgements
DFT calculations and MD simulations were performed on the HPC PURE cluster at the Biophysics Department of the Ruhr-University Bochum. Docking calculations were performed on the Partner Institute for Computational Biology High-Performance Computing cluster. S.W is funded by a Chinese Academy of Sciences Fellowship for Young International Scientists, and acknowledges a travel grant from the MERCATOR foundation. N.J. is funded by the Heinrich-and-Alma-Vogelsang-Stiftung. This work was supported by grants to H.H and K.G from the Deutsche Forschungsgemeinschaft (SFB642) and to S.W. from the National Natural Science Foundation of China Young Scientist Fund (grant no. 31250110070). We thank H. Matsunami (Duke University Medical Center, Durham, N.C.) for the kind donation of Hana3A cells, and M. Bamberg for help in preparing homology models. All molecular figures were prepared with PyMOL (Schrödinger, LLC).
Author information
Authors and Affiliations
Contributions
S.W., L.G., H.H., and K.G. conceived and coordinated the study, and wrote the paper. S.W. and S.B. performed theoretical calculations. N.J. and L.G. performed experiments. All authors reviewed the results and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wolf, S., Jovancevic, N., Gelis, L. et al. Dynamical Binding Modes Determine Agonistic and Antagonistic Ligand Effects in the Prostate-Specific G-Protein Coupled Receptor (PSGR). Sci Rep 7, 16007 (2017). https://doi.org/10.1038/s41598-017-16001-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-16001-4
This article is cited by
-
Copper-mediated thiol potentiation and mutagenesis-guided modeling suggest a highly conserved copper-binding motif in human OR2M3
Cellular and Molecular Life Sciences (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
