
Abstract
In human health, a fundamental challenge is the identification of disease-related genes. Bladder cancer (BC) is a worldwide malignant tumor, which has resulted in 170,000 deaths in 2010 up from 114,000 in 1990. Moreover, with the emergence of multi-omics data, more comprehensive analysis of human diseases become possible. In this study, we propose a multi-step approach for the identification of BC-related genes by using integrative Heterogeneous Network Modeling of Multi-Omics data (iHNMMO). The heterogeneous network model properly and comprehensively reflects the multiple kinds of relationships between genes in the multi-omics data of BC, including general relationships, unique relationships under BC condition, correlational relationships within each omics and regulatory relationships between different omics. Besides, a network-based propagation algorithm with resistance is utilized to quantize the relationships between genes and BC precisely. The results of comprehensive performance evaluation suggest that iHNMMO significantly outperforms other approaches. Moreover, further analysis suggests that the top ranked genes may be functionally implicated in BC, which also confirms the superiority of iHNMMO. In summary, this study shows that disease-related genes can be better identified through reasonable integration of multi-omics data.
Similar content being viewed by others

Introduction
Bladder cancer (BC) is a common malignant tumor, which is characterized by poor clinical outcome and frequent recurrence1,2,3. This malignancy is described as genetic disease, which is caused by multi-step accumulation of both epigenetic and genetic factors3. Although the treatment is greatly advanced, the prognosis of BC remains poor4. Therefore, there is an urgent need for researchers to identify genes related to BC, which can help uncover the mechanisms underlying this cancer and make an improvement in its diagnosis and therapy.
The identification of BC-related genes is an issue of prioritization of disease-related genes. The most common way to address this issue is to evaluate the similarities between known disease-related genes and given candidate genes. Various information can be used to calculate these similarities such as sequence5,6,7, functional annotation8 and protein-protein interactions (PPIs)9,10,11,12,13. Two famous methods are proposed by using PPIs: random walk14 and PRINCE15. In14, Kohler et al. prioritize genes related to disease by calculating the similarities of genes in PPI networks based on random walk analysis. In this process, the walkers that have the same initial probabilities transit to randomly selected neighbors from known disease-related genes14. Later, Vanunu et al. introduce prior information into the prioritization function and propose PRINCE15. Despite the great success of these two methods in identifying disease-related genes, they only employ the information of PPIs, which cannot reflect the unique relationships between genes under certain disease condition. Especially, rapid development of DNA sequencing technology promotes large projects such as ICGC16 and TCGA17, which produce enormous experimental data of different cancer in several omics including epigenomics, genomics and transcriptomics16,17. For example, more than 200 genomic rearrangements and segmental alterations per sample are detected in BC according to TCGA17. Analysis of these molecular aberrations in multiple omics can be very helpful for the improvement in diagnosis, treatment and prevention of cancer.
There are already many researches that use disease data of genes in multiple omics to identify BC-related genes. For example, Reinert et al. identify novel genes with tumor-specific differential methylation, which are shown to be promising cancer markers for early detection of BC, through a mapping of methylome18. Similarly, Zaravinos et al. find 17 differentially expressed genes that may be putative markers of BC by using genome microarrays19, i.e., gene expression data20,21,22. Besides, Zhang et al. suggest that susceptibility of BC can be predicted by the copy number variation (CNV) of GSTM1 by using multivariate logistic regression23. Although these studies can discover BC-related genes by making use of the disease data of one certain omics, a methodological limitation is the absence of efficient integration of different high-throughput experimental data, which may synergistically provide comprehensive and useful information about BC-related genes16,17. In our previous study24, we propose a method named HNP to identify BC-related genes. Although the data of three omics are integrated in HNP, the comprehensive information provided by these high-throughput data is not fully used in the algorithm24. In addition, more and more evidences indicate that microRNAs (miRNAs) can contribute to BC development25 and play the roles of suppressors or oncogenes26. Therefore, there is a great need to develop sophisticated methods that can efficiently integrate the heterogeneous data of both protein coding genes and non-coding miRNAs for identifying BC-related genes.
Here we propose a new method for the identification of BC-related genes by using integrative Heterogeneous Network Modeling of Multi-Omics data (iHNMMO). In iHNMMO, we make full use of known BC-related genes/miRNAs, gene expression profiles, miRNA expression profiles, CNV data, methylation data and PPIs. First, we perform a comprehensive literature curation for collecting known BC-related genes and miRNAs. Second, based on multi-omics data downloaded from TCGA, the correlational relationships of genes and miRNAs are extracted. These correlational relationships are further combined with PPIs to construct the networks of four omics. Third, the regulatory relationships between gene expression and other omics, which are used to connect the networks of different omics, are evaluated by linear regression model. Finally, based on the built heterogeneous network model, a modified propagation algorithm is implemented for the identification of BC-related genes. The comparison results show that iHNMMO achieves significantly better performance than other methods through integrating the information from different kinds of single-omics data. The predicted novel BC-related genes are also analyzed subsequently and the analysis results corroborate the superiority and effectiveness of the proposed method.
Methods
Multi-omics data of bladder cancer from TCGA
The multi-omics data used in this study are obtained from TCGA dataset, which provides tremendous experimental data of cancers17. Here the normalized data (‘level 3’ data) of four omics, i.e., CNV, gene expression, methylation and miRNA expression, are downloaded from TCGA Data Portal (https://tcga-data.nci.nih.gov/tcga). Specifically, gene expressions are derived from RNA sequencing data and the log2-transformed values are processed by quantile-normalized RSEM27 (RNA-Seq by Expectation Maximization). DNA methylation data used in this study are processed from Illumina HumanMethylation450 BeadChip. MiRNA expressions are log2-transformed RPMs (reads per million mapped) that calculated from sequencing data27. We then extract the common 377 patient samples of these four omics for follow-up studies. Since the CNV data in TCGA only contain the information of chromosome segments, we also download ‘refGene.txt’ that provides chromosomal locations of 44,914 genes from UCSC genome browser (http://genome.ucsc.edu/) and compute average CNV value of each gene accordingly. Finally, the data of four omics are normalized and transformed into four feature matrixes in which a column represents a patient sample and a row represents a gene/miRNA.
The collection of seeds in multiple omics
We perform a comprehensive literature curation for collecting known BC-related genes and miRNAs. For genes having aberrations in methylation, CNV and gene expression, we search the keywords: “bladder cancer” AND (“methylation” OR “CNV” OR “gene expression”) on Web of Science. The selected literatures are then ranked by their citations. After manually examining the full text of the top ranked literatures, 135 BC-related genes are finally obtained, which consist of 27, 9 and 99 genes with reported aberrations in methylation, CNV and gene expression, respectively. Meanwhile, we also extract 25 known BC-related miRNAs by the keywords: “bladder cancer” AND “miRNAs”. For convenience, known BC-related genes and miRNAs are collectively called seeds.
Pipeline of iHNMMO
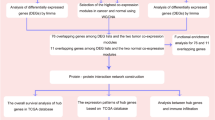
The proposed method begins with seeds, which are used as true positives later. Initially, we extract correlational relationships of these seeds based on the data of four omics. Then the weighted networks of each omics are constructed through the combination of correlational relationships and PPIs. Moreover, since miRNA expression, methylation and CNV can affect expression levels of genes18,23,26, regulatory relationships between gene expression and the other three omics are further evaluated by linear regression model and the corresponding coefficients are utilized to weight the edges connecting the networks of different omics. In this way, the heterogeneous network model of genes is constructed, in which not only general relationships and unique relationships under BC condition, but also correlational relationships within each omics as well as regulatory relationships between different omics are considered. Finally, a modified propagation algorithm28 is implemented on the model to identify BC-related genes. In this process, the information flow propagates from seeds to candidate genes iteratively and a score is obtained for each candidate gene when the propagation process ends. The final score is a measurement of how much a gene can be related to BC. The overall flowchart is shown in Fig. 1.

Flowchart of iHNMMO. The detailed process is described in Section “Pipeline of iHNMMO”.
Heterogeneous network model for the identification of BC-related genes
As a widespread use in measuring correlational relationships29, Pearson correlation coefficient (PCC) is employed to reflect the correlational relationships between seeds in different omics. Specifically, for a given seed in one omics, we calculate the PCCs as well as the corresponding t-test p-values between this seed and other genes/miRNAs appeared in the feature matrix of this omics (see Supplementary Section 1). Afterwards, four correlation matrixes M exp , M cnv , M methy and M mir are built based on the correlational relationships. The element M(i, j) represents absolute PCC between gene/miRNA i and j in a certain omics. These matrixes are then normalized30 respectively to \(\,\bar{{M}_{\exp }}\), \(\,\overline{{M}_{CNV}}\), \(\,\overline{{M}_{methy}}\) and \(\,\overline{{M}_{mir}}\) as follows30,31:
where E exp (i, i), E cnv (i, i), E methy (i, i) and E mir (i, i) are the entities in row i column i of diagonal matrixes E exp , E cnv , E methy and E mir , representing the sum of row i in M exp , M cnv , M methy and M mir , respectively.
Besides above unique relationships under BC condition, we also take advantage of PPIs, which represent general relationships of genes. Here 4,850,628 PPIs are downloaded from STRING database32 (version 9.1). The redundant PPIs that do not contain the genes in the omics of gene expression are removed and 524,348 PPIs are finally extracted in this study. Likewise, these PPIs are further normalized and transformed into a PPI matrix:
Then based on the correlational relationships and PPIs above, a weighted network33 of the omics of gene expression is constructed as follows:
where \({w}_{i,j}^{\exp }\) represents the weight of the edge in the network, \({m}_{i,j}^{Pcc}\) and \({m}_{i,j}^{PPI}\) are the elements in matrixes \(\,\bar{{M}_{\exp }}\) and \(\,\bar{{M}_{\exp }}\), respectively. Meanwhile, the networks of other three omics, i.e., CNV, methylation and miRNA expression, are also constructed by utilizing the correlational relationships in their omics.
Considering the influence on gene expression brought by miRNA expression, CNV and methylation18,23,26, we utilize liner regression model to evaluate the regulatory relationships between different omics. First, 17,197 regulatory relationships between genes in the omics of gene expression and miRNAs in the omics of miRNA expression are extracted from miRTarBase, which is a database of experimentally validated miRNA-gene interactions34. Here miRNAs that interact with a certain gene is called the miRNA regulators of this gene. Then, for a gene i with expression level Y i (y i1 , …, y in ), the relationships between its CNV level X i cnv (x i1 cnv, …, x in cnv), its methylation level X i methy (x i1 methy, …, x in methy) and expression levels of its miRNA regulators X i1 mir, …, X im mir (x im1 mir, …, x imn mir) (m and n are the number of miRNA regulators and the number of patient samples, respectively), are modeled using the following formula35 below:
where \(\beta \) represents regression coefficient and ε stands for noise. Finally, we use these coefficients to connect the networks of different omics and the heterogeneous network model is constructed, in which the edges properly and comprehensively reflect the complex relationships between nodes. Besides, the normalized weight matrix \(\bar{W}\) of the heterogeneous network is obtained, which denotes probability distribution of the information transition in the network. The overall process of the model construction is shown in Fig. 2.

Overall process of the heterogeneous network model construction. (A) The collection of seeds. (B) Extraction of correlational relationships. (C) Four networks within each omics. (D) The heterogeneous network model.
The modified propagation algorithm
In this study, we propose a modified propagation algorithm with resistance. Here a weighted graph model G = (V, E, w) is used to denote the heterogeneous network. In this graph model, nodes represent genes or miRNAs of four omics and edges represent the relationships between these genes or miRNAs. The weight w measures the confidence of the edge in the network. The goal of the algorithm is to score all candidate genes in V and the top-ranked genes are more probably to be BC-related genes.
First, for a node v ∈ V with direct neighbors N v , its prior information score D is calculated by following equation33:
where N v is the number of neighbors for v and n v represents the number of seeds in these neighbors. The parameter α is a threshold for N v and it is set to 50 in this study.
To evaluate the relationship between node v and BC, we then introduce a probability function S v based on the principle of “Guilt by Association”, which means that adjacent nodes in a network should share similar prior information and final scores36,37:
where \(\,\overline{{W}_{{\rm{uv}}}}\) is a component (row u column v) of |V| × |V| matrix \(\bar{W}\) and λ ∈ (0, 1) is set to 0.2 in this study. However, when meeting a hub node, the information flow will propagate to its neighbors with the same possibility, regardless of whether these neighbors are actually related to seeds or not. In order to suppress this bias, a small amount of resistance is incorporated into the propagation process28, which is described as the equation below:
where SR uv represents the new pwwrobability for the information flow transiting from u to v with an added resistance and is formulated by:
Here, є and β are respectively defined as |V|/|E|2 and 1/|E| according to28. Besides, θ is the threshold for S v and we set it to 0.005. Finally, the probability function S v can be further expressed in linear form:
Since SR is converted from S, the probability function can be computed through an iterative process15 as follows:
where S and D are both 1 × |V| matrixes, denoting the matrix of final scores and the matrix of prior information scores, respectively. Besides, \({S}^{1}:=D\). In this algorithm, the prior information is iteratively propagated from seeds to all other nodes in the heterogeneous network until the difference between S t and S t−1 is sufficiently small33, i.e., mean square error (MSE) between S t and S t−1 no larger than 1 × 10−5.
The performance evaluation
To evaluate the performance of the proposed method, leave-one-out cross-validation (LOOCV) is performed in the test process. In each round, we take one seed as test data and all other seeds as training data. To prevent potential bias on seeds in network modeling, when taking a seed as test data, its correlational relationships are re-evaluated in the same way as those non-seeds. Besides, the prior information score of this seed is also recalculated by equation (8). That is, in each CV run, the topology of the heterogeneous network changes and the matrix of prior information scores D together with the whole weight matrix are recomputed. Especially, to impartially evaluate the performance of iHNMMO in identifying BC-related genes, we only study the scores of genes in the results. Meanwhile, the max score of a certain gene in three omics is regarded as its final score. These scores of genes are further used for performance evaluation. Seed genes and candidate genes are respectively considered as golden standard positive (GSP) and golden standard negative (GSN). Due to the fact that the top k ranked genes predicted by our method are defined as BC-related genes in this study, the intersections of these genes with GSN and GSP are considered as false positive (FP) and true positive (TP). After removing these intersections, the rest of GSN and GSP are referred to as true negative (TN) and false negative (FN), respectively. Then specificity (Sp) value and sensitivity (Sn) value can be obtained by the following equation:
As a performance measurement, Receiver Operating Characteristic curves (ROC curves) are plotted, in which x axis and y axis represent 1-Sp and Sn, respectively. The area under this curve (AUC) is also computed. In addition, we use Rank Cutoff curves38 to evaluate the proportions of true positives in the top k% ranked genes (k varying from 0 to 20). Fold enrichment30 is also employed with the formula: fold enrichment = the number of candidate genes/2/the rank of the test gene. Here we utilize average fold enrichment of all test genes for assessment. Besides, the relationships between precision and recall with rank threshold in [100, 2000] are plotted based on the definitions:
Other network-based models using single-omics data
To verify the benefit from integration of multi-omics data, in this study we also examine simplified iHNMMO models with single-omics data for identifying BC-related genes, which only take advantage of the information in one omics among the multi-dimensional data of gene expression, CNV and methylation. For simplicity, these network-based models with single-omics data are thereafter named as NMSO-Expr, NMSO-CNV and NMSO-Meth, respectively.
Data availability
The datasets and source code can be downloaded from the following URL: http://hi.ustc.edu.cn/iHNMMO/index/.
Results
In this part, the performance of iHNMMO is evaluated systematically by comparing it with network-based models using single-omics data and other existing approaches.
Performance comparison between iHNMMO and network-based models using single-omics data
To verify the superiority of iHNMMO, we utilize several measurements to compare the performance of iHNMMO with network-based models using single-omics data. As shown in Fig. 3A, the ROC curve of iHNMMO is obviously above those of network-based models with single-omics data. Moreover, the AUC of iHNMMO is the largest among these methods, which is 11.3%, 28.0%, and 28.4% higher than that of NMSO-Expr, NMSO-CNV, and NMSO-Meth, respectively. Besides, at three stringent levels of Sp, the Sn values of iHNMMO are always the highest (Table 1). Specifically, at the high level of Sp, i.e., 99.0%, the Sn value of iHNMMO reaches 45.3%, which is much higher than those of other models. The huge promotion of Sn value corroborates the superiority of iHNMMO in improving the probability of detection. At the medium level of Sp, i.e., 95.0%, the Sn value of iHNMMO has a 29.1% growth and reaches 74.4%, while the Sn values of other models are 22.2%, 14.8% and 44.4%, respectively. As Sp level drops to 90.0%, the Sn value of iHNMMO rises to 86.3%, which is still higher than those of other methods.

Performance comparison of iHNMMO and network-based models with single-omics data. (A) ROC curves. The x axis and y axis represent 1-Sp and Sn, respectively. (B) Fold enrichment. (C) Rank cutoff curves.
Besides, the rank cutoff curves are plotted in Fig. 3B. Similar to Fig. 3A, the curve of iHNMMO is clearly above those of network-based models using single-omics data, which indicates a better performance of iHNMMO. The fraction enlarges as the threshold increases and the curve of iHNMMO rises most rapidly when the threshold varies from 0 to 5%. For the top 5% ranked genes, the fraction of true positives predicted by iHNMMO is 69.2% (Table 2), while the fractions of the other models are all less than half. When the threshold enlarges to top 10% and 15%, the fractions of iHNMMO are 84.6% and 94.0%, respectively, both of which are still the highest among these methods. Furthermore, the fraction of iHNMMO reaches 98.3% when the threshold is 20%, which is 21.5%, 76.1% and 61.3% higher than that of NMSO-Expr, NMSO-CNV and NMSO-Meth, respectively. This phenomenon suggests that iHNMMO can always predict the largest number of seed genes with different rank cutoffs. Since the numbers of seed genes are different in different methods, we further consider the fractions of seed genes in the network and calculate the hypergeometric-test p-values accordingly (Table 2). The p-values of iHNMMO are all statistically significant (<0.05) and consistently smaller than those of network-based models with single-omics data. In Fig. 3C, the average fold enrichment of iHNMMO and network-based models with single-omics data are 136, 5, 14 and 26, respectively, indicating that iHNMMO can better identify BC-related genes with higher rank. All these results suggest that iHNMMO significantly exceeds those network-based models using single-omics data and confirm the great advantage of the heterogeneous network model that constructed by integrating multi-omics data.
Performance comparison with existing approaches
To perform a comprehensive comparison of the proposed method with existing approaches, we implement four network-based approaches for identification of BC-related genes: PRINCE15, PageRank algorithm39, HNP24 and the original random walk algorithm14 (see Supplementary Section 1). Their performance are also comprehensively evaluated. As shown in Fig. 4A, the AUC value of iHNMMO is 95.9%, which is 4.3%, 6.4%, 6.1% and 8.0% higher than that of PRINCE, PageRank, HNP and Random walk, respectively. In addition, at three levels of Sp, iHNMMO always achieves the highest Sn value. Specifically, when Sp is 99%, the Sn value of iHNMMO is 45% while the Sn values of other four approaches are 39.7%, 13.5%, 25.5% and 8%, respectively. When Sp level decreases to 90%, the Sn values of PRINCE, PageRank, HNP and Random walk rise to 74.6%, 68.2%, 71.3% and 69%, which are 11.4%, 17.8%, 14.7% and 17% lower than that of iHNMMO, respectively. These results indicate a better accuracy of iHNMMO than other four approaches. In Fig. 4B, the precision-recall curve of iHNMMO is obviously above other four curves. Within the top 100 ranked genes, the precision of iHNMMO can even reach 42%, which is 8%, 37%, 22% and 38% higher than that of PRINCE, PageRank, HNP and Random walk, respectively. At the same time, the recall of iHNMMO and other four approaches are 36%, 27.0%, 4%, 20% and 3%, respectively. The higher recall also represents the better performance of iHNMMO in retrieving known BC-related genes by ranking them into top k. When k rises to 2000, the recall of iHNMMO can even reach 98%. Besides, from the rank cutoff curves shown in Fig. 4C, we can see that iHNMMO always achieves a higher fraction of seed genes than other four approaches in the whole range. When the threshold rises to top 3%, iHNMMO can recover more than half of seed genes. All the above results of performance comparison indicate that iHNMMO remarkably outperforms PRINCE, PageRank, HNP and Random walk in identifying BC-related genes. We also respectively apply the original random walk algorithm to the heterogeneous network model and implement the modified propagation algorithm on the PPI network model. The performance of these two approaches are evaluated and compared with iHNMMO in Supplementary Section 2.

Performance comparison of iHNMMO and existing approaches. (A) AUC values and Sn values at different levels of Sp. (B) Precision-recall curves. (C) The rank cutoff curves. The x and y axis respectively represents the threshold and the fraction of known BC-related genes.
Identifying novel BC-related genes
To analyze the predicted results of our method globally, the top 100 ranked genes that do not contain seed genes are picked up and functional enrichment analysis using DAVID are performed here. Interestingly, as shown in Table 3, functions: “GO:0042127~regulation of cell proliferation” (p-value = 1.5 × 10−2), “GO:0060548~negative regulation of cell death” (p-value = 1.6 × 10−2) and “GO:0043065~positive regulation of apoptotic process” (p-value = 2.6 × 10−4) appear in the results, which are common biological activities in human cancer40,41. Besides, many important pathways that related to cancer especially BC are listed in the table, e.g., “hsa04151: PI3K-Akt signaling pathway” (p-value = 2.8 × 10−9), “hsa05200: Pathways in cancer” (p-value = 1.0 × 10−4), “hsa04014: Ras signaling pathway” (p-value = 4.1 × 10−4), “hsa04350: TGF-beta signaling pathway” (p-value = 1.4 × 10−3), “hsa04010: MAPK signaling pathway” (p-value = 1.2 × 10−3), “GO:0007219~Notch signaling pathway” (p-value = 1.3 × 10−2) and “hsa04115: p53 signaling pathway” (p-value = 6.8 × 10−8). Among these pathways, some alterations of components AKT1, PTEN, TSC1 and PIK3CA in PI3K-Akt pathway of bladder cancer are observed to be remarkably related to tumor phenotype and clinical behavior42, Ras-mediated signaling pathway is expected to promote diagnostics and therapeutics of bladder cancer43, TGF-beta signaling pathway has been verified to have a possible involvement in the progression of BC44, new inactivating mutations of the components in Notch pathway are reported in more than 40% of BC45 and altered p53 pathway is expected to be an important prognostic factor on BC patient survival according to the study of46. All these studies indicate the potential relationships between the predicted genes and BC.
Furthermore, to explore the predicted genes in detail, we list the names and the normalized scores of the top 10 ranked genes predicted by iHNMMO in Table 4. In the latest literature47, the first-ranked gene CCNE2 is found to be a possible prognostic marker for BC patients47. At the same time, another latest literature48 reports that FSCN1 is implicated in the pathway of hsa-miR-145-ZEB1/2-FSCN1, which is used by lncRNA-UCA1 to reinforce cell migration and invasion of bladder cancer48. These new discoveries are good evidence for the reliability of our results. The third ranked gene KLK3 is a member of kallikrein-related peptidases, which are expressed aberrantly in many cancers49 such as prostate cancer, ovarian cancer50 and urogenital malignancies51. Besides, KLK3 has been found to be related to prostate cancer in several previous studies52,53. From these studies, we can see that although KLK3 is not directly related to BC, the functions in other cancers may imply its potential role in BC. The top ranked miRNAs are also analyzed in Supplementary Section 2.
Discussion
We present a multi-step method named iHNMMO to identify BC-related genes by constructing a heterogeneous network model based on the integration of multi-omics data. Commonly, network-based algorithms for the identification of disease-related genes are motivated by the discovery that genes closing to one another are more likely to lead to the same or similar diseases15. Therefore, whether the network model can reflect the relationships of genes suitably is critical to the method. In this study, we address this issue by integrating multi-omics data. According to the information provided by the data of methylation, miRNA expression, gene expression and CNV, we obtain both regulatory and correlational relationships of genes, which are further used to build and combine the networks of four omics. Besides, to fully reflect general relationships and unique relationships under BC condition, not only the correlations calculated by statistical analysis, but also PPIs downloaded from well-established database are utilized to generate the correlational relationships between genes. Thus, the heterogeneous network model that contains comprehensive information for the identification of BC-related genes is set up, which may be the most important factor leading to the success of iHNMMO. In addition, another factor that contributes to the superiority of iHNMMO is the modified propagation algorithm implemented on the model, which can score and rank candidate genes precisely. It is also important to note that the heterogeneous network model and propagation algorithm should be integrated properly to make sure their advantages could be fully used. For example, although our previous method HNP24 utilizes the data of three omics, it does not achieve a comparable performance with iHNMMO. This may be due to the fact that the comprehensive information provided by multi-omics data is only used at the beginning of HNP, i.e., the initialization of propagation, which does not sufficiently promote the whole process of propagation.
Although our method achieves an excellent performance in identifying BC-related genes, it will be better to utilize independent datasets to facilitate a fair performance assessment. However, we cannot perform more rigorous evaluation because of the lack of parallel data of complete four omics. Actually, this issue also occurs in many computational studies of multi-omics data36,54,55,56 in cancer. The insufficiency of data also leads to some limitations in the generalization of iHNMMO to other diseases. For example, since the heterogeneous network is constructed based on seeds, iHNMMO cannot be generalized to those diseases that have no known disease-related genes. In this case, other information such as the similarities between diseases will be introduced to the method to make comparison of candidate genes and the genes that are known to be related to similar diseases. Similarly, iHNMMO cannot be performed on the researches where multi-omics data are incomplete even unavailable. However, high-throughput technologies with reduced cost such as next generation sequencing and microarrays develop rapidly, and many researches of multi-omics data are underway now. It is believed that we can obtain more comprehensive data of different omics in the future. Moreover, the functions of long non-coding RNAs (lncRNAs) are explored in more and more cancer studies29,33 and these information should be incorporated properly into the heterogeneous model to reflect the molecular mechanism of disease better. Despite the difficulties listed above in the generalization of iHNMMO, our method can still be well applied to identify other disease-related genes as long as relevant data meets the requirement in this study. Here we take glioblastoma (GBM) as an additional example and the known GBM-related genes and multi-omics data of GBM processed in our previous study57 are utilized. The results of performance evaluation are shown in Supplementary Section 2, which indicate the good generalization ability of iHNMMO.
References
Lozano, R. et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. The Lancet 380, 2095–2128 (2013).
Rothman, N. et al. A multi-stage genome-wide association study of bladder cancer identifies multiple susceptibility loci. Nature genetics 42, 978–984 (2010).
Sanchez-Carbayo, M., Socci, N. D., Lozano, J., Saint, F. & Cordon-Cardo, C. Defining molecular profiles of poor outcome in patients with invasive bladder cancer using oligonucleotide microarrays. Journal of Clinical Oncology 24, 778–789 (2006).
Abbosh, P. H., McConkey, D. J. & Plimack, E. R. Targeting signaling transduction pathways in bladder cancer. Current oncology reports 17, 58 (2015).
George, R. A. et al. Analysis of protein sequence and interaction data for candidate disease gene prediction. Nucleic acids research 34, e130–e130 (2006).
Yu, H.-J. & Huang, D.-S. Normalized feature vectors: a novel alignment-free sequence comparison method based on the numbers of adjacent amino acids. IEEE/ACM Transactions on Computational Biology and Bioinformatics (TCBB) 10, 457–467 (2013).
Deng, S.-P. & Huang, D.-S. SFAPS: an R package for structure/function analysis of protein sequences based on informational spectrum method. Methods 69, 207–212 (2014).
Perez-Iratxeta, C., Bork, P. & Andrade-Navarro, M. A. Update of the G2D tool for prioritization of gene candidates to inherited diseases. Nucleic acids research 35, W212–W216 (2007).
Oti, M., Snel, B., Huynen, M. A. & Brunner, H. G. Predicting disease genes using protein–protein interactions. Journal of medical genetics 43, 691–698 (2006).
Xia, J.-F., Han, K. & Huang, D.-S. Sequence-based prediction of protein-protein interactions by means of rotation forest and autocorrelation descriptor. Protein and Peptide Letters 17, 137–145 (2010).
You, Z.-H., Lei, Y.-K., Gui, J., Huang, D.-S. & Zhou, X. Using manifold embedding for assessing and predicting protein interactions from high-throughput experimental data. Bioinformatics 26, 2744–2751 (2010).
Zhu, L., You, Z.-H. & Huang, D.-S. Increasing the reliability of protein–protein interaction networks via non-convex semantic embedding. Neurocomputing 121, 99–107 (2013).
Huang, D.-S. et al. Prediction of protein-protein interactions based on protein-protein correlation using least squares regression. Current Protein and Peptide Science 15, 553–560 (2014).
Köhler, S., Bauer, S., Horn, D. & Robinson, P. N. Walking the interactome for prioritization of candidate disease genes. The American Journal of Human Genetics 82, 949–958 (2008).
Vanunu, O., Magger, O., Ruppin, E., Shlomi, T. & Sharan, R. Associating genes and protein complexes with disease via network propagation. PLoS Comput Biol 6, e1000641 (2010).
Hudson, T. J. et al. International network of cancer genome projects. Nature 464, 993–998 (2010).
Network, C. G. A. R. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 507, 315–322 (2014).
Reinert, T. et al. Comprehensive genome methylation analysis in bladder cancer: identification and validation of novel methylated genes and application of these as urinary tumor markers. Clinical Cancer Research 17, 5582–5592 (2011).
Zaravinos, A., Lambrou, G. I., Boulalas, I., Delakas, D. & Spandidos, D. A. Identification of common differentially expressed genes in urinary bladder cancer. PloS one 6, e18135 (2011).
Huang, D. S. The Study of Data Mining Methods for Gene Expression Profiles, Science Press of China, March 2009.
Huang, D.-S. & Zheng, C.-H. Independent component analysis-based penalized discriminant method for tumor classification using gene expression data. Bioinformatics 22, 1855–1862 (2006).
Zheng, C.-H., Huang, D.-S., Zhang, L. & Kong, X.-Z. Tumor clustering using nonnegative matrix factorization with gene selection. IEEE Transactions on Information Technology in Biomedicine 13, 599–607 (2009).
Zhang, X. et al. Association between GSTM1 copy number, promoter variants and susceptibility to urinary bladder cancer. Int J Mol Epidemiol Genet 3, 228–236 (2012).
Peng, C., Li, A., Feng, H. & Wang, M. In Natural Computation, Fuzzy Systems and Knowledge Discovery (ICNC-FSKD), 2016 12th International Conference on. 1396–1401 (IEEE).
Yoshino, H. et al. Aberrant expression of microRNAs in bladder cancer. Nature Reviews Urology 10, 396–404 (2013).
Han, Y. et al. MicroRNA expression signatures of bladder cancer revealed by deep sequencing. PloS one 6, e18286 (2011).
Ding, Z., Zu, S. & Gu, J. Evaluating the molecule-based prediction of clinical drug responses in cancer. Bioinformatics 32, 2891–2895 (2016).
Lei, C. & Ruan, J. A novel link prediction algorithm for reconstructing protein–protein interaction networks by topological similarity. Bioinformatics 29, 355–364 (2013).
Liao, Q. et al. Large-scale prediction of long non-coding RNA functions in a coding–non-coding gene co-expression network. Nucleic acids research 39, 3864–3878 (2011).
Chen, X., Liu, M.-X. & Yan, G.-Y. Drug–target interaction prediction by random walk on the heterogeneous network. Molecular BioSystems 8, 1970–1978 (2012).
Wang, W., Yang, S., Zhang, X. & Li, J. Drug repositioning by integrating target information through a heterogeneous network model. Bioinformatics 30, 2923–2930 (2014).
Franceschini, A. et al. STRINGv9. 1: protein-protein interaction networks, with increased coverage and integration. Nucleic acids research 41, D808–D815 (2013).
Guo, X. et al. Long non-coding RNAs function annotation: a global prediction method based on bi-colored networks. Nucleic acids research, gks967 (2012).
Hsu, S.-D. et al. miRTarBase update 2014: an information resource for experimentally validated miRNA-target interactions. Nucleic acids research 42, D78–D85 (2014).
Peng, C., Wang, M., Shen, Y., Feng, H. & Li, A. Reconstruction and analysis of transcription factor–miRNA co-regulatory feed-forward loops in human cancers using filter-wrapper feature selection. PloS one 8, e78197 (2013).
Chen, Y., Jiang, T. & Jiang, R. Uncover disease genes by maximizing information flow in the phenome–interactome network. Bioinformatics 27, i167–i176 (2011).
Zhou, D., Bousquet, O., Lal, T. N., Weston, J. & Schölkopf, B. Learning with local and global consistency. NIPS 16, 321–328 (2003).
Linghu, B., Snitkin, E. S., Hu, Z., Xia, Y. & DeLisi, C. Genome-wide prioritization of disease genes and identification of disease-disease associations from an integrated human functional linkage network. Genome biology 10, R91 (2009).
Brin, S. & Page, L. Reprint of: The anatomy of a large-scale hypertextual web search engine. Computer Networks 56, 3825–3833 (2012).
Dyrskjøt, L. et al. Genomic profiling of microRNAs in bladder cancer: miR-129 is associated with poor outcome and promotes cell death in vitro. Cancer Research 69, 4851–4860 (2009).
Cohen, S. & Ellwein, L. Cell proliferation in carcinogenesis. (1990).
Knowles, M. A., Platt, F. M., Ross, R. L. & Hurst, C. D. Phosphatidylinositol 3-kinase (PI3K) pathway activation in bladder cancer. Cancer and Metastasis Reviews 28, 305–316 (2009).
Oxford, G. & Theodorescu, D. The role of Ras superfamily proteins in bladder cancer progression. The Journal of urology 170, 1987–1993 (2003).
Hung, T.-T., Wang, H., Kingsley, E. A., Risbridger, G. P. & Russell, P. J. Molecular profiling of bladder cancer: involvement of the TGF-β pathway in bladder cancer progression. Cancer letters 265, 27–38 (2008).
Rampias, T. et al. A new tumor suppressor role for the Notch pathway in bladder cancer. Nature medicine 20, 1199–1205 (2014).
Lu, M.-L. et al. Impact of alterations affecting the p53 pathway in bladder cancer on clinical outcome, assessed by conventional and array-based methods. Clinical Cancer Research 8, 171–179 (2002).
Matsushita, R. et al. Tumour-suppressive microRNA-144-5p directly targets CCNE1/2 as potential prognostic markers in bladder cancer. British journal of cancer 113, 282–289 (2015).
Xue, M. et al. Long non-coding RNA urothelial cancer-associated 1 promotes bladder cancer cell migration and invasion by way of the hsa-miR-145-ZEB1/2-FSCN1 pathway. Cancer science 107, 18–27 (2016).
Kryza, T., Silva, M., Loessner, D., Heuzé-Vourc’h, N. & Clements, J. A. The kallikrein-related peptidase family: dysregulation and functions during cancer progression. Biochimie 122, 283–299 (2016).
Fuhrman-Luck, R. A. et al. Proteomic and other analyses to determine the functional consequences of deregulated kallikrein-related peptidase (KLK) expression in prostate and ovarian cancer. PROTEOMICS-Clinical Applications 8, 403–415 (2014).
Dorn, J. et al. Clinical utility of kallikrein-related peptidases (KLK) in urogenital malignancies. Thrombosis and haemostasis 110, 408–422 (2013).
Lai, J. et al. Analysis of androgen and anti-androgen regulation of KLK-related peptidase 2, 3, and 4 alternative transcripts in prostate cancer. Biological chemistry 395, 1127–1132 (2014).
Zambon, C.-F. et al. Effectiveness of the combined evaluation of KLK3 genetics and free-to-total prostate specific antigen ratio for prostate cancer diagnosis. The Journal of urology 188, 1124–1130 (2012).
Zhang, S. et al. Discovery of multi-dimensional modules by integrative analysis of cancer genomic data. Nucleic acids research 40, 9379–9391 (2012).
Chen, Y. et al. Identifying potential cancer driver genes by genomic data integration. Scientific reports 3, 3538 (2013).
Vaske, C. J. et al. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics 26, i237–i245 (2010).
Peng, C., Shen, Y., Ge, M., Wang, M. & Li, A. Discovering key regulatory mechanisms from single-factor and multi-factor regulations in glioblastoma utilizing multi-dimensional data. Molecular BioSystems 11, 2345–2353 (2015).
Acknowledgements
This work is supported by National Natural Science Foundation of China [Grant Nos 61571414, 61471331, 31100955, 61702371, 61520106006, 31571364, 61532008, U1611265, 61672382 and 61402334], and China Postdoctoral Science Foundation (Grant No. 2017M611619).
Author information
Authors and Affiliations
Contributions
C.P. and A.L. conceived and designed the method, C.P. conducted the experiments and wrote the manuscript text. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Peng, C., Li, A. & Wang, M. Discovery of Bladder Cancer-related Genes Using Integrative Heterogeneous Network Modeling of Multi-omics Data. Sci Rep 7, 15639 (2017). https://doi.org/10.1038/s41598-017-15890-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-15890-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
