
Abstract
Breakdown of the blood-retinal barrier (BRB), as occurs in diabetic retinopathy and other chronic retinal diseases, results in vasogenic edema and neural tissue damage, causing vision loss. Vasoinhibins are N-terminal fragments of prolactin that prevent BRB breakdown during diabetes. They modulate the expression of some transient receptor potential (TRP) family members, yet their role in regulating the TRP vanilloid subtype 4 (TRPV4) remains unknown. TRPV4 is a calcium-permeable channel involved in barrier permeability, which blockade has been shown to prevent and resolve pulmonary edema. We found TRPV4 expression in the endothelium and retinal pigment epithelium (RPE) components of the BRB, and that TRPV4-selective antagonists (RN-1734 and GSK2193874) resolve BRB breakdown in diabetic rats. Using human RPE (ARPE-19) cell monolayers and endothelial cell systems, we further observed that (i) GSK2193874 does not seem to contribute to the regulation of BRB and RPE permeability by vasoinhibins under diabetic or hyperglycemic-mimicking conditions, but that (ii) vasoinhibins can block TRPV4 to maintain BRB and endothelial permeability. Our results provide important insights into the pathogenesis of diabetic retinopathy that will further guide us toward rationally-guided new therapies: synergistic combination of selective TRPV4 blockers and vasoinhibins can be proposed to mitigate diabetes-evoked BRB breakdown.
Similar content being viewed by others
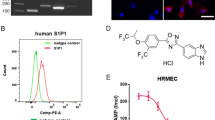

Introduction
Diverse conditions, including diabetic retinopathy and macular edema, are associated with exacerbated leakage through the blood-retinal barrier (BRB)1,2. The BRB is comprised of inner and outer components that mainly refer to vascular endothelial and retinal pigment epithelial (RPE) cells, respectively1. Although high glucose conditions predominantly affect retinal capillaries, the damage to RPE cells has been increasingly recognized to play a major role in the progression of these diseases3,4. Nevertheless, its regulation has been less studied than that of retinal capillaries in the context of diabetes. Additionally, that most clinical therapies address symptoms rather than the molecular pathophysiology of diabetic retinopathies5,6 indicates that many molecular and cellular mechanisms underlying damage to the BRB by high glucose levels remain to be characterized. More particularly, advances in understanding the key role of endogenous cytokines, their conate receptors and ion channels in BRB regulation may lead to the development of novel therapeutic options for rationally-targeted treatment of diabetic retinopathy and macular edema.
Vasoinhibins, derived from prolactin cleavage, are endogenous regulators of angiogenesis and vascular function that occur naturally in the retina7. It has been shown that patients with diabetic retinopathy have lower levels of circulating vasoinhibins than nondiabetic patients8. Increasing ocular levels of vasoinhibins were reported to protect against the pathological increase in BRB permeability associated with diabetes9,10,11,12. Vasoinhibins were recently shown to reduce BRB permeability by targeting both its main inner and outer components13; however, their action mechanisms have been best described in vasculature. Vasoinhibins regulate endothelial cell permeability by lowering NO production10,13,14 and stabilizing the actin cytoskeleton13. Vasoinhibins reduce NO production by limiting endothelial NOS (eNOS) activation through phosphorylation and Ca2+/calmodulin binding15. Vasoinhibins have been indeed shown to abrogate Ca2+ entry through both capacitative16,17 and receptor-operated pathways16 in endothelial cells. Further evidence supports the idea that vasoinhibins regulate Ca2+ homeostasis by interfering with the activity of the Ca2+-permeable transient receptor potential (TRP) family members, decreasing the expression of canonical subfamily member 5 protein (TRPC5) mRNA in endothelial cells16.
Among the 26 members of the mammalian TRP family, all of which are present in the retina18, the vanilloid subfamily member 4 protein (TRPV4) uniquely regulates the capillary endothelial barrier19. TRPV4 is a non-selective cation channel permeable to Ca2+ that was originally identified as an osmotically activated channel20,21,22, but it is also activated by ligands such as phorbol derivatives23. TRPV4 has been demonstrated to participate in both capacitative24 and receptor-operated Ca2+ entry25,26,27,28,29,30,31, and Ca2+ entry through TRPV4 promotes the formation of Ca2+-calmodulin complexes, which can bind to TRPV4 enhancing channel activity32,33. Ca2+ entry through TRPV4 has been also shown to increase lung endothelial cell permeability by disrupting cell-cell or cell-matrix adhesion34,35. A mechanism through which TRPV4 activation evokes the reorganization of actin cytoskeleton that associates with increased permeability may involve NO release36,37. Inversely, blockage of TRPV4 channels inhibits eNOS activation by phosphorylation38 and mitigates pulmonary edema39.
Functional expression of TRPV4 has been reported in retinal mouse capillaries40,41 and TRPV4 protein in primary cultures of human fetal RPE42. Importantly, in this context we do not know about its expression in adult RPE nor about its participation as a regulator of BRB permeability. We therefore tested this possibility and further postulated that vasoinhibins may regulate BRB permeability by blocking TRPV4. This novel concept is rooted in the fact that vasoinhibins exert effects opposite to the ones induced by TRPV4 activation to regulate capillary endothelial barrier (i.e., intracellular Ca2+ rise, eNOS phosphorylation, NO release, cytoskeleton reorganization36,43,44,45,46).
Here, we identified hitherto under-appreciated TRPV4 retinal distribution and observed that TRPV4 is also expressed by both retinal endothelia and RPE. Given that potent and selective TRPV4 antagonists are available39,47, we evaluated the outcome of TRPV4 antagonist intravitreal injection after enhanced BRB breakdown due to experimental diabetes induced by streptozotocin and found that TRPV4 antagonism mitigated BRB breakdown to similar levels than vasoinhibins. We explored the possibility that vasoinhibins and TRPV4 antagonism may synergize to regulate BRB permeability, using ARPE-19 cell monolayers and microvascular endothelial cells in hyperglycemic and normoglycemic conditions, mimicking the diabetic and non-diabetic conditions, respectively. We showed that TRPV4 antagonism and vasoinhibins synergize by activating complementary pathways to counteract the diabetes-like effects on RPE permeability. In addition, vasoinhibins can block the exogenously activated TRPV4 to regulate BRB and endothelial permeability, likely by interfering with the TRPV4/Ca2+/NO/cytoskeletal reorganization cascade.
Results
TRPV4 is expressed in both the inner and outer BRB, and its pharmacological inhibition resolves the streptozotocin-induced increase of BRB permeability similarly to vasoinhibins in rats
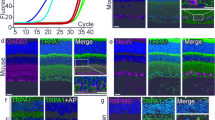
We determined whether the BRB expresses TRPV4 in situ. The reliability of TRPV4 detection by the polyclonal sc-98592 antibody was addressed using Trpv4 −/− mice48 as a negative control, and by comparing the sc-98592 staining to the one obtained with the anti-TRPV4 LS-C94498 antibody, the specificity of which has previously been confirmed40. The histology of Trpv4 −/− retinas is similar to that of wild-type mice except for the outer limiting membrane that is not visible in Trpv4 −/− retinas (supplemental Fig. 1A). Layer thickness was similar in wild-type and Trpv4 −/− retinas (supplemental Fig. 1B). Representative images of transversal retinal sections from wild-type mice stained with the sc-98592 antibody showed uniform cytoplasmic immunoreactivity (Fig. 1A). TRPV4 immunostaining was distributed throughout the retina, including cell bodies in the ganglion cell layer; radial processes within the inner plexiform and outer nuclear layers, and outer limiting membrane; cell bodies and capillaries in the inner nuclear layers; and RPE, marked with RPE-65. This immunopositive pattern for TRPV4 was absent in retinal sections from Trpv4 −/− mice (Fig. 1A). In parallel, we have repeated the immunohistochemistry on the same tissues using the LS-C94498 antibody (Fig. 1A) and confirmed the previously described immunostaining pattern40. Our data further showed a positive staining in the RPE. Projections in z showed that TRPV4 localizes to the basal outer non-pigmented part of the RPE, while RPE-65 is in the cytosol (Fig. 1B). The colocalization of TRPV4 with a fluorescein isothiocyanate, used as blood vessel marker49 can be appreciated in Fig. 1C. Immunohistochemistry using the LS-C94498 anti-TRPV4 antibody labeled TRPV4 in whole mounts of mouse RPE, whereas no specific signal appeared in Trpv4 −/− RPE (Fig. 1D). These data show that TRPV4 is expressed in both the inner and the outer main component of the BRB.

TRPV4 is expressed in both the inner and outer BRB, and its pharmacological inhibition prevents the streptozotocin-induced increase of BRB permeability in rats, similar to the effect of vasoinhibins. (A) Representative confocal stack image of transverse sections of Trpv4 +/+ and Trpv4 −/− mouse retinas showing TRPV4 (red), RPE-65 (green), and merge immunofluorescence. The anti-TRPV4 sc-98592 and LS-C94498 antibodies (Ab) were used as indicated and cell nuclei were stained with DAPI. Magnification bars were as indicated. Retinal pigment epithelium (RPE), outer segments (OS), outer nuclear layer (ONL), outer limiting membrane (OLM), outer plexiform layer (OPL), inner nuclear layer (INL), inner plexiform layer (IPL), and ganglion cell layer (GCL). Images were captured in three different regions of the same retina section (n = 3). Both retinas of three animals per group were analyzed. (B) Representative confocal image and corresponding projection in z-x (Z1, right) and z-y (Z2, bottom) of transverse sections of wild-type choroid-RPE (full-size image is shown in supplemental Fig. 3) showing RPE-65 (green) and TRPV4 (red) antibody immunofluorescence. Projections in z correspond to the area indicated by the yellow lines and show that TRPV4 and RPE-65 did not colocalize. (C) Representative confocal image and projection in z-x (Z1, right) and z-y (Z2, bottom) of transverse sections of wild-type OPL and INL (full-size image is shown in supplemental Fig. 3) stained with the anti-TRPV4 sc-98592 or LS-C94498 antibodies (red), the anti-mouse antibody coupled to fluorescein isothiocyanate as a marker of blood vessels (green), as previously reported49, and DAPI (blue). (D) Representative images of whole mounts of Trpv4 +/+ and Trpv4 −/− mouse RPE stained with the LS-C94498 anti-TRPV4 antibody and DAPI. Projection in z-x (right) and z-y (bottom) is also shown. (E) Evaluation of the Evans blue dye content in retinas from control rats intravitreously injected with PBS, the TRPV4 antagonists RN1734 (100 µM) and GSK2193874 (100 nM) or vasoinhibins (Vi, 1 µM) for 24 h and from streptozotocin (STZ)-induced diabetic rats intravitreously injected with PBS, RN1734, GSK2193874 or Vi 24 h before the end of the 4 weeks of diabetes. Values are mean ± s.e.m. normalized to the control (n = 8–14 per group; *P < 0.05). n.s., not significant.
Given this compelling localization of the channel, the functional role of TRPV4 in the BRB was then investigated. Previous studies have shown that TRPV4 inhibition protects against rupture of the endothelial barrier in the lung46 and that expression levels of TRPV4 are altered in both macrovascular50 and retinal microvascular41 vessels from streptozotocin-induced diabetic rats. Taken together with the fact that BRB breakdown is a feature of diabetes, this information prompted us to examine whether TRPV4 inhibition would eliminate the excessive increase in BRB permeability induced by a diabetic metabolic situation. In the streptozotocin rat preclinical model of diabetes, BRB breakdown occurs as early as 5 days and up to 10 weeks post-streptozotocin treatment9,11,12,51,52,53,54,55. Here we confirmed the BRB breakdown in this diabetes model by showing that retinal accumulation of Evans blue-stained albumin doubled at 4 weeks after streptozotocin injection (Fig. 1E). The selective TRPV4-channel blockers, RN173456 and GSK219387439, were injected intravitreally, and 24 h later inhibition of the streptozotocin-induced BRB breakdown was observed (Fig. 1E). In controls without streptozotocin, RN1734 and GSK2193874 alone did not modify BRB permeability (Fig. 1E). As previously reported10, vasoinhibins prevented the streptozotocin-induced increase in BRB permeability, and this effect was similar to that of RN1734 and GSK2193874 (Fig. 1E), and vasoinhibins did not modify the basal transport through the BRB (Fig. 1E). Taken together, these data suggest that vasoinhibins may block excessive BRB permeability associated with diabetes by inhibiting TRPV4.
TRPV4 is expressed in human ARPE-19 monolayers and its protein levels and cellular localization are not altered by exposure to high glucose
To study whether vasoinhibins mitigate the streptozotocin-induced increase of BRB permeability by acting directly on the RPE and to test if TRPV4 inhibition mediates this effect, we first provided evidence for monolayer formation and TRPV4 expression in ARPE-19 cells. Phase-contrast microscopy images on cross-sections of ARPE-19 cultures showed that these cells displayed the characteristic features of RPE, including defined cell borders and overall “cobblestone” appearance, and DAPI staining showed that they formed a relatively uniform monolayer (Fig. 2A). Immunocytochemistry showed that TRPV4 is localized throughout the cell (Fig. 2A). Immunolabelling with the sc-98592 antibody showed a pattern resembling the one previously described in cultured capillary endothelial cells28,41, while the LS-C94498 antibody preferentially stained cell cytoplasm.

TRPV4 expression in ARPE-19 monolayers. (A) Representative phase-contrast microscopy and confocal images of ARPE-19 cultured on Transwell membrane inserts with pore sizes of 0.4 µm (see Methods) stained with DAPI and anti-TRPV4 sc-98592 and LS-C94498 antibodies (Ab), under control conditions (day 0) and after exposure to high glucose (D-glucose) for 28 days. Projection in z-x (right) and z-y (bottom) showed nucleus alignment and TRPV4 localization. Magnification bar was as indicated. (B) ARPE-19 cells cultured in 5.5 mM D-glucose (control), 5.5 mM D-glucose plus 19.5 mM mannitol (mannitol), and 25 mM D-glucose (D-glucose) for 28 days were analyzed for TRPV4 protein. Total β-actin served as loading control. Extracts from three independent ARPE-19 cell cultures in each condition were analyzed (N = 3); MWM, molecular weight markers. (C) Densitometric analysis of the TRPV4 fragment normalized to β-tubulin (control) expressed in arbitrary units (AU). Values correspond to mean ± s.e.m. for three independent experiments. n.s., not significant.
To simulate blood glucose found in the streptozotocin rat model of diabetes, ARPE-19 cells were subjected to basolateral administration of a high glucose concentration (25 mM). After a 28-day exposure to high glucose, ARPE-19 cells conserved defined cell borders and overall “cobblestone” appearance but were flattened, and DAPI staining was indicative of monolayer (Fig. 2A). Projection in z showed that TRPV4 localization was overall similar in ARPE-19 cells exposed or not for 28 days to 25 mM of D-glucose (Fig. 2A). Western blot showed a unique band of 98 kDa (Fig. 2B). We observed that levels of TRPV4 protein were not modified after a 4-week exposure to high glucose in culture (Fig. 2B and C).
Exposure to high glucose increases and decreases the resistance of ARPE-19 monolayers after 3 and 28 days, respectively
Since inconsistent results were obtained when the trans-electrical resistance (TER) was measured in ARPE-19 cells treated with high glucose concentrations57,58,59,60, we examined the TER through monolayers of ARPE-19 cells over a period of 4 weeks. Under control conditions, TER was maintained at ∼65 ± 7 Ohm.cm2 the whole time (Fig. 3A). The resistance of ARPE-19 monolayers increased after 3 days of exposure to high glucose (Fig. 3A). This increase was transient since TER values at days 0, 7, 14, and 21 were similar in all conditions. At day 28, high glucose associated with a TER decrease, compared with levels under mannitol, which was used as an osmotic control (Fig. 3A). Additionally, some previous studies showed that high glucose conditions associate with cytotoxicity in ARPE-19 cells61,62,63,64,65,66, while others did not59,67. We found no change in ARPE-19 cell viability after a 4-week exposure to high glucose (Fig. 3B). Thus, prolonged cultures preserve stable TER and viability in ARPE-19 monolayers, allowing us to explore the effect of TRPV4 antagonism and vasoinhibins on ARPE-19 permeability under hyperglycemic-mimicking conditions.

Dual contribution of TRPV4 antagonism and vasoinhibins to regulate the high glucose-induced effects on ARPE-19 monolayer resistance. (A) Time course of trans-electrical resistance (TER) in ARPE-19 cell monolayers cultured in 5.5 mM D-glucose (control), 5.5 mM D-glucose plus 19.5 mM mannitol (mannitol), and 25 mM D-glucose (D-glucose) over 28 days. (B) Quantification of viability levels in ARPE-19 monolayers treated as previously indicated at day 28. ARPE-19 cells were cultured on inserts with pore sizes of 0.4 µm. TER and MTT signals were normalized to the untreated condition. (B and D) Quantification of TER values in ARPE-19 monolayers after 3 (C) and 28 (D) days of culture in control, mannitol, and D-glucose conditions, in the presence and in the absence of GSK2193874 (50 nM), vasoinhibins (Vi, 10 nM) or both. *P < 0.05 from 3 independent experiments. n.s., not significant.
TRPV4 antagonism and vasoinhibins activate additive pathways to inhibit the high glucose-related effects on ARPE-19 monolayer resistance
We previously reported that vasoinhibins regulate the RPE barrier in the human ARPE-19 cell line13,68. We therefore studied the role of TRPV4 inhibition in this process. GSK2193874 did not prevent the high glucose-induced increase in TER at day 3 (Fig. 3C). Alone or in the presence of mannitol, GSK2193874 did not modify the resistance of ARPE-19 monolayers at day 3 (Fig. 3C). In contrast, vasoinhibins enhanced the high glucose-induced increase in TER after 3 days of culture (Fig. 3C). Vasoinhibins had no effect on the resistance of ARPE-19 monolayers alone or in the presence of mannitol (Fig. 3C). When GSK2193874 was applied concomitantly with vasoinhibins under high glucose, ARPE-19 resistance reached similar values than in the presence of vasoinhibins alone (Fig. 3C). GSK2193874 and vasoinhibin co-administration had no effect on the resistance of ARPE-19 monolayers under control and mannitol conditions.
At day 28, GSK2193874 prevented the high glucose-induced reduction in ARPE-19 TER (Fig. 3D). Vasoinhibins also prevented the high glucose-induced decrease in TER (Fig. 3D). Alone or in the presence of mannitol, neither GSK2193874 nor vasoinhibins modified the resistance of ARPE-19 monolayers (Fig. 3D). When co-administered with vasoinhibins under high glucose, GSK2193874 increased ARPE-19 TER by additional 17% (Fig. 3D).
Taken together, these data show that vasoinhibins regulate ARPE-19 permeability exposed to high glucose through a mechanism that is independent from TRPV4 blockage.
Vasoinhibins can protect BRB from increased endothelial permeability evoked by TRPV4-activation under non-diabetic conditions
Expression levels of endogenous TRPV4 have been shown to be altered in tissues from diabetic rodents, including the retina41,50,69. We therefore tested whether vasoinhibins regulate BRB permeability by inhibiting the activation of TRPV4 under non-diabetic conditions. Injection of the TRPV4 agonists RN-1747 and GSK1016790A39,70 into adult male Wistar rats caused BRB breakdown, as shown by the retinal accumulation of Evans blue-stained albumin (Fig. 4A). Coinjection of vasoinhibins with either RN-1747 or GSK1016790A reduced the BRB changes induced by both TRPV4 agonists (Fig. 4A). These data show that endogenous TRPV4 channels in rat retina activated with selective TRPV4 agonists, are inhibited by vasoinhibins to maintain BRB permeability. Because most regulatory effects of vasoinhibins and TRPV4 activation on permeability have been defined in microvascular endothelial cells36,43,44,45,46, and the inner BRB involves the tight junctions between vascular endothelial cells forming retinal capillaries1,71, we tested whether vasoinhibins regulate BRB permeability by blocking TRPV4 in microvascular endothelial cells. First, we performed ratiometric Ca2+ imaging in TRPV4-transfected human microvascular endothelial cells (HMEC) loaded with Fura-2 AM. TRPV4 expression and distribution at the membrane levels were confirmed72 (Fig. 4B, supplemental Fig. 4). Application of the synthetic phorbol ester 4α-phorbol 12-myristate 13-acetate didecanoate (4α-PDD), a TRPV4 agonist that binds directly to the transmembrane region of the protein and selectively activates TRPV4 in non-neuronal cells23,25,73,74,75,76,77,78, induced transient increases in intracellular Ca2+ levels in 87 ± 7% of the cells (Fig. 4C). Pretreatment with vasoinhibins inhibited the 4α-PDD-induced Ca2+ oscillations (Fig. 4C) and reduced the percentage of HMEC that responded to 4α-PDD to 37 ± 16% (Fig. 4D). Second, in freshly isolated mouse retinal capillary endothelial cells (MRCEC) that were previously shown to present stable resistance over time13, Western blot analysis demonstrated expression of TRPV4 (Fig. 4E). In MRCEC, 4α-PDD induced a transient decrease in TER that was maximal at ∼5 min, lasted about 100 sec, and was fully prevented by vasoinhibins (Fig. 4F). Vasoinhibins alone had no effect (Fig. 4F). To complement the TER evaluations, we examined the tight junction-associated actin microfilaments, as they are known to regulate endothelial cell permeability37,79. Treatment with 4α-PDD induced stress fiber formation with a uniform polarity (Fig. 5A) that coincided with increased actin fluorescence (Fig. 5B) in MRCEC. These effects were blocked by vasoinhibins (Fig. 5A and B), which were previously shown to have no effect alone13. Figure 5B shows quantification of changes in actin fluorescence.

Vasoinhibins block TRPV4-induced BRB breakdown in rat retinas and TRPV4-induced Ca2+ transients and TER reduction in endothelial cell systems in endothelial cell systems. (A) Evaluation of the Evans blue dye content in retinas of control rats intravitreously injected with PBS or one of the TRPV4 agonists RN1747 (100 µM) and GSK1016790A (100 nM) in the presence and in the absence of vasoinhibins (Vi, 1 µM) for 4 h. Values are mean ± s.e.m. normalized to the control (n = 8–14 per group; *P < 0.05). (B) Epifluorescence and confocal images from HMVECs transfected with TRPV4-eGFP, where one can appreciate that TRPV4 is located at the membrane level. Magnification bars, 10 µm. (C) Representative measurements of the change in intracellular Ca2+ measured by the change in the fluorescence ratio (R340/380) in TRPV4-transfected HMEC with or without 10 µM 4α-PDD and 10 nM vasoinhibins (Vi). (D) Corresponding distribution of responding cells (% ± s.e.m.). *P < 0.05. (E) MRCEC cells were analyzed for TRPV4 protein. Total β-actin served as loading control. Extracts from three independent MRCEC cell cultures were analyzed (N = 3); MWM, molecular weight markers. (F) Time course of TER in MRCEC monolayers cultured in complete medium (control) with or without 10 µM 4α-PDD and 10 nM Vi. n.s., not significant.

Vasoinhibins block TRPV4-induced actin cytoskeleton redistribution in MRCEC cells in a NO-dependent manner. (A) MRCEC were cultured in complete medium (control) with or without 10 µM 4α-PDD and 10 mM L-NAME, and with the NO donor DETANONOate (10 µM) in the presence and in the absence of 4α-PDD and Vi (10 nM) for 5 min (corresponding to the peak TER values measured in panel D in the presence of 4α-PDD), and then actin cytoskeleton (F-actin) distribution was determined using rhodamine-phalloidin. Representative fields are shown. Scale bar, 10 µm. (B) Corresponding quantification of phalloidin/DAPI fluorescence. Values are mean ± s.d. (*P < 0.05). n.s., not significant.
Vasoinhibins inhibit the TRPV4/Ca2+/NO/cytoskeletal reorganization cascade
That TRPV4 C-terminal binding protein microtubule-associated protein 7 has been proposed to link TRPV4 to cytoskeletal microfilaments80 suggests that TRPV4 may directly mediate permeability increase. However, it is long known that Ca2+ oscillations associate with elevated permeability by targeting many intracellular targets that include intercellular junctions and/or cytoskeletal proteins37. Among the major pathways linking cytosolic Ca2+ level increases and reorganization of actin cytoskeleton are protein kinase C (PKC), Ca2+/calmodulin-dependent protein kinase II (CaMKII), and NADPHoxidase/reactive oxygen species (ROS)37. CaMKII stands out because its activation promotes cytoskeletal reorganization through increased nitric oxide (NO) production via endothelial NO synthase (eNOS)-Ser11,79 phosphorylation81 and CaMKII antagonism prevents TRPV4-mediated effects82, including eNOS activation70. Vasoinhibins block eNOS activation by abrogating Ca2+ transients in endothelial cells10,14,16, thereby regulating the BRB13. For these reasons, we explored the role of NOS and NO in mediating the inhibition of TRPV4 by vasoinhibins. We observed that coadministration of 4α-PDD and the NOS inhibitor L-NAME83 eliminated the 4α-PDD-induced stress fiber formation, but L-NAME alone had no effect (Fig. 5A). Both the NO donor DETANONOate and 4α-PDD increased actin fluorescence (Fig. 5B), but DETANONOate induced the formation of actin stress fibers that show apparently random orientation with respect to each other (Fig. 5A) and 4α-PDD induced a larger increase in actin fluorescence than DETANONOate (Fig. 5B). 4α-PDD and DETANONOate together stimulated actin to levels that overcame their individual effects (Fig. 5B). We then asked whether DETANONOate reverts the action of vasoinhibins in the presence of 4α-PDD. Exogenous NO prevented vasoinhibin-mediated inhibition of 4α-PDD-induced F-actin rearrangement in MRCEC (Fig. 5A). All together, these data suggest that NO partly mediates the 4α-PDD-induced increase in endothelial cell monolayer actin cytoskeleton redistribution, and that vasoinhibins block the NO-dependent component of these actions by acting upstream of NO production, as previously reported13,14,16.
Discussion
Microaneurysms, hemorrhages, and retinal edema in diabetic retinopathy can critically imperil vision, thus greatly contributing to disease-related disability. These retinal changes ultimately reflect BRB breakdown. Our previous work showed that vasoinhibins have significant therapeutic potential for the control of diabetic retinopathy and macular edema since they are natural inhibitors of angiogenesis and BRB breakdown9,10,11,12,49. Here, we unveil two novel concepts in the pathogenesis of diabetic retinopathy that will further guide us toward rationally-guided new therapies: (i) vasoinhibins can inhibit TRPV4 to maintain BRB and endothelial permeability, (ii) TRPV4 blockage does not seem to contribute to the regulation of BRB and RPE permeability by vasoinhibins under diabetic or hyperglycemic-mimicking conditions; importantly, the TRPV4 antagonist GSK2193874 resolves BRB breakdown associated with diabetes.
Specificity of TRPV4 detection
TRPV4 is present in mouse retina18,40. We detected TRPV4 protein in the inner and outer main components of BRB in adult mice. Our data complemented previous studies41,42,84 by detecting TRPV4 protein in ARPE-19 cells and mouse RPE flat mounts, and in primary cultures of mouse retinal microvascular endothelial cells. Because Trpv4 −/− retinas lacked immunopositive pattern for TRPV4 when probed with sc-98592 and LS-C94498, both antibodies can be considered specific. The expression pattern revealed by the two antibodies concurred in that cell bodies located in the ganglion cell layer, radial processes of Müller glia, and RPE were immune-positive. However, both antibodies did not recognize a strictly identical TRPV4 pattern: sc-98592 detected immunoreactivity in inner nuclear layer cell bodies that was not present with LS-C94498; sc-98592 immunoreactivity was uniform compared to the LS-C94498 one; and sc-98592 recognized intraretinal capillaries, while LS-C94498 did not. The differential pattern of immunopositive staining for TRPV4 may be due to the recognition of distinct epitopes of the channel protein by the two antibodies. The sc-98592 antibody, also known as H-79, targets an epitope corresponding to amino acids 62–134 mapping near the N-terminus of TRPV4. Immunoblots from several cell types showed a primary band of 132 kDa, and in few cases, a secondary band at ~90 kDa which probably corresponded to N-glycosylated and unglycosylated forms of TRPV4. In our hands, Western blot of ARPE-19 cells and MRCECs showed a unique band at ~100 kDa. In contrast, the anti-TRPV4 LS-C94498 antibody recognized a primary band of 85 kDa and a secondary band at 105 kDa (manufacturer and ref.40). This antibody was raised against synthetic peptide from 1st cytoplasmic domain of mouse TRPV4 conjugated to an immunogenic carrier protein. The differences we observed in the expression pattern, in keeping with previous reports, also suggest the existence of TRPV4 isoforms85,86,87,88.
TRPV4 as a regulator of BRB?
Although we observed that the TRPV4 antagonists RN1734 and GSK2193874 alone did not alter BRB permeability, intravitreal administration of the TRPV4 agonists RN-1747 and GSK1016790A promoted the accumulation of Evans blue dye in rat retinas, indicating that stimulation of endogenous TRPV4 increases BRB permeability. These data are consistent with the observations that intravenous administration of TRPV4 activators can lead to circulatory collapse, which at least is partly explained by increased capillary permeability and fluid loss in the lungs46,70,89,90. Also, 4α-PDD exposure decreases TER in microvascular endothelial cells, including primary culture of retinal capillary endothelial cells, which is in agreement with previous studies19,34,44,91. Further analysis should include trpv4−/− mice to define the precise role of endogenous TRPV4 channels in BRB transport.
TRPV4 inhibitors resolve the BRB breakdown associated with streptozotocin-induced diabetes. This effect is new for TRPV4 antagonists in the retina. HC-067047 has been recently shown to reduce brain water content and Evans blue extravasation at 48 h after middle cerebral artery occlusion in mice92. In view that Trpv4 −/− and wild-type mice treated with a TRPV4 antagonist did not develop insulin resistance under high-fat diet93 and that this diet associates with retinal dysfunction94, our findings implicate that (i) TRPV4 blockage disables one or more signaling pathways that participate in diabetes-induced alterations of the retinal neurovascular complex5,66 and/or that (ii) TRPV4 activity is excessive in the diabetic retina44. Observations reporting consistent TRPV4 down-regulation during diabetes challenge this view41,50,69. We did not observe any change in TRPV4 protein levels of ARPE-19 cells exposed basolaterally to high glucose. Nevertheless, that high glucose decreased ARPE-19 permeability after 3 days is consistent with the decreased mRNA and protein levels of TRPV4 in retinal bovine capillary endothelial cells (RBCECs) after 3 days of high glucose41. Additionally, we found that at day 3, TRPV4 antagonism does not prevent the high glucose-induced increase in ARPE-19 TER, which is consistent with a loss of 4-α PDD-induced Ca2+ response in RBCECs under hyperglucemic-mimicking conditions for 3 days41. Reduced channel expression is likely to play a role in this effect41; however, direct inhibition of TRPV4 channels under high glucose may also contribute to silenced TRPV4 function in ARPE-19 cells.
Reduced TRPV4 channel expression has been suggested as an underlying reason for the dysfunction of vascular function during diabetes, but it may be a compensatory effect in diabetes. TRPV4 antagonism in diabetic rats controled excessive BRB permeability and GSK2193874 protected against the high glucose-induced decrease of ARPE-19 resistance. This further indicates that after 4 weeks of diabetes or high glucose, some TRPV4 is still functional. In agreement with this conclusion, faint yet detectable TRPV4 immunostaining in retinal arterioles and capillaries is seen after 12 weeks of streptozotocin-induced diabetes in rats41. We also detected TRPV4 protein in ARPE-19 treated for 28 days with high glucose.
Reduced TRPV4 expression may not always be accompanied by a reduced function because a limited number of functional channels expressed by a cell can completely maintain function. The diabetic milieu modifies many parameters as an early compensatory response, including factors that modulate TRPV4, e.g., generation of arachidonic and epoxyeicosatrienoic acids95 (both of them being endogenous metabolites that activate TRPV4), glycosylation levels96, and Ca2+ homeostasis97. Long-term exposure to these mediators associates with diabetic complications that may include the excessive permeability of BRB.
In each tested microvascular beds, the relative sensitivity of TRPV4 to stimulation/inhibition varied; their use therefore only provides qualitative data. The latency (300 sec) and duration (100 sec) of Ca2+ response is comparable to the response observed in endothelial cells89 and in other cell systems44,77,98, and the transient decrease in TER coincides with the rapid (within 5 min) decrease in resistance of mouse mammary gland cell line HC1144. In comparing kinetics of 4α-PDD-evoked responses in endothelial cells from similar microvascular beds24,72,99 and other cell types24, we see that 4α-PDD induced oscillatory [Ca2+]i changes and that Ca2+ transients preceed and are concomitant to TER decrease, suggesting that both events may be causally linked. This possibility is plausible since several studies established that a rise in cytosolic Ca2+ levels is sufficient to activate key signaling pathways that mediate cytoskeletal reorganization (through myosin light chain-dependent contraction) and VE-cadherin disassembly at the adherens junctions, which in turn increase endothelial permeability100. Simultaneous recording of [Ca2+]i and actin dynamics confirmed that TRPV4-induced stress fiber formation takes place during the Ca2+ elevation101. As already mentioned, TRPV4 -mediated Ca2+ entry may be sufficient to reorganize actin80, but Ca2+ is also a potent activator of NOS and we observed that L-NAME blocked the effect of 4α-PDD on actin reorganization. The existence of a TRPV4/Ca2+/NO/cytoskeletal reorganization cascade is consistent with studies showing that 4α-PDD increases NO production36,102,103,104 and that responses to TRPV4 depend on NO production70,105,106. Furthermore, eNOS has been reported to colocalize with TRPV4107, and eNOS expression is up-regulated under mechanic stretch, which associates with TRPV4-gated Ca2+ influx101,108. Nevertheless, our data showing that both 4α-PDD and DETANONOate increased actin fluorescence have to be taken carefully since DETANONOate appeared to induce the formation of randomly orientated actin stress fibers, while 4α-PDD induced stress fiber formation with a uniform polarity. These differences may be due to pharmacological considerations but they also suggest that 4α-PDD may activate additional pathways than DETANONOate to reorganize actin. The canonical phospholipase A2 and ROS pathways may be involved as previously described91,101,109.
Additionally, 4α-PDD induces a transient decrease in TER, as previously reported44. Currently, it is unclear as to what ends the TRPV4-mediated regulation of epithelial permeability. The transient decrease in TER may be due to the Ca2+-mediated negative feedback inhibition on TRPV4 channels24,86,110,111; TRPV4-mediated Ca2+ oscillations were previously reported to end rapidly24. Also, the surface expression of TRPV4 channels is tightly controlled. TRPV4 proteins may be retrieved from plasma membrane112, which would abrogate the signal promoting decrease in TER. It is also important to mention that the large permeability changes observed at days 3 and 28 under high glucose cannot be compared to the acute regulatory effect of TRPV4 stimulation on cell permeability. Usually physiological signals (i.e., bradykinin) transiently regulate barrier permeability. This does not contradict that these same signals induce an exacerbated response under pathological conditions like diabetes, due to alterations in their whole signaling pathway113,114 and in the amount and/or nature of TRPV4 endogenous agonist(s), glycosylation levels, and basal levels of Ca2+ 95,96,97.
Molecular crosstalk between TRPV4 and vasoinhibins?
Our data suggest that vasoinhibins under high glucose promote ARPE-19 resistance through a mechanism that is independent from TRPV4 blockage but that under normoglycemic conditions, vasoinhibins can block TRPV4 to maintain BRB and endothelial permeability. So far, vasoinhibins have not been related to any stimuli known to activate TRPV4 (i.e., cell swelling, low pH, mechanical stress, and temperature115,116,117,118,119), and TRPV4 has not been related to the few described binding sites for vasoinhibins120,121, but vasoinhibins may indirectly regulate TRPV4 by interfering with both receptor-operated Ca2+ entry14,16,17 and NO production10,13,14,122.
On the one hand, vasoinhibins may act upstream of TRPV4 channels. Indeed, vasoinhibins have been demonstrated to prevent activation of phospholipase C by G-protein-coupled receptors, which diminishes intracellular Ca2+ release and plasma membrane Ca2+ channels activation16,17; La3+, an inhibitor of receptor-operated Ca2+ entry, prevented vasoinhibin effects16, and TRPV4 has been demonstrated to participate in the receptor-operated Ca2+ entry25. On the other hand, vasoinhibins may interfere with TRPV4 activity through Ca2+/calmodulin signaling. By abrogating Ca2+ transients in endothelial cells, vasoinhibins have been shown to block eNOS activation14,16 that mainly depends on Ca2+/calmodulin binding15. In contrast, Ca2+ entry through TRPV4 promotes the formation of Ca2+-calmodulin complexes, which binding to TRPV4 enhance channel activity32,33.
Vasoinhibins reduce NO production by limiting eNOS activation through Ca2+/calmodulin, and also by causing its dephosphorylation by protein phosphatase 2A10,13,14,122. NO has been shown to inactivate TRPV4 by two mechanisms: (i) inducing TRPV4 S-nitrosylation on the Cys8,53 residue, which reduces the channel sensitivity to 4-αPDD and its interaction with calmodulin123, and (ii) increasing cGMP that inhibits TRPV4 by PKG-mediated phosphorylation124. In our study, a pretreatment with vasoinhibins would prevent the NO-mediated inhibition of TRPV4. The lack of this negative feedback inhibition would initially increase TRPV4-mediated Ca2+ entry and, based on the fact Ca2+-mediated negative feedback inhibition on TRPV4 channels has been well documented in vascular endothelial cells86,110,111, a similar inhibitory mechanism could occur in our conditions. In this feedback mechanism, Ca2+ influx would stimulate a NO-cGMP-PKG and/or NO-S-nitrosylation cascade(s), resulting in inactivation of TRPV4 channels, which would explain the reduced Ca2+ transients in response to 4-αPDD in TRPV4-transfected HMECs pretreated with vasoinhibins. We propose that vasoinhibins reduce TRPV4 activity by promoting a microenvironment where the non-capacitative Ca2+ entry is depressed and where the Ca2+-mediated negative feedback inhibition on TRPV4 channels is enhanced via the abrogation of NO production.
In conclusion, our novel data indicate that TRPV4-signaling can be recruited as a novel target to combat diabetic retinopathy, which is characterized by permeability changes that can be attenuated by endogenous vasoinhibins, which combine synergistically with selective TRPV4 blockers. Interestingly, these two therapeutically-beneficial approaches function via non-overlapping signaling pathways in diabetic conditions. Our findings elevate understanding of the blinding consequences of diabetic retinopathy and pave the way for successfully treating this dreaded and disabling diabetes complication, which is on the rise world-wide.
Methods
Reagents
Recombinant human vasoinhibins (corresponding to a 14-kDa fragment of prolactin) used in cell culture experiments were generated by site-directed mutagenesis as previously described125. TRPV4 agonists (RN1747, GSK1016790A) and antagonists (RN1734, GSK2193874), mannitol, D-glucose, 4alpha-phorbol-didecanoate (4α-PDD) were purchased from Sigma-Aldrich (St Louis, MO). Rhodamine-phalloidin was purchased from Thermo Fisher Scientific Inc. (Waltham, MA). Anti-TRPV4 (LS-C94498, Lifespan Biosciences, Seattle, WA and sc-98592, Santa Cruz Biotechnology, Dallas, TX) and anti-β-tubulin (ZYMED from Life Technologies; #22833) antibodies were purchased as specified.
Ethics statement
All experiments were approved by the Bioethics Committee of the Institute of Neurobiology at the National Autonomous University of Mexico (UNAM, protocol #74) and methods were carried out in accordance with the rules and regulations of the Society for Neuroscience: Policies on the Use of Animals and Humans in Neuroscience Research. All efforts were made to minimize the number of animals used and their suffering.
Animal care and retinal tissue
Male albino rats (Wistar, 250–300 g) and C57BL/6J mice of either sex (4–6 months old) were obtained from commercial suppliers, whereas Trpv4 −/− mice were a kind gift from Dr. Wolfgang Liedtke (University). The animals were fed ad libitum and reared in normal cyclic light conditions (12 h light: 12 h dark) with an ambient light level of approximately 400 lux. Animals were sacrificed by CO2 inhalation and decapitation. Eyes were enucleated and processed by the Evans blue method and immunohistochemistry. Diabetes was induced in Wistar rats with a single intraperitoneal injection of streptozotocin (60 mg/kg)126. Animals with glucose levels greater than 250 mg/dl were used 4 weeks after diabetes induction.
Intravitreal injections
Rats were injected intravitreously as reported49. The final injection volume was 5 µl. In one group, one eye was injected with RN1747 (2.4 µg per eye, corresponding to 100 µM as the estimated volume of rat vitreous is 60 µl127) or GSK1016790A (1.6 ng, 40 nM) and the contra-lateral eye with RN1747 or GSK1016790A combined with vasoinhibins (1 µg, 1 µM). Controls for PBS and injected protein itself are shown in SI. The control groups consisted of rat eyes injected with vehicle (PBS), and the contralateral eye received RN1747 (100 µM) or GSK1016790A (40 nM). The second group consisted of rat eyes injected with vehicle (PBS), and the contralateral eye received RN1734 (2.1 µg, 100 µM), GSK2193874 (1.7 ng, 40 nM) or vasoinhibins (1 µg, 1 µM).
Quantification of BRB permeability
The Evans blue dye permeation assay was performed as previously described55. It is, however, worth mentioning that in experiments performed in Fig. 4A, Evans blue dye was administered 2 h after the intravitreous injections and left in the circulation for an additional 2 h.
Histology
See SI.
Immunohistochemistry
Eyes were fixed in 4% paraformaldehyde. Then, they were washed overnight in 0.1 M PBS, immersed in 15% sucrose at room temperature, oriented and frozen in Tissue-Tek (Sakura Finetek, Torrance, CA), and sectioned (12 µm thick) along the sagittal axis of the eye or dissected for a whole mount of the RPE as described128. Retina cryosections were incubated in PBS with 1% SDS for 5 min, rinsed three times with PBS for 5 min, blocked in PBS containing 10% normal goat serum and 0.1% Triton X-100 for 1 h. Double labeling was performed with both anti-TRPV4 and anti-RPE cell specific antibodies [anti-TRPV4 polyclonal sc-98592 antibody, diluted 1:150; anti-TRPV4 LS-C94498 antibody from Lifespan Biosciences, diluted 1:200; anti-RPE65, at 0.08 µg/ml]. After incubation with the primary antibodies, samples were rinsed 3 times in PBS and labeled for 2 h with Alexa-594-conjugated goat anti-rabbit IgG (diluted 1:100), or Alexa-488-goat anti-mouse IgG (diluted 1:500) obtained from Molecular Probes (Eugene, OR). Extensive controls for the anti-TRPV4 LS-C94498 antibody were performed previously40, and given that staining with the sc-98592 anti-TRPV4 antibody showed a pattern of TRPV4 immunoreactivity in retina sections similar to that observed with the LS-C94498 antibody (data not shown), we used the sc-98592 antibody. Labeled samples were examined with a laser scanning confocal microscope (Zeiss Axiovert 200 LSM 510 Meta, Carl Zeiss International, Oberkochen, Germany). Images were prepared using the Zeiss LSM Image Examiner.
ARPE-19 cell cultures
The ARPE-19 human cell line was purchased from ATCC (Number: CRL-2302)68 and was grown in Dulbecco’s Modified Eagle’s Medium (DMEM)/nutrient mixture F12 supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Cultures were seeded at an initial density of 106 cells and maintained at 37 °C and 5% CO2. Cells were used between passages 10–13, reaching approximately 5 weeks in culture.
Primary cultures of mouse retinal capillary endothelial cells (MRCEC)
We isolated MRCEC from the mouse retina using collagenase digestion and magnetic-activated cell sorting (MACS), ensuring a preparation of >90% purity, as previously described13. CD31-positive cells from retinal populations were isolated by MACS as previously described129.
Obtention of TRPV4-transfected human microvascular endothelial cells (HMEC)
HMECs were obtained from derma and were isolated using an anti-CD105 antibody coupled to magnetic beads by magnetic cell sorting using the MACS system (Miltenyi Biotec, Auburn, CA) as previously described130,131. HMECs from the derma were immortalized by the infection of primary cultures with a replication-defective adeno-5/SV40 virus as previously described132,133. HMECs were grown in complete EndoGRO-MV-VEGF (Millipore) supplemented with 50 μg/mL gentamicin (Cambrex). Cells were used at passages 3 to 15. Periodically, cells were characterized by their morphology and expression of a panel of endothelial antigens such as CD105, CD31, Muc-18 (CD146), CD44, and VEGF receptor 2.
Because HMECs endogenously express low levels of TRPV472,134, they were transfected with the Amaxa Basic Nucleofector Kit for mammalian endothelial cells according to the instructions of the manufacturer using the M-003 program (Lonza) and 2 μg TRPV4-GFP as previously described72. Epifluorescence and confocal images from HMVECs transfected with TRPV4-eGFP showed TRPV4 expression (supplemental Fig. 4).
Immunoblotting
Protein samples from ARPE-19 and MRCEC cultures were resuspended in lysis buffer (0.5% Igepal, 0.1% SDS, 50 mM Tris, 150 mM NaCl, 1 μg/ml aprotinin, and 100 μg/ml PMSF, pH 7.0) and subjected to SDS/PAGE. Total protein (50 μg) was blotted and probed overnight with a 1:500 dilution of the sc-98592 anti-TRPV4 or a 1:1000 dilution of anti-β-actin antibodies. Primary antibodies were detected using an alkaline phosphatase-coupled secondary antibody and a colorimetric detection kit (Bio-Rad).
Measurement of TER
ARPE-19 cells were seeded on 1.12-cm2 Transwell clear polyester membrane inserts (Corning Inc., Corning, NY) with pore sizes of 0.4 µm at an initial density of 150,000 cells per well in DMEM/Nutrient Mixture F-12 Ham medium. After 24 h, TER measured by the EVOM2 Epithelial Voltohmmeter (World Precision Instrument) reached 55 to 75 Ω.cm2 and treatments started. They included control (DMEM containing 5.5 mM D-glucose), mannitol (19.5 mM mannitol added in DMEM), and D-glucose (19.5 mM of D-glucose added in DMEM) conditions. Treatments were applied on the lower chamber of membrane inserts, which are in contact with the basolateral side of cultured cells68. TER values were expressed as percent of control (complete medium) at time 0.
Viability assay
The reduction of 3-(4, 5-dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide (MTT) was used to assess ARPE-19 viability. Cells were seeded in 96-well collagen-coated flat bottom microculture plates (Corning) at an initial density of 2,500 cells/well and treated for 28 days with the previously described control, mannitol, and D-glucose conditions. Next, cells were incubated with MTT (500 mg/mL, Sigma-Aldrich) at 37 °C for 3 h, and the formazan precipitate was solubilized with 0.4 N HCl containing 10% SDS for 30 min at room temperature and quantified by measuring absorbance at 570 nm.
Ca2+ Measurements using Fura-2 AM
Prior to fluorescence measurements, HMECs were trypsinized and plated onto glass coverslips. The medium was replaced every 48 h. Cells were used 3 days after trypsinization. The culture medium was replaced by HBSS solution containing 142 mM NaCl, 5.6 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 0.34 mM Na2HPO4, 0.44 mM KH2PO4, 10 mmol/L HEPES, and 5.6 mM glucose. The osmolarity and pH of this solution were adjusted to 310 mOsm/L and 7.4, respectively. Dye loading was achieved by transferring the cells into a standard HBSS solution containing 1 μM Fura-2 acetoxymethyl ester (Calbiochem, San Diego, CA) and loaded for 40 min at 37 °C. Subsequently, cells were washed three times with HBSS without dye. Observations were performed at 37 °C on an Eclipse Ti microscope using an S Fluor 20x°—/0.75 NA objective lens (both from Nikon). Images were collected through a Rolera EM-C2 charge-coupled device (CCD) camera (QImaging, Surrey, Canada) controlled with Metafluor software (Molecular Devices, Sunnyvale, CA) and analyzed with Igor Pro software (WaveMetrics Inc., Lake Oswego, OR). >94% of HMECs respond to TRPV4 agonist.
Filamentous F-Actin staining
TRITC-labeled phalloidin (Molecular Probes) staining was performed and the mean fluorescence intensity ratio (phalloidin/DAPI) was determined as previously described13.
Statistical analysis
All results were replicated in three or more independent experiments. Data are reported as mean ± S.E.M.; all data showed normal distribution or equal variance according to D’Agostino-Pearson omnibus and Levene’s tests, respectively. Differences between two groups were evaluated by a two-tailed Student’s t-test. Comparisons between different groups were determined by ANOVA followed by Bonferroni’s post-hoc comparison test (Sigma Stat 7.0, Systat Software Inc., San Jose, CA). Differences in means with P < 0.05 were considered statistically significant.
Change history
20 June 2018
A correction to this article has been published and is linked from the HTML and PDF versions of this paper. The error has not been fixed in the paper.
References
Klaassen, I., Van Noorden, C. J. & Schlingemann, R. O. Molecular basis of the inner blood-retinal barrier and its breakdown in diabetic macular edema and other pathological conditions. Prog Retin Eye Res 34, 19–48, https://doi.org/10.1016/j.preteyeres.2013.02.001 (2013).
Gardner, T. W., Antonetti, D. A., Barber, A. J., LaNoue, K. F. & Levison, S. W. Diabetic retinopathy: more than meets the eye. Surv Ophthalmol 47(Suppl 2), S253–262 (2002).
Simo, R., Villarroel, M., Corraliza, L., Hernandez, C. & Garcia-Ramirez, M. The retinal pigment epithelium: something more than a constituent of the blood-retinal barrier–implications for the pathogenesis of diabetic retinopathy. J Biomed Biotechnol 2010, 190724, https://doi.org/10.1155/2010/190724 (2010).
Xu, H. Z. & Le, Y. Z. Significance of outer blood-retina barrier breakdown in diabetes and ischemia. Invest Ophthalmol Vis Sci 52, 2160–2164, https://doi.org/10.1167/iovs.10-6518 (2011).
Stem, M. S. & Gardner, T. W. Neurodegeneration in the pathogenesis of diabetic retinopathy: molecular mechanisms and therapeutic implications. Curr Med Chem 20, 3241–3250 (2013).
Chistiakov, D. A. Diabetic retinopathy: pathogenic mechanisms and current treatments. Diabetes Metab Syndr 5, 165–172, https://doi.org/10.1016/j.dsx.2012.02.025 (2011).
Clapp, C., Aranda, J., Gonzalez, C., Jeziorski, M. C. & Martinez de la Escalera, G. Vasoinhibins: endogenous regulators of angiogenesis and vascular function. Trends Endocrinol Metab 17, 301–307, https://doi.org/10.1016/j.tem.2006.08.002 (2006).
Triebel, J., Huefner, M. & Ramadori, G. Investigation of prolactin-related vasoinhibin in sera from patients with diabetic retinopathy. Eur J Endocrinol 161, 345–353, https://doi.org/10.1530/EJE-09-0130 (2009).
Arnold, E. et al. High levels of serum prolactin protect against diabetic retinopathy by increasing ocular vasoinhibins. Diabetes 59, 3192–3197, https://doi.org/10.2337/db10-0873 (2010).
Garcia, C. et al. Vasoinhibins prevent retinal vasopermeability associated with diabetic retinopathy in rats via protein phosphatase 2A-dependent eNOS inactivation. J Clin Invest 118, 2291–2300, https://doi.org/10.1172/JCI34508 (2008).
Diaz-Lezama, N. et al. Diabetes enhances the efficacy of AAV2 vectors in the retina: therapeutic effect of AAV2 encoding vasoinhibin and soluble VEGF receptor 1. Lab Invest 96, 283–295, https://doi.org/10.1038/labinvest.2015.135 (2016).
Ramirez, M. et al. Vasoinhibin gene transfer by adenoassociated virus type 2 protects against VEGF- and diabetes-induced retinal vasopermeability. Invest Ophthalmol Vis Sci 52, 8944–8950, https://doi.org/10.1167/iovs.11-8190 (2011).
Arredondo Zamarripa, D. et al. Vasoinhibins regulate the inner and outer blood-retinal barrier and limit retinal oxidative stress. Front Cell Neurosci 8, 333, https://doi.org/10.3389/fncel.2014.00333 (2014).
Gonzalez, C. et al. 16K-prolactin inhibits activation of endothelial nitric oxide synthase, intracellular calcium mobilization, and endothelium-dependent vasorelaxation. Endocrinology 145, 5714–5722, https://doi.org/10.1210/en.2004-0647 (2004).
Wu, K. K. Regulation of endothelial nitric oxide synthase activity and gene expression. Ann N Y Acad Sci 962, 122–130 (2002).
Thebault, S. et al. Vasoinhibins Prevent Bradykinin-Stimulated Endothelial Cell Proliferation by Inactivating eNOS via Reduction of both Intracellular Ca2+ Levels and eNOS Phosphorylation at Ser1179. Pharmaceuticals 4, doi:https://doi.org/10.3390/ph4071052 (2011).
Putney, J. W. Jr. Recent breakthroughs in the molecular mechanism of capacitative calcium entry (with thoughts on how we got here). Cell Calcium 42, 103–110, https://doi.org/10.1016/j.ceca.2007.01.011 (2007).
Gilliam, J. C. & Wensel, T. G. TRP channel gene expression in the mouse retina. Vision Res 51, 2440–2452, https://doi.org/10.1016/j.visres.2011.10.009 (2011).
Alvarez, D. F. et al. Transient receptor potential vanilloid 4-mediated disruption of the alveolar septal barrier: a novel mechanism of acute lung injury. Circ Res 99, 988–995, https://doi.org/10.1161/01.RES.0000247065.11756.19 (2006).
Liedtke, W. et al. Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell 103, 525–535 (2000).
Strotmann, R., Harteneck, C., Nunnenmacher, K., Schultz, G. & Plant, T. D. OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nat Cell Biol 2, 695–702, https://doi.org/10.1038/35036318 (2000).
Wissenbach, U., Bodding, M., Freichel, M. & Flockerzi, V. Trp12, a novel Trp related protein from kidney. FEBS Lett 485, 127–134 (2000).
Watanabe, H. et al. Activation of TRPV4 channels (hVRL-2/mTRP12) by phorbol derivatives. J Biol Chem 277, 13569–13577, https://doi.org/10.1074/jbc.M200062200 (2002).
Dunn, K. M., Hill-Eubanks, D. C., Liedtke, W. B. & Nelson, M. T. TRPV4 channels stimulate Ca2+-induced Ca2+ release in astrocytic endfeet and amplify neurovascular coupling responses. Proc Natl Acad Sci USA 110, 6157–6162, https://doi.org/10.1073/pnas.1216514110 (2013).
Lorenzo, I. M., Liedtke, W., Sanderson, M. J. & Valverde, M. A. TRPV4 channel participates in receptor-operated calcium entry and ciliary beat frequency regulation in mouse airway epithelial cells. Proc Natl Acad Sci USA 105, 12611–12616, https://doi.org/10.1073/pnas.0803970105 (2008).
Fiorio Pla, A., Avanzato, D., Munaron, L. & Ambudkar, I. S. Ion channels and transporters in cancer. 6. Vascularizing the tumor: TRP channels as molecular targets. Am J Physiol Cell Physiol 302, C9–15, https://doi.org/10.1152/ajpcell.00280.2011 (2012).
Fiorio Pla, A. & Gkika, D. Emerging role of TRP channels in cell migration: from tumor vascularization to metastasis. Front Physiol 4, 311, https://doi.org/10.3389/fphys.2013.00311 (2013).
Hatano, N., Suzuki, H., Itoh, Y. & Muraki, K. TRPV4 partially participates in proliferation of human brain capillary endothelial cells. Life Sci 92, 317–324, https://doi.org/10.1016/j.lfs.2013.01.002 (2013).
Schierling, W. et al. Cerebral arteriogenesis is enhanced by pharmacological as well as fluid-shear-stress activation of the Trpv4 calcium channel. Eur J Vasc Endovasc Surg 41, 589–596, https://doi.org/10.1016/j.ejvs.2010.11.034 (2011).
Troidl, C. et al. Calcium-dependent signalling is essential during collateral growth in the pig hind limb-ischemia model. J Mol Cell Cardiol 49, 142–151, https://doi.org/10.1016/j.yjmcc.2010.03.021 (2010).
Troidl, C. et al. Trpv4 induces collateral vessel growth during regeneration of the arterial circulation. J Cell Mol Med 13, 2613–2621, https://doi.org/10.1111/j.1582-4934.2008.00579.x (2009).
Shi, M. et al. Glial cell-expressed mechanosensitive channel TRPV4 mediates infrasound-induced neuronal impairment. Acta Neuropathol 126, 725–739, https://doi.org/10.1007/s00401-013-1166-x (2013).
Masuyama, R. et al. Calcium/calmodulin-signaling supports TRPV4 activation in osteoclasts and regulates bone mass. J Bone Miner Res 27, 1708–1721, https://doi.org/10.1002/jbmr.1629 (2012).
Villalta, P. C., Rocic, P. & Townsley, M. I. Role of MMP2 and MMP9 in TRPV4-induced lung injury. Am J Physiol Lung Cell Mol Physiol 307, L652–659, https://doi.org/10.1152/ajplung.00113.2014 (2014).
Yin, J. et al. Role of Transient Receptor Potential Vanilloid 4 in Neutrophil Activation and Acute Lung Injury. Am J Respir Cell Mol Biol 54, 370–383, https://doi.org/10.1165/rcmb.2014-0225OC (2016).
Randhawa, P. K. & Jaggi, A. S. TRPV4 channels: physiological and pathological role in cardiovascular system. Basic Res Cardiol 110, 54, https://doi.org/10.1007/s00395-015-0512-7 (2015).
De Bock, M. et al. Endothelial calcium dynamics, connexin channels and blood-brain barrier function. Prog Neurobiol 108, 1–20, https://doi.org/10.1016/j.pneurobio.2013.06.001 (2013).
Adapala, R. K. et al. PKCalpha mediates acetylcholine-induced activation of TRPV4-dependent calcium influx in endothelial cells. Am J Physiol Heart Circ Physiol 301, H757–765, https://doi.org/10.1152/ajpheart.00142.2011 (2011).
Thorneloe, K. S. et al. An orally active TRPV4 channel blocker prevents and resolves pulmonary edema induced by heart failure. Sci Transl Med 4, 159ra148, https://doi.org/10.1126/scitranslmed.3004276 (2012).
Ryskamp, D. A. et al. The polymodal ion channel transient receptor potential vanilloid 4 modulates calcium flux, spiking rate, and apoptosis of mouse retinal ganglion cells. J Neurosci 31, 7089–7101, https://doi.org/10.1523/JNEUROSCI.0359-11.2011 (2011).
Monaghan, K. et al. Hyperglycemia and Diabetes Downregulate the Functional Expression of TRPV4 Channels in Retinal Microvascular Endothelium. PLoS One 10, e0128359, https://doi.org/10.1371/journal.pone.0128359 (2015).
Zhao, P. Y. et al. TRP Channels Localize to Subdomains of the Apical Plasma Membrane in Human Fetal Retinal Pigment Epithelium. Invest Ophthalmol Vis Sci 56, 1916–1923, https://doi.org/10.1167/iovs.14-15738 (2015).
Clapp, C. et al. Regulation of blood vessels by prolactin and vasoinhibins. Adv Exp Med Biol 846, 83–95, https://doi.org/10.1007/978-3-319-12114-7_4 (2015).
Reiter, B. et al. TRPV4-mediated regulation of epithelial permeability. FASEB J 20, 1802–1812, https://doi.org/10.1096/fj.06-5772com (2006).
Villalta, P. C. & Townsley, M. I. Transient receptor potential channels and regulation of lung endothelial permeability. Pulm Circ 3, 802–815, https://doi.org/10.1086/674765 (2013).
Cioffi, D. L., Lowe, K., Alvarez, D. F., Barry, C. & Stevens, T. TRPing on the lung endothelium: calcium channels that regulate barrier function. Antioxid Redox Signal 11, 765–776, https://doi.org/10.1089/ARS.2008.2221 (2009).
Vincent, F. & Duncton, M. A. TRPV4 agonists and antagonists. Curr Top Med Chem 11, 2216–2226 (2011).
Liedtke, W. & Friedman, J. M. Abnormal osmotic regulation in trpv4−/− mice. Proc Natl Acad Sci USA 100, 13698–13703, https://doi.org/10.1073/pnas.1735416100 (2003).
Aranda, J. et al. Prolactins are natural inhibitors of angiogenesis in the retina. Invest Ophthalmol Vis Sci 46, 2947–2953, https://doi.org/10.1167/iovs.05-0173 (2005).
Ma, X. et al. Functional role of TRPV4-KCa2.3 signaling in vascular endothelial cells in normal and streptozotocin-induced diabetic rats. Hypertension 62, 134–139, https://doi.org/10.1161/HYPERTENSIONAHA.113.01500 (2013).
Kusari, J., Zhou, S. X., Padillo, E., Clarke, K. G. & Gil, D. W. Inhibition of vitreoretinal VEGF elevation and blood-retinal barrier breakdown in streptozotocin-induced diabetic rats by brimonidine. Invest Ophthalmol Vis Sci 51, 1044–1051, https://doi.org/10.1167/iovs.08-3293 (2010).
Zhang, J. et al. Intravitreal injection of erythropoietin protects both retinal vascular and neuronal cells in early diabetes. Invest Ophthalmol Vis Sci 49, 732–742, https://doi.org/10.1167/iovs.07-0721 (2008).
Shi, X. et al. Hesperidin prevents retinal and plasma abnormalities in streptozotocin-induced diabetic rats. Molecules 17, 12868–12881, https://doi.org/10.3390/molecules171112868 (2012).
Shyong, M. P. et al. Reduction of experimental diabetic vascular leakage by delivery of angiostatin with a recombinant adeno-associated virus vector. Mol Vis 13, 133–141 (2007).
Xu, Q., Qaum, T. & Adamis, A. P. Sensitive blood-retinal barrier breakdown quantitation using Evans blue. Invest Ophthalmol Vis Sci 42, 789–794 (2001).
Vincent, F. et al. Identification and characterization of novel TRPV4 modulators. Biochem Biophys Res Commun 389, 490–494, https://doi.org/10.1016/j.bbrc.2009.09.007 (2009).
Villarroel, M., Garcia-Ramirez, M., Corraliza, L., Hernandez, C. & Simo, R. Effects of high glucose concentration on the barrier function and the expression of tight junction proteins in human retinal pigment epithelial cells. Exp Eye Res 89, 913–920, https://doi.org/10.1016/j.exer.2009.07.017 (2009).
Pavan, B., Capuzzo, A. & Forlani, G. High glucose-induced barrier impairment of human retinal pigment epithelium is ameliorated by treatment with Goji berry extracts through modulation of cAMP levels. Exp Eye Res 120, 50–54, https://doi.org/10.1016/j.exer.2013.12.006 (2014).
Wang, S., Du, S., Wu, Q., Hu, J. & Li, T. Decorin Prevents Retinal Pigment Epithelial Barrier Breakdown Under Diabetic Conditions by Suppressing p38 MAPK Activation. Invest Ophthalmol Vis Sci 56, 2971–2979, https://doi.org/10.1167/iovs.14-15874 (2015).
Trudeau, K. et al. Fenofibric acid reduces fibronectin and collagen type IV overexpression in human retinal pigment epithelial cells grown in conditions mimicking the diabetic milieu: functional implications in retinal permeability. Invest Ophthalmol Vis Sci 52, 6348–6354, https://doi.org/10.1167/iovs.11-7282 (2011).
Chen, X. L. et al. Involvement of HMGB1 mediated signalling pathway in diabetic retinopathy: evidence from type 2 diabetic rats and ARPE-19 cells under diabetic condition. Br J Ophthalmol 97, 1598–1603, https://doi.org/10.1136/bjophthalmol-2013-303736 (2013).
Lim, S. K. et al. Hyperglycemia induces apoptosis via CB1 activation through the decrease of FAAH 1 in retinal pigment epithelial cells. J Cell Physiol 227, 569–577, https://doi.org/10.1002/jcp.22756 (2012).
Kim, D. I. et al. High-glucose-induced CARM1 expression regulates apoptosis of human retinal pigment epithelial cells via histone 3 arginine 17 dimethylation: role in diabetic retinopathy. Arch Biochem Biophys 560, 36–43, https://doi.org/10.1016/j.abb.2014.07.021 (2014).
Song, M. K., Roufogalis, B. D. & Huang, T. H. Reversal of the Caspase-Dependent Apoptotic Cytotoxicity Pathway by Taurine from Lycium barbarum (Goji Berry) in Human Retinal Pigment Epithelial Cells: Potential Benefit in Diabetic Retinopathy. Evid Based Complement Alternat Med 2012, 323784, https://doi.org/10.1155/2012/323784 (2012).
Yuan, Z. et al. p38MAPK and ERK promote nitric oxide production in cultured human retinal pigmented epithelial cells induced by high concentration glucose. Nitric Oxide 20, 9–15, https://doi.org/10.1016/j.niox.2008.09.001 (2009).
Xie, P., Fujii, I., Zhao, J., Shinohara, M. & Matsukura, M. A novel polysaccharide compound derived from algae extracts protects retinal pigment epithelial cells from high glucose-induced oxidative damage in vitro. Biol Pharm Bull 35, 1447–1453 (2012).
Shi, H. et al. Inhibition of autophagy induces IL-1beta release from ARPE-19 cells via ROS mediated NLRP3 inflammasome activation under high glucose stress. Biochem Biophys Res Commun 463, 1071–1076, https://doi.org/10.1016/j.bbrc.2015.06.060 (2015).
Dunn, K. C., Aotaki-Keen, A. E., Putkey, F. R. & Hjelmeland, L. M. ARPE-19, a human retinal pigment epithelial cell line with differentiated properties. Exp Eye Res 62, 155–169, https://doi.org/10.1006/exer.1996.0020 (1996).
Hills, C. E., Bland, R. & Squires, P. E. Functional expression of TRPV4 channels in human collecting duct cells: implications for secondary hypertension in diabetic nephropathy. Exp Diabetes Res 2012, 936518, https://doi.org/10.1155/2012/936518 (2012).
Willette, R. N. et al. Systemic activation of the transient receptor potential vanilloid subtype 4 channel causes endothelial failure and circulatory collapse: Part 2. J Pharmacol Exp Ther 326, 443–452, https://doi.org/10.1124/jpet.107.134551 (2008).
Campbell, M. & Humphries, P. The blood-retina barrier: tight junctions and barrier modulation. Adv Exp Med Biol 763, 70–84 (2012).
Fiorio Pla, A. et al. TRPV4 mediates tumor-derived endothelial cell migration via arachidonic acid-activated actin remodeling. Oncogene 31, 200–212, https://doi.org/10.1038/onc.2011.231 (2012).
Vriens, J., Owsianik, G., Janssens, A., Voets, T. & Nilius, B. Determinants of 4 alpha-phorbol sensitivity in transmembrane domains 3 and 4 of the cation channel TRPV4. J Biol Chem 282, 12796–12803, https://doi.org/10.1074/jbc.M610485200 (2007).
Clark, A. L., Votta, B. J., Kumar, S., Liedtke, W. & Guilak, F. Chondroprotective role of the osmotically sensitive ion channel transient receptor potential vanilloid 4: age- and sex-dependent progression of osteoarthritis in Trpv4-deficient mice. Arthritis Rheum 62, 2973–2983, https://doi.org/10.1002/art.27624 (2010).
Gevaert, T. et al. Deletion of the transient receptor potential cation channel TRPV4 impairs murine bladder voiding. J Clin Invest 117, 3453–3462, https://doi.org/10.1172/JCI31766 (2007).
Hamanaka, K. et al. TRPV4 channels augment macrophage activation and ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol 299, L353–362, https://doi.org/10.1152/ajplung.00315.2009 (2010).
Mihara, H., Boudaka, A., Sugiyama, T., Moriyama, Y. & Tominaga, M. Transient receptor potential vanilloid 4 (TRPV4)-dependent calcium influx and ATP release in mouse oesophageal keratinocytes. J Physiol 589, 3471–3482, https://doi.org/10.1113/jphysiol.2011.207829 (2011).
Shen, J. et al. Functional expression of transient receptor potential vanilloid 4 in the mouse cochlea. Neuroreport 17, 135–139 (2006).
Dudek, S. M. & Garcia, J. G. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol (1985) 91, 1487–1500 (2001).
Suzuki, M., Hirao, A. & Mizuno, A. Microtubule-associated [corrected] protein 7 increases the membrane expression of transient receptor potential vanilloid 4 (TRPV4). J Biol Chem 278, 51448–51453, https://doi.org/10.1074/jbc.M308212200 (2003).
Park, J. H. et al. Far-infrared radiation acutely increases nitric oxide production by increasing Ca(2+) mobilization and Ca(2+)/calmodulin-dependent protein kinase II-mediated phosphorylation of endothelial nitric oxide synthase at serine 1179. Biochem Biophys Res Commun 436, 601–606, https://doi.org/10.1016/j.bbrc.2013.06.003 (2013).
Li, L. et al. Activation of Transient Receptor Potential Vanilloid 4 Increases NMDA-Activated Current in Hippocampal Pyramidal Neurons. Front Cell Neurosci 7, 17, https://doi.org/10.3389/fncel.2013.00017 (2013).
Rees, D. D., Palmer, R. M., Hodson, H. F. & Moncada, S. A specific inhibitor of nitric oxide formation from L-arginine attenuates endothelium-dependent relaxation. Br J Pharmacol 96, 418–424 (1989).
Cordeiro, S., Seyler, S., Stindl, J., Milenkovic, V. M. & Strauss, O. Heat-sensitive TRPV channels in retinal pigment epithelial cells: regulation of VEGF-A secretion. Invest Ophthalmol Vis Sci 51, 6001–6008, https://doi.org/10.1167/iovs.09-4720 (2010).
Ma, X. et al. Heteromeric TRPV4-C1 channels contribute to store-operated Ca(2+) entry in vascular endothelial cells. Cell Calcium 50, 502–509, https://doi.org/10.1016/j.ceca.2011.08.006 (2011).
Yao, X., Kwan, H. Y., Chan, F. L., Chan, N. W. & Huang, Y. A protein kinase G-sensitive channel mediates flow-induced Ca(2+) entry into vascular endothelial cells. FASEB J 14, 932–938 (2000).
Du, J. et al. TRPV4, TRPC1, and TRPP2 assemble to form a flow-sensitive heteromeric channel. FASEB J 28, 4677–4685, https://doi.org/10.1096/fj.14-251652 (2014).
Ma, X. et al. Functional role of vanilloid transient receptor potential 4-canonical transient receptor potential 1 complex in flow-induced Ca2+ influx. Arterioscler Thromb Vasc Biol 30, 851–858, https://doi.org/10.1161/ATVBAHA.109.196584 (2010).
Wu, S. et al. Ca2+ entry via alpha1G and TRPV4 channels differentially regulates surface expression of P-selectin and barrier integrity in pulmonary capillary endothelium. Am J Physiol Lung Cell Mol Physiol 297, L650–657, https://doi.org/10.1152/ajplung.00015.2009 (2009).
Gao, F. & Wang, D. H. Hypotension induced by activation of the transient receptor potential vanilloid 4 channels: role of Ca2+-activated K+ channels and sensory nerves. J Hypertens 28, 102–110, https://doi.org/10.1097/HJH.0b013e328332b865 (2010).
Suresh, K. et al. Hydrogen peroxide-induced calcium influx in lung microvascular endothelial cells involves TRPV4. Am J Physiol Lung Cell Mol Physiol 309, L1467–1477, https://doi.org/10.1152/ajplung.00275.2015 (2015).
Jie, P. et al. Blockage of transient receptor potential vanilloid 4 inhibits brain edema in middle cerebral artery occlusion mice. Front Cell Neurosci 9, 141, https://doi.org/10.3389/fncel.2015.00141 (2015).
Ye, L. et al. TRPV4 is a regulator of adipose oxidative metabolism, inflammation, and energy homeostasis. Cell 151, 96–110, https://doi.org/10.1016/j.cell.2012.08.034 (2012).
Chang, R. C. et al. High-Fat Diet-Induced Retinal Dysfunction. Invest Ophthalmol Vis Sci 56, 2367–2380, https://doi.org/10.1167/iovs.14-16143 (2015).
Cohen, G. et al. Signaling properties of 4-hydroxyalkenals formed by lipid peroxidation in diabetes. Free Radic Biol Med 65, 978–987, https://doi.org/10.1016/j.freeradbiomed.2013.08.163 (2013).
Xu, H., Fu, Y., Tian, W. & Cohen, D. M. Glycosylation of the osmoresponsive transient receptor potential channel TRPV4 on Asn-651 influences membrane trafficking. Am J Physiol Renal Physiol 290, F1103–1109, https://doi.org/10.1152/ajprenal.00245.2005 (2006).
Arruda, A. P. & Hotamisligil, G. S. Calcium Homeostasis and Organelle Function in the Pathogenesis of Obesity and Diabetes. Cell Metab 22, 381–397, https://doi.org/10.1016/j.cmet.2015.06.010 (2015).
Butenko, O. et al. The increased activity of TRPV4 channel in the astrocytes of the adult rat hippocampus after cerebral hypoxia/ischemia. PLoS One 7, e39959, https://doi.org/10.1371/journal.pone.0039959 (2012).
Fernandes, J. et al. IP3 sensitizes TRPV4 channel to the mechano- and osmotransducing messenger 5′-6′-epoxyeicosatrienoic acid. J Gen Physiol 131, i2, https://doi.org/10.1085/JGP1315OIA2 (2008).
Tiruppathi, C., Minshall, R. D., Paria, B. C., Vogel, S. M. & Malik, A. B. Role of Ca2+ signaling in the regulation of endothelial permeability. Vascul Pharmacol 39, 173–185 (2002).
Ryskamp, D. A. et al. TRPV4 regulates calcium homeostasis, cytoskeletal remodeling, conventional outflow and intraocular pressure in the mammalian eye. Sci Rep 6, 30583, https://doi.org/10.1038/srep30583 (2016).
Cabral, P. D. & Garvin, J. L. TRPV4 activation mediates flow-induced nitric oxide production in the rat thick ascending limb. Am J Physiol Renal Physiol 307, F666–672, https://doi.org/10.1152/ajprenal.00619.2013 (2014).
Ding, X. L. et al. Involvement of TRPV4-NO-cGMP-PKG pathways in the development of thermal hyperalgesia following chronic compression of the dorsal root ganglion in rats. Behav Brain Res 208, 194–201, https://doi.org/10.1016/j.bbr.2009.11.034 (2010).
Takeda-Nakazawa, H. et al. Hyposmotic stimulation-induced nitric oxide production in outer hair cells of the guinea pig cochlea. Hear Res 230, 93–104 (2007).
Pankey, E. A., Zsombok, A., Lasker, G. F. & Kadowitz, P. J. Analysis of responses to the TRPV4 agonist GSK1016790A in the pulmonary vascular bed of the intact-chest rat. Am J Physiol Heart Circ Physiol 306, H33–40, https://doi.org/10.1152/ajpheart.00303.2013 (2014).
Sukumaran, S. V. et al. TRPV4 channel activation leads to endothelium-dependent relaxation mediated by nitric oxide and endothelium-derived hyperpolarizing factor in rat pulmonary artery. Pharmacol Res 78, 18–27, https://doi.org/10.1016/j.phrs.2013.09.005 (2013).
Fichna, J. et al. Transient receptor potential vanilloid 4 inhibits mouse colonic motility by activating NO-dependent enteric neurotransmission. J Mol Med (Berl) 93, 1297–1309, https://doi.org/10.1007/s00109-015-1336-5 (2015).
Pu, J. et al. [Role of TRPV4 channels in regulation of eNOS expression in brain microvascular endothelial cells under the condition of mechanical stretch]. Zhong Nan Da Xue Xue Bao Yi Xue Ban 40, 960–966, https://doi.org/10.11817/j.issn.1672-7347.2015.09.003 (2015).
Bubolz, A. H. et al. Activation of endothelial TRPV4 channels mediates flow-induced dilation in human coronary arterioles: role of Ca2+ entry and mitochondrial ROS signaling. Am J Physiol Heart Circ Physiol 302, H634–642, https://doi.org/10.1152/ajpheart.00717.2011 (2012).
Kwan, H. Y., Leung, P. C., Huang, Y. & Yao, X. Depletion of intracellular Ca2 + stores sensitizes the flow-induced Ca2+ influx in rat endothelial cells. Circ Res 92, 286–292 (2003).
Yao, X. & Huang, Y. From nitric oxide to endothelial cytosolic Ca2+: a negative feedback control. Trends Pharmacol Sci 24, 263–266, https://doi.org/10.1016/S0165-6147(03)00122-6 (2003).
Kottgen, M. et al. Trafficking of TRPP2 by PACS proteins represents a novel mechanism of ion channel regulation. EMBO J 24, 705–716, https://doi.org/10.1038/sj.emboj.7600566 (2005).
Bhat, M., Pouliot, M., Couture, R. & Vaucher, E. The kallikrein-kinin system in diabetic retinopathy. Prog Drug Res 69, 111–143 (2014).
Gao, B. B. et al. Extracellular carbonic anhydrase mediates hemorrhagic retinal and cerebral vascular permeability through prekallikrein activation. Nat Med 13, 181–188, https://doi.org/10.1038/nm1534 (2007).
Guilak, F., Leddy, H. A. & Liedtke, W. Transient receptor potential vanilloid 4: The sixth sense of the musculoskeletal system? Ann N Y Acad Sci 1192, 404–409, https://doi.org/10.1111/j.1749-6632.2010.05389.x (2010).
Liedtke, W. & Kim, C. Functionality of the TRPV subfamily of TRP ion channels: add mechano-TRP and osmo-TRP to the lexicon! Cell Mol Life Sci 62, 2985–3001, https://doi.org/10.1007/s00018-005-5181-5 (2005).
Liedtke, W. Molecular mechanisms of TRPV4-mediated neural signaling. Ann N Y Acad Sci 1144, 42–52, https://doi.org/10.1196/annals.1418.012 (2008).
McNulty, A. L., Leddy, H. A., Liedtke, W. & Guilak, F. TRPV4 as a therapeutic target for joint diseases. Naunyn Schmiedebergs Arch Pharmacol 388, 437–450, https://doi.org/10.1007/s00210-014-1078-x (2015).
White, J. P. et al. TRPV4: Molecular Conductor of a Diverse Orchestra. Physiol Rev 96, 911–973, https://doi.org/10.1152/physrev.00016.2015 (2016).
Clapp, C. & Weiner, R. I. A specific, high affinity, saturable binding site for the 16-kilodalton fragment of prolactin on capillary endothelial cells. Endocrinology 130, 1380–1386, https://doi.org/10.1210/endo.130.3.1311239 (1992).
Bajou, K. et al. PAI-1 mediates the antiangiogenic and profibrinolytic effects of 16K prolactin. Nat Med 20, 741–747, https://doi.org/10.1038/nm.3552 (2014).
Garcia, C. et al. Requirement of phosphorylatable endothelial nitric oxide synthase at Ser-1177 for vasoinhibin-mediated inhibition of endothelial cell migration and proliferation in vitro. Endocrine 45, 263–270, https://doi.org/10.1007/s12020-013-9964-4 (2014).
Lee, E. J., Shin, S. H., Hyun, S., Chun, J. & Kang, S. S. Mutation of a putative S-nitrosylation site of TRPV4 protein facilitates the channel activates. Animal Cells Syst (Seoul) 15, 95–106, https://doi.org/10.1080/19768354.2011.555183 (2011).
Du, J., Wong, W. Y., Sun, L., Huang, Y. & Yao, X. Protein kinase G inhibits flow-induced Ca2+ entry into collecting duct cells. J Am Soc Nephrol 23, 1172–1180, https://doi.org/10.1681/ASN.2011100972 (2012).
Galfione, M. et al. Expression and purification of the angiogenesis inhibitor 16-kDa prolactin fragment from insect cells. Protein Expr Purif 28, 252–258 (2003).
Navaratna, D., McGuire, P. G., Menicucci, G. & Das, A. Proteolytic degradation of VE-cadherin alters the blood-retinal barrier in diabetes. Diabetes 56, 2380–2387, https://doi.org/10.2337/db06-1694 (2007).
Guo, L. et al. Targeting amyloid-beta in glaucoma treatment. Proc Natl Acad Sci USA 104, 13444–13449, https://doi.org/10.1073/pnas.0703707104 (2007).
Claybon, A. & Bishop, A. J. Dissection of a mouse eye for a whole mount of the retinal pigment epithelium. J Vis Exp. https://doi.org/10.3791/2563 (2011).
Smith, A. J. et al. Isolation and characterization of resident endogenous c-Kit+ cardiac stem cells from the adult mouse and rat heart. Nat Protoc 9, 1662–1681, https://doi.org/10.1038/nprot.2014.113 (2014).
Grange, C. et al. Isolation and characterization of human breast tumor-derived endothelial cells. Oncol Rep 15, 381–386 (2006).
Bussolati, B., Deambrosis, I., Russo, S., Deregibus, M. C. & Camussi, G. Altered angiogenesis and survival in human tumor-derived endothelial cells. FASEB J 17, 1159–1161, https://doi.org/10.1096/fj.02-0557fje (2003).
Cassoni, P. et al. Oxytocin induces proliferation and migration in immortalized human dermal microvascular endothelial cells and human breast tumor-derived endothelial cells. Mol Cancer Res 4, 351–359, https://doi.org/10.1158/1541-7786.MCR-06-0024 (2006).
Fonsato, V. et al. PAX2 expression by HHV-8-infected endothelial cells induced a proangiogenic and proinvasive phenotype. Blood 111, 2806–2815, https://doi.org/10.1182/blood-2007-04-085555 (2008).
Abbott, N. J. Permeability and transport of glial blood-brain barriers. Ann N Y Acad Sci 633, 378–394 (1991).
Acknowledgements
We thank Ataúlfo Martínez Torres, Xarubet Ruiz Cabrera and Yazmín Macotela Guzmán for technical advice for MACS, Dulce María Soría Lara for TEER measurements, Nicole Marilu Hernández-Godínez and Ana Patricia Juárez Mercado for help with Trpv4 −/− mouse genotyping, and Liora Shoshani for providing us ARPE-19 cells. D. Arredondo Zamarripa is a Doctoral student from the Programa de Posgrado en Ciencias, Universidad Nacional Autónoma de México (UNAM) and received fellowships from National Council of Science and Technology of Mexico (CONACYT). We thank E. Espino, M. Ramírez Romero, M. García, A. Castilla, and E. N. Hernández Ríos for their technical assistance, and D. D. Pless and Jessica Norris for critically editing the manuscript. This study was supported by the UNAM grant IN201814 (ST), the CONACYT grant 247246 (ST), the Ministère de l'Education Nationale, the Institut National de la Santé et de la Recherche Medicale (INSERM), and by the Lille I University.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: S.T., D.G., A.F.P., N.P., and C.C. Performed the experiments: D.A.Z., R.N.I., A.F.P., A.M.B.C., M.B., and S.T. Analyzed the data: D.A.Z., R.N.I., D.G., A.M.B.C., and S.T. Interpreted the data: W.L., C.C., and S.T. Contributed reagents/materials/analysis tools: A.F.P., D.G., N.P., F.L.C., W.L., C.C., and S.T. Wrote the paper: S.T. Critical revision for intellectual content: D.G., A.F.P., N.P., W.L., and C.C. All authors finally approved the submitted version.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Arredondo Zamarripa, D., Noguez Imm, R., Bautista Cortés, A.M. et al. Dual contribution of TRPV4 antagonism in the regulatory effect of vasoinhibins on blood-retinal barrier permeability: diabetic milieu makes a difference. Sci Rep 7, 13094 (2017). https://doi.org/10.1038/s41598-017-13621-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-13621-8
This article is cited by
-
TRPV4-Rho GTPase complex structures reveal mechanisms of gating and disease
Nature Communications (2023)
-
Channels that Cooperate with TRPV4 in the Brain
Journal of Molecular Neuroscience (2020)
-
The effect of spleen tyrosine kinase inhibitor R406 on diabetic retinopathy in experimental diabetic rats
International Ophthalmology (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
