
Abstract
The interplay between inflammation and lymphangiogenesis is mediated by various cytokines. However, most of these molecules and their associated mechanism are yet to be defined. Here, we explored the role of IL-33 in modulating inflammation-induced lymphangiogenesis (ILA) and its underlying mechanisms using an ILA mouse model and a lymphatic endothelial cell (LEC) line. Our results show that IL-33 promoted the proliferation, migration and tube formation of LECs and ILA in vivo. The pro-lymphangiogenic activity of IL-33 was abolished by ST2 blockage. In mechanisms, IL-33 induced the phosphorylation of Akt/eNOS to produce NO in LECs. The IL-33-induced Akt/eNOS activation was suppressed by the PI3K-specific-inhibitor wortmannin, and NO-production was inhibited by both wortmannin and the NO synthase-inhibitor NMA. Knock-down of ST2 or TRAF6 suppressed Akt/eNOS phosphorylation and NO production. The reduction of NO treated with wortmannin or NMA abolished the promoting effects of IL-33 on the chemotactic motility and tube formation of HDLECs. In vivo, IL-33-induced ILA was also impaired in eNOS−/− mice. In conclusion, our study is the first to show that IL-33 promotes inflammation-induced lymphangiogenesis via a ST2/TRAF6-mediated Akt/eNOS/NO signalling pathway. This findings may provide us more opportunities to treat inflammation and lymphangiogenesis associated diseases.
Similar content being viewed by others


Introduction
Lymphangiogenesis has been implicated in many inflammatory processes1. Modulating lymphangiogenesis may provide more possibilities to treat inflammation-associated diseases2. Interplay between inflammation and lymphangiogenesis is mediated by various cytokines. However, most of these molecules and their associated mechanism are yet to be defined3.
IL-33 is a multifunctional cytokine and is the 11th identified IL-1 family member4. IL-33 is localized in cell nucleus but also functions as a cell-free cytokine, and in this way is similar to HMGB1 and IL-1α5,6,7,8. IL-33 signals via the ST2 receptor, an orphan receptor that does not bind to IL-1α, IL-1β or other IL-1 family members9. IL-33 is mainly involved in type-2 immunity and inflammation and plays a key role in some pathological processes7. Although IL-33 was historically isolated from endothelial cells, epithelial cells, and keratinocytes, it is now recognized to be released from various tissues as a pro-inflammatory cytokine4, 8, 10.
Nitric oxide (NO) plays a crucial role in regulating the growth and function of lymphatic vessels11, 12. NO production in lymphatic endothelial cells (LECs) relies on the endothelial NO synthase (eNOS) activity, which is regulated by multiple inflammatory factors11,12,13,14,15. According to a recent study, eNOS inhibition blocks NO production and lymphangiogenesis11. Although the mechanisms by which NO regulates lymphangiogenesis are not completely understood, NO has emerged as an important mediator of physiological and pathological lymphangiogenesis and inflammation.
Since the identification of IL-33 as a functional ligand of ST2, the immunomodulatory and inflammatory roles of IL-33 have been well studied. However, the role of IL-33 in inflammation-associated lymphangiogenesis has not been determined. IL-33 is a pro-inflammatory cytokine and also induces eNOS/NO activation. Inflammation and eNOS/NO activation are closely related to lymphangiogenesis. Thus, we hypothesized that IL-33 has a regulatory function in inflammation-associated lymphangiogenesis via the eNOS/NO signalling pathway. To test this hypothesis, we used a mouse model of ILA and a LEC cell line. The results showed that IL-33 promoted the proliferation, migration and tube formation of LECs in vitro and inflammation-induced lymphangiogenesis (ILA) in vivo, which were regulated by PI3K/Akt/eNOS-mediated NO production. ST2/TRAF6 was required for IL-33-induced Akt/eNOS activation and NO production.
Our results indicate for the first time that IL-33 promotes inflammation-induced lymphangiogenesis via the ST2/TRAF6-mediated Akt/eNOS/NO signalling pathway. This findings may provide us more opportunities to treat inflammation and lymphangiogenesis associated diseases.
Results
IL-33 promotes the proliferation, migration and tube formation of LECs via the ST2 receptor
To explore the lymphangiogenic activity of IL-33, we first determined whether IL-33 promoted LECs proliferation in vitro. Our results showed that IL-33 promoted VEGF-C–induced HDLECs proliferation in a dose-dependent manner, with a maximal effect at 20 ng/mL (Fig. 1A). Knock-down of ST2 with an ST2-specific siRNA significantly abolished the promoting effect of IL-33 (Fig. 1B), indicating that ST2 mediates IL-33-induced LECs proliferation.

IL-33 promotes LECs proliferation, migration and tube formation via the ST2 receptor. (A) IL-33 promotes IL-33-induced HDLECs proliferation in a dose-dependent manner. (B) The ST2 receptor mediates IL-33-induced HDLECs proliferation. (C) The ST2 receptor mediates IL-33-induced HDLECs chemotactic mobility. (D) The ST2 receptor mediates IL-33-induced HDLECs tube formation. Three independent experiments were performed in duplicate. *p < 0.05, **p < 0.01, ***p < 0.001.
Then, we tested the roles of IL-33 and ST2 in LECs chemotactic mobility and tube formation. The results showed that IL-33 promoted chemotactic mobility and increased the tube length, and these effects were inhibited by ST2 knock-down (Fig. 1C and D).
IL-33 itself (without additional VEGF-C stimulation) also induced LECs proliferation and tube formation (Figure S2). In addition, IL-33 didn’t up-regulate the expression of pro-lymphangiogenic factors (VEGF-C/D) by LECs (Figure S2).
So, we can confirm that it is IL-33 itself, not indirect VEGF-C/D, that takes the main effects on LECs in the test.
IL-33 promotes ILA in the mouse cornea via the ST2 receptor
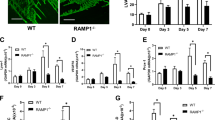
In vivo, a lower concentration (10 μg/mL) of IL-33 treatment promoted the ILA in mouse corneas (Fig. 2A and B). A higher concentration (40 μg/mL) of IL-33 induced more ILA. ST2 knock-out decreased ILA in both the presence (low or high concentrations) and absence of IL-33. These results indicate that IL-33/ST2 signalling does take effects in promoting ILA.

IL-33 promotes ILA in the mouse cornea via the ST2 receptor. (A,B) Representative images and quantification of LYVE-1-labelled corneal lymphangiogenesis in different groups showing that the ST2 receptor mediates IL-33-associated ILA. Three independent experiments were performed in duplicate. *p < 0.05, **p < 0.01. The scale bars represent 300 μm.
IL-33 stimulates NO production in LECs via the PI3K/Akt/eNOS signalling pathway
NO plays a crucial role in regulating the growth and function of lymphatic vessels11, 12. NO production in the lymphatic system is usually regulated by PI3K/Akt/eNOS signaling16,17,18,19. Therefore, we tested whether IL-33 induces PI3K/Akt/eNOS activation to produce NO in LECs. Western blotting showed that IL-33 promoted the phosphorylation of Akt and eNOS, with a maximal effect at 20 ng/mL (Fig. 3A). IL-33-induced Akt and eNOS phosphorylation began to increase significantly at 10 min after treatment and was sustained for at least 50 min (Fig. 3B).

IL-33 stimulates NO production in LECs via the PI3K/Akt/eNOS signalling pathway. (A–C) IL-33-induced (20 ng/mL) Akt and eNOS phosphorylation was determined by Western blotting. (A) HDLECs were stimulated with various concentrations of IL-33 for 30 min. (B) HDLECs were stimulated with IL-33 (20 ng/mL) for the indicated times. (C) HDLECs were pre-treated with wortmannin (100 nmol/L, 30 min) and then stimulated with IL-33 (20 ng/mL, 30 min). (D) HDLECs were pre-treated with wortmannin (100 nmol/L, 30 min) or NMA (1 mmol/L) and then treated with IL-33 (20 ng/mL) for 4 hours. LECs were incubated with DAF-FM DA for 1 hour at 37 °C. The relative intracellular NO levels were determined from the fluorescence intensity of DAF-FM. Three independent experiments were performed in duplicate. *p < 0.05, **p < 0.01, ***p < 0.001. Full-length blots are presented in Supplementary Figure S3.
Further, we investigated whether PI3K was required for the activation of Akt/eNOS using wortmannin (a PI3K-specific inhibitor). As a result, the wortmannin treatment (100 nmol/L, 30 min) limited IL-33-induced Akt and eNOS phosphorylation to a very low levels, indicating that PI3K is required for IL-33-induced Akt/eNOS activation (Fig. 3C).
IL-33-induced NO production was also suppressed by the wortmannin or NMA (a NO synthase inhibitor) treatment (Fig. 3D).
ST2/TRAF6 is required for IL-33-induced Akt/eNOS activation and NO production
TRAF6 has been reported to mediate Akt/eNOS activation and is modulated by ST220,21,22. Our results showed that the elevated ST2 or TRAF6 expression induced by IL-33 increased Akt/eNOS phosphorylation (Fig. 4A and B). On the other hand, the knock-down of ST2 or TRAF6 by an ST2- or TRAF6-specific siRNA suppressed Akt/eNOS phosphorylation and NO production (Fig. 4A–D). Thus, the results suggest that ST2 and TRAF6 are upstream regulators of IL-33-induced Akt/eNOS activation.

IL-33 induces Akt/eNOS activation and NO production via ST2/TRAF6. (A,B) After transfection with an ST2- or TRAF6-specific siRNA, HDLECs were treated with IL-33 (20 ng/mL, 30 minutes). Akt and eNOS phosphorylation were detected by Western blotting. (C,D) After transfection with the ST2- or TRAF6-specific siRNA, HDLECs were treated with IL-33 (20 ng/mL, 4 hours) and incubated with DAF-FM DA for 1 hour. Relative intracellular NO levels were determined from the fluorescence intensity of DAF-FM. Three independent experiments were performed in duplicate. **p < 0.01, ***p < 0.001. Full-length blots are presented in Supplementary Figure S4.
Taken together, the above results demonstrate that IL-33 promotes the NO production in LECs via a ST2/TRAF6-PI3K/Akt/eNOS signalling pathway.
PI3K/Akt/eNOS-mediated NO production is required for IL-33-induced ILA
To evaluate the role of PI3K/Akt/eNOS-mediated NO production in IL-33-induced ILA, HDLECs were treated with wortmannin or NMA before IL-33 stimulation and then the chemotactic motility and tube formation of HDLECs were assessed. The reduction of NO production following treatment with wortmannin or NMA abolished the promoting effects of IL-33 on HDLECs chemotactic motility and tube formation (Fig. 5A and B). In vivo, IL-33-induced ILA was also impaired in eNOS−/− mice compared with WT mice (Fig. 5C). These results show that PI3K/Akt/eNOS-mediated NO production is required for IL-33-induced ILA.

PI3K/Akt/eNOS-mediated NO production is required for IL-33-induced ILA. (A,B) HDLECs were pre-treated with or without wortmannin (100 nmol/L) or NMA (1 mmol/L) for 30 minutes before stimulation with IL-33 (20 ng/mL). (A) Chemotaxis was quantified after a 4 hour incubation. (B) HDLECs tube formation was quantified after a 16 hour incubation. (C) After IL-33 (10 μg/mL) treatment, ILA was quantified in WT and eNOS KO mice in vivo. Three independent experiments were performed in duplicate. *p < 0.05.
Discussion
In the present study, we explored the role of IL-33 in inflammation-induced lymphangiogenesis and its associated mechanisms. For the first time, we show that IL-33 directly activates LECs, resulting in promoting inflammation-induced lymphangiogenesis. Inflammation and lymphangiogenesis are associated with multiple diseases; therefore, our findings may provide us more opportunities to treat inflammation and lymphangiogenesis associated diseases.
Firstly, we find that IL-33 is involved in ILA (Figure S1). Both mRNA and protein of IL-33 are significantly increased in the inflamed corneas after the ILA surgery. This finding is consistent with the results reported by Hazlett LD, who showed IL-33 mRNA levels were significantly up-regulated in both BALB/c and B6 mouse corneas after infection, and immuno-staining used to localize IL-33 in the cornea showed qualitatively intense IL-33-positive staining23. Therefore, a topical blockade of IL-33 would be a possible treatment for corneal lymphangiogenesis-associated diseases.
To determine the lymphangiogenic activity of IL-33, we tested whether IL-33 directly activated LECs. Our in vitro tests verified that IL-33 does not up-regulate the expression of pro-lymphangiogenic factors (VEGF-C/D) in LECs, and IL-33 itself promotes the proliferation, migration and tube formation of LECs through a VEGF-C/D-independent mechanism (Figs 1 and S2). Most importantly, our further in vivo tests ascertain that IL-33 promotes inflammation-induced lymphangiogenesis in the mouse cornea. IL-33 takes effect mainly via the ST2 receptors5, 24, 25. When we blocked ST2, most of the pro-lymphangiogenic activity of IL-33 was abolished both in vitro and in vivo, which confirms the pro-lymphangiogenic role of IL-33.
Our repeated tests show that the effects of IL-33 on LECs were reduced when cells were treated with concentrations greater than 20 ng/mL. A similar phenomenon was also observed in some other studies21, 26. According to Choi YS, et al., IL-33 stimulated the proliferation, chemotactic motility and tube formation of HUVECs, with a maximal effect at 20 ng/mL21, and these effects decreased at 50 ng/mL. As shown in the study by Hayashi H, et al., IL-33 enhanced fibrocyte proliferation, with a maximal effect at 10 ng/mL, and the number of viable fibrocytes decreased at 100 ng/mL26. Higher concentrations of IL-33 than the concentrations that exert the maximal effects on LECs may induce inhibitory mechanisms. For example, Hazlett LD, et al. showed that overexpression of IL-33 markedly decreased the expression of pro-inflammatory cytokines (IL-1, MIP-2, IL-6, and TNF-alpha)23. Other as yet unknown mechanisms may also contribute. We may explore these mechanisms in the future.
We did not notice a substantial difference in ILA between untreated WT group and IL-33 treated ST2−/− groups. This result suggests that ST2 knock-out only blocks partial, but not all, effects of IL-33 on ILA. The study by Luzina IG reported a similar phenomenon and their results showed that ST2 knock-out attenuated only the size of, but did not completely abrogate, mature-mouse-IL-33-induced inflammatory infiltrates25. Their further tests certificates that some of the effects of IL-33 on the lung were mediated by ST2-independent mechanisms, such as up-regulating the expression of CCL2 (MCP-1), IL-6, and matrix metalloproteinases (MMPs) 3, 10, and 13 genes. These ST2-independent mechanisms may have been responsible for the lack of a difference in ILA between untreated WT group and IL-33 treated ST2−/− groups. We may verify this hypothesis in the future.
During these experiments, we observed a interesting phenomenon that exogenous IL-33 up-regulated ST2 expression in LECs. Furthermore, some other researches showed the similar phenomenon. As shown in the study by Hazlett LD, increased ST2 staining is associated with microphages in IL-33–treated mice, and ST2 mRNA is significantly up-regulated in microphages in vitro after LPS stimulation in the presence of exogenous IL-3323. According to Choi YS, exogenous IL-33 up-regulates ST2 expression in HUVECs21. We still do not know the exact mechanisms underlying this phenomenon. However, we have proposed some possible hypotheses. For example, some positive feedback mechanisms that promote inflammation will occur in the early-stage of inflammation. The gradually increased inflammation-associated factors stimulates LECs to increase ST2 expression. The phenomenon could be completely illustrated in the future.
Several previous studies have reported a role for IL-33 in mediating angiogenesis21, 27,28,29,30,31. Most of these studies support the hypothesis that IL-33 promotes angiogenesis directly through its pro-angiogenic effects21 or indirectly by up-regulating pro-angiogenic factors27,28,29,30, such as VEGF, angiogenin, von Willebrand factor (vWF), etc. Only one paper draws the opposite conclusion that IL-33 inhibits angiogenesis, but the investigators did not show the exact mechanisms. Although researchers hold different opinions, all agree that ST2 is dependent for IL-33-mediated angiogenesis. In our study, IL-33 promotes lymphangiogenesis in a VEGF-C/D-independent manner, consistent with the results reported by Choi YS21.
In addition, accumulating evidence indicates that IL-33/ST2 has emerged as an intercellular signalling pathway that participates in various processes, such as autoimmunity, antigen/allergen responses, organ fibrosis, cardiac injury and a variety of other inflammatory conditions5, 24. Efforts to clarify the role of IL-33/ST2 in the interaction between end-organ effector cells and their environment may result in the discovery of novel therapeutic targets for the modulation of the above pathological conditions.
NO plays crucial role not only in regulating blood vessel growth and function but also in regulating lymphangiogenesis11, 21. NO donors stimulate lymphatic endothelial cell proliferation, migration and tube formation11, 12, 14, 15. NOS inhibition blocks lymphangiogenesis in a dermal regeneration mouse-tail model11. NO production during lymphangiogenesis is usually regulated by PI3K/Akt/eNOS signaling16,17,18,19. Therefore, we performed additional experiments to explore the PI3K/Akt/eNOS signalling associated mechanisms in IL-33-induced lymphangiogenesis. Our results showed that IL-33 induced Akt/eNOS activation to produce NO in LECs. IL-33-induced Akt and eNOS phosphorylation was suppressed by the PI3K-specific inhibitor wortmannin. Furthermore, NO production was inhibited by both wortmannin and the NO synthase inhibitor NMA. These findings suggests that IL-33 stimulates NO production in LECs via the PI3K/Akt/eNOS signalling pathway.
TNF-receptor associated factor 6 (TRAF6) is a critical signal transducer in the IL-33 signalling pathway21, 22. TRAF6 inhibition suppressed Akt/eNOS activation and NO production in endothelial cells (ECs)20, 21. TRAF6 is also expressed in LECs32. Therefore, the possibility is raised that IL-33 may induce LECs activation via TRAF6-mediated eNOS/NO pathway. Our present study shows that the elevated TRAF6 expression induced by IL-33 increased the Akt/eNOS phosphorylation, and the knock-down of TRAF6 with a TRAF6-specific siRNA suppressed Akt/eNOS phosphorylation and NO production, which confirms that TRAF6 is a crucial bridge that mediates IL-33/ST2 and PI3K/Akt/eNOS/NO signalling in LECs.
Finally, we performed additional experiments to evaluate the role of PI3K/Akt/eNOS-mediated NO production in IL-33-induced ILA. Our in vitro tests showed that the reduction of NO by using PI3K-specific inhibitor wortmannin or NOS inhibitor NMA abolished the effect of IL-33 on HDLECs chemotactic motility and tube formation. Our in vivo tests showed that the knock-out of eNOS suppressed ILA induced by IL-33. These results confirms that PI3K/Akt/eNOS-mediated NO production is required for IL-33-induced ILA.
In conclusion, to our knowledge, the present study provides the first evidence that IL-33 promotes inflammation-induced lymphangiogenesis via ST2/TRAF6-mediated Akt/eNOS/NO signaling pathway. Consequently, the administration of modulating inflammation-induced lymphangiogenesis through IL-33 and its downstream signalling TRAF6-Akt/eNOS/NO may provide us more possibilities of treating inflammation and lymphangiogenesis associated diseases.
Materials and Methods
Ethics statement and animals
The study strictly adhered to the Association for Research in Vision and Ophthalmology’s Statement for the Use of Animals in Ophthalmic and Vision Research. All animal experiments were conducted with the approval and supervision of the Institutional Animal Care and Use Committee of Hebei Provincial Eye Hospital. Eight-week-old male C57BL/6 mice, ST2−/− mice on the C57BL/6 background and eNOS−/− mice on the C57BL/6 background were obtained from the Model Animal Research Center of Nanjing University. All surgeries were performed under chloral hydrate anaesthesia.
Antibodies and reagents
The antibodies, including polyclonal anti-lymphatic vessel endothelial hyaluronan receptor-1 (LYVE-1), monoclonal anti-Akt, monoclonal anti-phosphorylated Akt, monoclonal anti-eNOS, and monoclonal anti-phosphorylated eNOS antibodies, were obtained from Abcam (Hong Kong, China). Other antibodies, including the monoclonal anti-β-actin antibody, horseradish peroxidase (HRP)-conjugated anti-mouse secondary antibody, HRP-conjugated anti-rabbit secondary antibody, and Alexa Fluor 555-conjugtaed goat anti-rabbit secondary antibody, were purchased from Cell Signaling Technology Inc. (Danvers, MA). Recombinant mouse IL-33 was purchased from Alexis Biochemicals (San Diego, USA). VEGF-C was obtained from Sigma (St. Louis, MO, USA). Wortmannin and NG-Monomethyl-L-arginine (NMA) were obtained from Abcam (Hong Kong, China).
Cell culture
Human dermal lymphatic endothelial cells (HDLECs) were purchased from ScienCell (Carlsbad, CA, USA) and grown in endothelial cell basal medium-2 with growth supplements (EBM-2 MV) at 37 °C in a humidified 95%/5% (vol/vol) air/CO2 atmosphere.
LEC proliferation assay
For the proliferation assay, HDLECs were incubated with different doses of IL-33 (0, 10, 20, 50 or 100 ng/mL) with or without VEGF-C (10 ng/mL) for 24 hours. Then, 100 μL of cells from each well were transferred to a new 96-well plate and incubated with 10 μL of Cell Counting Kit-8 solution (Dojindo Laboratories, Kumamoto, Japan). The absorbance was measured at 450 nm using a microplate reader.
LEC migration assay
Transwell chambers (Corning Costar) were used to assay the chemotatic motility of HDLECs. The 6.5 mm diameter polycarbonate filters (8 μm pore size) were used. The lower surface of the filter was coated with 10 μg of gelatine. Fresh EBM-2 MV medium containing IL-33 (20 or 50 ng/mL) was placed in the lower wells. The cells were suspended at a final density of 106 cells/mL in EBM-2 MV medium. Cell suspensions (100 μL) were loaded into each of the upper wells and incubated at 37 °C for 4 hours. The cells were fixed and stained with haematoxylin and eosin. Cells on the upper surface of the filter were removed, and chemotaxis was quantified by counting the cells that had migrated to the lower side of the filter under an optical microscope.
Tube formation assay
Tube formation was assayed using a previously described method33, 34. Mixtures of 30 μL of Matrigel (BD Biosciences) and 20 μL of EBM-2 were polymerized in each well of a 96-well plate. Approximately 6 × 103 HDLECs in 100 μL of ECM were placed on the Matrigel layer in each well. After a 16-hour incubation at 37 °C, tube morphogenesis was assessed using phase contrast microscopy. The total tube length was quantified using ImageJ software (National Institutes of Health).
Knock-down of ST2 or TRAF6 by siRNA transfection
HDLECs cultured in a 6-well plate were transfected with 180 pmol of ST2- or TRAF6-specific siRNAs (Santa Cruz Biotechnology, Inc., Santa Cruz, USA). After 48 hours of transfection, cells were used for further experiments.
ILA model and treatment
An ILA model was established using previously described methods35,36,37. Briefly, a 2-mm corneal trephine was used to mark a circle on the centre of the cornea in the right eye. Three “8”-shaped intrastromal sutures were performed using 11-0 nylon sutures (Jiahe Inc., Taibei, Taiwan, China; NT0411), and each suture extended to approximately 120° of the corneal circumference. Then, the ILA model mice were randomly divided into each group (n = 5) and were administered a topical treatment of 5 μL IL-33 (10 or 40 μg/mL) or vehicle (sterile saline solution) in the right eye twice a day for 10 days. The in vivo experiments were independently repeated 3 times.
Immunostaining and quantification
Whole-mount corneal LYVE-1 immunostaining was performed as previously described35, 38. Briefly, the mice were sacrificed on the 10th day. The eyeballs were fixed with 4% (wt/vol) paraformaldehyde for 16 hours at 4 °C. The excised corneas were blocked with 3% bovine serum albumin (BSA) in PBST (0.3% (vol/vol) Triton X-100 in PBS) for 2 hours, followed by an overnight incubation at 4 °C with the anti-LYVE-1 antibody (1:200). On the second day, the tissue was incubated with an Alexa Fluor 555-conjugated goat anti-rabbit antibody (1:400) for 2 hours at room temperature. The cornea was flat-mounted on microscope slides with Vectashield mounting medium and then examined under a fluorescent microscope. Lymphatic vessels were quantified using Adobe Photoshop CC software, as previously described35.
Western blot analysis
HDLEC or cornea lysates (50 μg of total protein) were separated on a polyacrylamide-SDS gel and electro-blotted onto a nitrocellulose membrane (Bio-Rad, Hercules, CA, USA). After blocking with 5% non-fat milk, the membrane was incubated with antibodies against Akt, P-Akt, eNOS, P-eNOS, ST2, TRAF6 and β-actin (1:1000), followed by an incubation with an HRP-conjugated secondary antibody (1:3000). Signals were visualised using enhanced chemiluminescence detection (Pierce, Rockford, IL).
Intracellular NO assay
Intracellular NO levels were assayed using a previously described method21. Briefly, HDLECs were treated with 20 ng/mL IL-33 for 4 hours and then incubated with 5 μmol/L 3-amino,4-aminomethyl-2′,7′-difluorescein, diacetate (DAF-FM DA) (Beyotime Biotechnology, China) for 1 hour at 37 °C. After the excess probe was removed, cells were incubated for an additional 20 minutes. Fluorescence images were captured from at least 5 randomly selected fields per dish using a fluorescence microscope. The intracellular NO level was determined from the fluorescence of DAF-FM.
Statistical analysis
Data are presented as means ± SD. Statistical analyses were performed using SPSS 21.0 software (SPSS Inc., Chicago, IL). The normal distribution of the data was tested with Kolmogorov-Smirnov tests. Statistical comparisons between two groups were performed using Student’s t-tests. A p < 0.05 was considered statistically significant.
Data Availability
The datasets generated and/or analysed during the current study are available from the corresponding author upon reasonable request.
References
Liao, S. & der Weid, P. Y. V. Inflammation-induced lymphangiogenesis and lymphatic dysfunction. Angiogenesis. 17, 325–334 (2014).
Tammela, T. & Alitalo, K. Lymphangiogenesis: Molecular mechanisms and future promise. Cell. 140, 460–476 (2010).
Kataru, R. P. et al. Critical role of CD11b+ macrophages and VEGF in inflammatory lymphangiogenesis, antigen clearance, and inflammation resolution. Blood. 113, 5650–5659 (2009).
Villarreal, D. O. & Weiner, D. B. Interleukin 33: a switch-hitting cytokine. Curr Opin Immunol. 28, 102–106 (2014).
Molofsky, A. B., Savage, A. K. & Locksley, R. M. Interleukin-33 in Tissue Homeostasis, Injury, and Inflammation. Immunity. 42, 1005–1019 (2015).
Carriere, V. et al. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc Natl Acad Sci USA 104, 282–287 (2007).
Garlanda, C., Dinarello, C. A. & Mantovani, A. The interleukin-1 family: back to the future. Immunity. 39, 1003–1018 (2013).
Moussion, C., Ortega, N. & Girard, J. P. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin’. PLoS One. 3, e3331 (2008).
Schmitz, J. et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 23, 479–490 (2005).
Baekkevold, E. S. et al. Molecular characterization of NF-HEV, a nuclear factor preferentially expressed in human high endothelial venules. Am J Pathol. 163, 69–79 (2003).
Lahdenranta, J. et al. Endothelial nitric oxide synthase mediates lymphangiogenesis and lymphatic metastasis. Cancer Res. 69, 2801–2808 (2009).
Kajiya, K., Huggenberger, R., Drinnenberg, I., Ma, B. & Detmar, M. Nitric oxide mediates lymphatic vessel activation via soluble guanylate cyclase alpha1beta1-impact on inflammation. FASEB J. 22, 530–537 (2008).
Coso, S., Zeng, Y., Opeskin, K. & Williams, E. D. Vascular endothelial growth factor receptor-3 directly interacts with phosphatidylinositol 3-kinase to regulate lymphangiogenesis. PLoS One. 7, e39558 (2012).
Prangsaengtong, O., Koizumi, K., Senda, K., Sakurai, H. & Saiki, I. eNOS and Hsp90 interaction directly correlates with cord formation in human lymphatic endothelial cells. Lymphat Res Biol. 9, 53–59 (2011).
Hammer, T. et al. IL-20 activates human lymphatic endothelial cells causing cell signalling and tube formation. Microvasc Res. 78, 25–32 (2009).
Nojiri, H. & Ohhashi, T. Immunolocalization of nitric oxide synthase and VEGF receptors in cultured lymphatic endothelial cells. Microcirculation. 6, 75–78 (1999).
Marchetti, C. et al. Endothelin and nitric oxide synthase in lymphatic endothelial cells: immunolocalization in vivo and in vitro. Anat Rec. 248, 490–497 (1997).
Marchetti, C. et al. Immunolocalization of endothelin and nitric oxide-synthase in lymphatic vessels and cultured lymphatic endothelial cells. Microvasc Res. 53, 282–285 (1997).
Dimmeler, S. et al. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 399, 601–605 (1999).
Min, J. K. et al. Receptor activator of nuclear factor (NF)-kappaB ligand (RANKL) increases vascular permeability: impaired permeability and angiogenesis in eNOS-deficient mice. Blood. 109, 1495–1502 (2007).
Choi, Y. S. et al. Interleukin-33 induces angiogenesis and vascular permeability through ST2/TRAF6-mediated endothelial nitric oxide production. Blood. 114, 3117–3126 (2009).
Funakoshi-Tago, M. et al. TRAF6 is a critical signal transducer in IL-33 signaling pathway. Cell Signal. 20, 1679–1686 (2008).
Hazlett, L. D. et al. IL-33 shifts macrophage polarization, promoting resistance against Pseudomonas aeruginosa keratitis. Invest Ophthalmol Vis Sci. 51, 1524–1532 (2010).
Kakkar, R. & Lee, R. T. The IL-33/ST2 pathway: therapeutic target and novel biomarker. Nat Rev Drug Discov. 7, 827–840 (2008).
Luzina, I. G. et al. Full-length IL-33 promotes inflammation but not Th2 response in vivo in an ST2-independent fashion. J Immunol. 189, 403–410 (2012).
Hayashi, H. et al. IL-33 enhanced the proliferation and constitutive production of IL-13 and IL-5 by fibrocytes. Biomed Res Int. 2014, 738625 (2014).
Maywald, R. L. et al. IL-33 activates tumor stroma to promote intestinal polyposis. Proc Natl Acad Sci USA 112, E2487–2496 (2015).
Shan, S. et al. Nasal administration of interleukin-33 induces airways angiogenesis and expression of multiple angiogenic factors in a murine asthma surrogate. Immunology. 148, 83–91 (2016).
He, R. et al. IL-33 improves wound healing through enhanced M2 macrophage polarization in diabetic mice. Mol Immunol. 90, 42–49 (2017).
Zhang, Y. et al. IL-33 promotes growth and liver metastasis of colorectal cancer in mice by remodeling the tumor microenvironment and inducing angiogenesis. Mol Carcinog. 56, 272–287 (2017).
Theodoropoulou, S. et al. Interleukin-33 regulates tissue remodelling and inhibits angiogenesis in the eye. J Pathol. 241, 45–56 (2017).
Sawa, Y. et al. LPS-induced IL-6, IL-8, VCAM-1, and ICAM-1 expression in human lymphatic endothelium. J Histochem Cytochem. 56, 97–109 (2008).
Liu, D. et al. SIX1 promotes tumor lymphangiogenesis by coordinating TGFβ signals that increase expression of VEGF-C. Cancer Res. 74, 5597–5607 (2014).
Lin, C. C. et al. WISP-1 promotes VEGF-C-dependent lymphangiogenesis by inhibiting miR-300 in human oral squamous cell carcinoma cells. Oncotarget. (2016).
Han, L. et al. Doxycycline inhibits inflammation-induced lymphangiogenesis in mouse cornea by multiple mechanisms. PLoS One. 9, e108931 (2014).
Cursiefen, C. et al. Thrombospondin 1 inhibits inflammatory lymphangiogenesis by CD36 ligation on monocytes. J Exp Med. 208, 1083–1092 (2011).
Han, L. et al. High Mobility Group Box-1 Promotes Inflammation-Induced Lymphangiogenesis via Toll-Like Receptor 4-Dependent Signalling Pathway. PLoS One. 11, e0154187 (2016).
Cao, R. et al. Mouse corneal lymphangiogenesis model. Nat Protoc. 6, 817–826 (2011).
Author information
Authors and Affiliations
Contributions
Conception and design of this study: L.H. and Minglian Z. Collection, analysis and explanation of the experimental data: L.H., Minglian Z., X.L., X.J., J.J. and Y.F. Drafting and critical revision of this article: L.H., Minglian Z., X.L., X.J., J.J., and Miying Z. All authors approved the final submission.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Han, L., Zhang, M., Liang, X. et al. Interleukin-33 promotes inflammation-induced lymphangiogenesis via ST2/TRAF6-mediated Akt/eNOS/NO signalling pathway. Sci Rep 7, 10602 (2017). https://doi.org/10.1038/s41598-017-10894-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-10894-x
This article is cited by
-
Endothelial cell dynamics in sepsis-induced acute lung injury and acute respiratory distress syndrome: pathogenesis and therapeutic implications
Cell Communication and Signaling (2024)
-
Exogenous interleukin 33 enhances the brain’s lymphatic drainage and toxic protein clearance in acute traumatic brain injury mice
Acta Neuropathologica Communications (2023)
-
Role of IL-33-ST2 pathway in regulating inflammation: current evidence and future perspectives
Journal of Translational Medicine (2023)
-
IL-33 enhances Jagged1 mediated NOTCH1 intracellular domain (NICD) deubiquitination and pathological angiogenesis in proliferative retinopathy
Communications Biology (2022)
-
Sprouty2 limits intestinal tuft and goblet cell numbers through GSK3β-mediated restriction of epithelial IL-33
Nature Communications (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
