
Abstract
A recently presented taxonomical arrangement of the moss genus Orthotrichum Hedw. s.l. substantially changed the traditional view of the taxon that had been accepted throughout the twentieth century. This paper provides the results of mitogenomic studies that strongly support the new taxonomical concept. Comparative analyses presented in this study confirmed the stable structure of moss mitogenomes. Moreover, 17 complete mitogenome sequences were used to identify the major evolutionary groups, including 11 newly sequenced ones, for this study. The analysis of mitochondrial hotspots revealed intron 4 of the cox1 gene to be the most variable non-coding region. The most variable protein-coding genes in the tribe Orthotricheae were ccmFC and tatC. The intergenic and intronic hotspots of Orthotrichum s.l. identified in the present study do not correspond to those described in vascular plant mitogenomes.
Similar content being viewed by others

Introduction
The genera Nyholmiella Holmen & Warncke, Lewinskya F. Lara, Garilleti & Goffinet and Orthotrichum Hedw. represent a group of approximately 167 species widely distributed subcosmopolitan mosses of the family Orthotrichaceae1,2,3,4,5,6,7,8,9,10,11,12,13. Species of these genera are found in several biomes, with the exception of deserts and wet tropical forests. Orthotrichum species are predominantly epiphytes that grow on tree trunks and branches, and they are less often found in saxicolous habitats at high altitudes, up to 5,000 m a.s.l.14. The subdivision within this genus has been the subject of continuing debate since the late 19th century. Specific taxa have been repeatedly included in and removed from Orthotrichum s.l. in an attempt to identify infrageneric taxa, including subgenera and sections. The history of Orthotrichum s.l. taxonomic classification was described in detail by Lewinsky and Hedenäs14, 15, and several phylogenetic analyses revealed that the genus was polyphyletic16,17,18,19. Nuclear and chloroplastic molecular markers were used to identify four groups within Orthotrichum s.l. with variable reproductive systems and localization of stomata17, 18. Based on these molecular results, a new taxonomical arrangement was proposed for the group, in which Orthotrichum s.l. was divided into the genera Lewinskya, Nyholmiella, Orthotrichum and Pulvigera Plášek, Sawicki & Ochyra20, 21.
Moreover, previous research demonstrated that the Orthotrichum s.l. evolutionary and taxonomic debate would not be resolved without the inclusion of species from the genus Ulota Mohr., which is closely related to the genus Lewinskya 16, 17, 19. Ulota contains approximately 60 species22, more than half of which are found in the southern hemisphere. The genus is characterized by superficial stomata, highly differentiated basal leaf cells, crisped leaves, and a lack of brood bodies on the leaf lamina. Based on results of molecular studies17, 19, the dioecious U. phyllantha Brid. was transferred to the separate monospecific genus Plenogemma Plášek, Sawicki & Ochyra20. The genera mentioned above, together with Sehnemobryum Lewinsky-Haapasaari & Hedenäs and Stoneobryum D.H. Norris & H. Rob., are classified in the tribe Orthotricheae17.
The genera of the tribe Orthotricheae comprise monoecious (Lewinskya, Orthotrichum s.s., Sehnemobryum, Ulota) and dioecious (Nyholmiella, Plenogemma, Pulvigera and Stoneobryum) taxa, which raises questions about the ancestry and monophyly of taxa representing the different sexual systems14. The tribe Zygodonteae, sister to Orthotricheae, is characterized by dioecy, which suggests dioecy as the ancestral state of Orthotricheae. The sexual system in Orthotricheae is usually correlated with the position of stomata; dioecious taxa are characterized by phaneroporous stomata (Nyholmiella, Plenogemma, and Pulvigera), except Stoneobryum, which is the only known dioecious genus with cryptoporous stomata. Remaining cryptoporous species are monoecious and are classified into the monophyletic genus Orthotrichum s.s. The phylogenetic position of Stoneobryum will determine whether the transformation from phaneroporous to cryptoporous stomata independently appeared only once or twice during the evolution of the tribe.
Molecular markers and phylogenetic analyses have been effectively utilized to distinguish between the genera of the tribe Orthotricheae, and these relationships have been supported with high bootstrap values and significant Bayesian posterior probabilities17, 18, 23. However, despite the strong statistical support, data poorly support the determination of mutual phylogenetic relationships, because phylogenetic trees often differ depending on the sequences analyzed.
Next-generation sequencing supports phylogenetic reconstruction based on the complete sequences of organellar genomes24, 25. In animals, complete mitochondrial genomes containing tens of thousands of base pairs are commonly used in phylogenetic reconstruction26. The mitochondrial genomes of vascular plants are characterized by highly diverse structures, but these genomes are rather conservative in mosses27. However, plant phylogenetic analyses mostly rely on plastid gene sequences, which are characterized by a relatively stable structure and size at lower taxonomic levels and a higher rate of evolution that increases the number of parsimony informative mutations28. Moreover, the absence of structural changes in the mitochondrial genomes of mosses facilitates the analysis of these genomes and their phylogenetic reconstruction. To date, mitochondrial genomes have rarely been used to investigate the evolution of lower taxonomic plant groups. Although mitochondrial genomes (hereafter referred to as ‘mitogenomes’) evolve more slowly than plastomes and nuclear genomes, they may be useful for the resolution of phylogenetic relationships in some cases where traditional methods fail. For instance, studies on the broadly understood genus Racomitrium Brid. commonly use nuclear ITS regions that are too variable for intergeneric studies, resulting in controversial alignments29. Furthermore, commonly used plastid regions (trnH-psbA, matK) have revealed a rather small number of parsimony informative sites29, 30. However, recent Racomitrium studies that used complete mitogenomes supported splitting this genus into five distinct clades27.
The main goal of the current study was to provide molecular support for the recent taxonomical rearrangements of the tribe Orthotricheae17, 19, 20 and to evaluate the usefulness of mitogenomes for evolutionary research at the genus level. Comparative genomics will provide information about mitogenomic hotspots for further phylogenetic studies.
Results
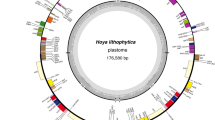
The developed libraries were sequenced to produce approximately 3–6 million paired-end reads, of which 6–10% were mapped to the mitogenome (see Supplementary Table S1). The sizes of the obtained mitogenomes ranged from 104,351 bp in Stoneobryum mirum D.H. Norris & H. Rob. to 104,785 bp in Orthotrichum callistomum Bruch & Schimp. (see Supplementary Table S1). The newly sequenced mitogenomes contained 40 protein-coding genes, three ribosomal RNA (rRNA) sequences, and 24 transfer RNA (tRNA) sequences. The order and localization of these genes are illustrated in Fig. 1.

Gene map of the mitogenome of Orthotrichum pulchellum. Genes inside and outside the outer circle are transcribed in counterclockwise and clockwise directions, respectively. The genes are color coded based on their function. The inner circle visualizes the G/C content.
Orthotricheae phylogenetic reconstruction and mitogenomic evolution
Alignments of 92 regions extracted from complete mitogenomes were used to construct phylogenetic trees using the ML method. Among them, 64 resolved at least one significantly supported (85% or more bootstrap support) clade, and those trees were used as input trees for SplitsTree. The super network analysis resolved all genera of the Orthotricheae as monophyletic (Fig. 2). Single-region trees differed in the relationships among these genera, but the lack of direct parallelism between OTUs suggested a lack of genes that could be transferred via hybridization and introgression.

A supernetwork of 64 of 92 Orthotrichaceae mitochondrial single-locus trees constructed using SplitsTree. Parallelograms indicate incongruence among single-locus trees, and gray-shaded areas encompass the competing splits among major Orthotrichaceae lineages.
Phylogenetic reconstruction based on the ML and BI methods produced trees with similar topologies (Fig. 3), and two well-supported clades were identified in all analyses. The first clade (A) comprised Ulota species and large taxa of the genus Orthotrichum s.l. with phaneroporous stomata, namely, Lewinskya and Pulvigera 20, 21. Dioecious Plenogemma phyllantha (Brid.) Plášek, Sawicki & Ochyra did not form a monophyletic clade with the monoecious species, and all phylogenetic analyses suggested that this taxon should be considered as sister to Pulvigera and Lewinskya species (maximum BS and BI support). The monoecious Lewinskya incana (Müll. Hal.) F. Lara, Garilleti & Goffinet and L. speciosa (Nees) F. Lara, Garilleti & Goffinet formed a monophyletic, well-supported clade (0.99 PP and 86% BS) with dioecious Pulvigera lyellii (Hook. & Taylor) Plášek, Sawicki & Ochyra. The second main clade (B) comprised the genera Orthotrichum s.s., Nyholmiella and Stoneobryum, but the phylogenetic relationships among them were only partially resolved. The ML and BI analyses differed in the support of the Stoneobryum clade as sister to Orthotrichum s.s. In the case of the ML tree, Stoneobryum was resolved as sister to Orthotrichum s.s., with significant (85.2%) bootstrap support indicating Nyholmiella as a basal genus for clade B (Fig. 3A). However, the BI did not support this topology, leaving the Nyholmiella-Stoneobryum relationship unresolved (Fig. 3B). Both analyses were congruent in resolving relationships among species of Orthotrichum s.s., where both clades had maximum BS and BI support. The first clade contained O. rogeri Brid., which was resolved as sister to O. diaphanum Brid. and O. macrocephalum F. Lara, Garilleti & Mazimpaka. The second clade of Orthotrichum s.s. contained O. callistomum, which was resolved as sister to O. pulchellum Brunt. and O. stellatum Brid.

The phylogenetic relationships of 17 Orthotrichaceae species based on complete mitogenome sequences: A - Maximum Likelihood (ML), B - Bayesian Interference (BI). The bootstrap values (ML) and posterior probabilities for BI trees lower than the maximum values (100 and 1.0, respectively) are shown at the nodes. The underlined species have cryptoporous stomata, and taxa in bold are dioecious.
The analysis of the complete mitochondrial genome clarified numerous doubts concerning the evolution of taxa from clade B, which included cryptopore-containing taxa from Nyholmiella, Stoneobryum, and the small monoecious Orthotrichum s.s. genus. The results of this phylogenomic analysis indicated that the Nyholmiella, represented by two species (N. obtusifolia (Brid.) Holmen & Warncke and N. gymnostoma (Brid.) Holmen & Warncke), is sister to the monoecious species of Orthotrichum s.l. with immersed stomata, including O. pulchellum, O. rogeri, O. stellatum, and O. callistomum (which represents the separate subgenus Callistoma).
The phylogenetic analyses of partitioned data sets (CDS, 20 most variable CDS, non-coding regions and 20 most variable non-coding regions) resulted in poorly resolved trees without any incongruences with trees based on the complete mitogenomes (see Supplementary Figs SF1–4).
Hotspots of variation within non-coding regions
A total of 174 regions were identified in the mitogenomes of the species analyzed, including 90 coding regions (CDSs, tRNAs, and rRNAs), 27 introns, and 57 spacers. Among the non-coding regions longer than 100 bp (for bias elimination), the highest number of polymorphic sites (8.06%) occurred in intron 4 of the cox1 gene. In this 1798 bp-long fragment, 64 single nucleotide polymorphisms (SNPs) and 81 indels were identified (Fig. 4, see Supplementary Table S2). The intergenic spacer between the cob and nad9 genes (1,982 bp) was the second most variable mitogenome fragment, with 6.81% of polymorphic sites, including 104 SNPs and 31 indels (see Supplementary Table S2). Only slightly lower variation (6.58% of polymorphic sites) was detected in the more-than-five-times-shorter (365 bp) nad1-cob spacer, including 14 SNPs and 10 indels. Among introns, the highest variation was found in the introns of the cox1 gene in the aforementioned intron 4 and intron 2. The latter included 15 SNPs and six indels (5.29% of polymorphic sites). The third most variable intron was the 1556-bp-long intron 3 of atp9 (4.63% of polymorphic sites), which included 58 SNPs and 14 indels. The highest number of mutations (227 SNPs and 77 indels) was found in the sdh3-rpl10 spacer, which was the longest intergenic spacer in the analyzed mitogenomes (10,761 bp). The variation of all identified intergenic spacers is shown in Supplementary Table S2.

The pattern of the SNP/indel variation of analyzed Orthotricheae mitogenomes.
Hotspots of variation within coding regions
Mutations (427 SNPs and six indels) were detected in 40 of 90 coding regions (see Supplementary Table S2), and single 3-bp-long indels were found in atp4, rps1, ccmFN, sdh4, rps7, and rps14. The most variable genes in the tribe Orthotricheae were ccmFC, which is responsible for cytochrome c biogenesis, and tatC, which encodes a TatC protein essential for the correct membrane translocation mechanism (38 and 19 SNPs, respectively). The remaining genes involved in cytochrome c biogenesis were less variable, including nine SNPs found in ccmB (one nonsynonymous) and 12 SNPs found in ccmC (eight nonsynonymous). By contrast, the ccmFN gene, with 2.11% of polymorphic sites, was fifth among the most variable coding regions (26 of 39 SNPs were nonsynonymous), after the sdh3 (nine SNPs, five nonsynonymous) and rps3 (35 SNPs, 19 nonsynonymous) genes. More than 10 SNPs were detected in 15 genes. The variability of those genes and others not discussed here is presented in Supplementary Table S2.
The dN/dS was also calculated for each gene, and ratios >1 were detected in the following genes: ccmFC, ccmFN, rps1, rpl2, tatC, cob and nad1. Several genes (nad3, cox2, rps12, nad4 L, nad7, rps19, rpl6, and rps11) only exhibited synonymous mutations, but these genes were also characterized by a low number of SNPs (1–5 SNPs per gene).
Discussion
The complete sequences of mitogenomes confirmed previous hypotheses that postulated the structural stability of moss genomes27, 30. The gene content and order of the Lewinskya, Nyholmiella, Orthotrichum, Plenogemma, Pulvigera, Stoneobryum, Ulota, and Zygodon Hook. & Taylor mitogenomes (Fig. 1) were identical to those in previously researched Orthotrichaceae species27, 31, 32. Phylogenetic analyses based on complete mitogenomes were partially congruent with those of previous studies. For instance, the results of previous molecular studies17,18,19 indicated that Nyholmiella was a sister group to Orthotrichum species with immersed stomata. Orthotrichum callistomum, which belongs to the separate monospecific subg. Callistoma (according to Lewinsky)14, was not analyzed in previous studies; however, based on its diagnostic character, which is an unusual configuration of the endostome14, 15, O. callistomum is the most distinct species within Orthotrichum. This result was, however, not significantly reflected in the infrageneric arrangement of Orthotrichum s.l. in other studies33.
The endostome segments of O. callistomum are joined above to form a ring; they are also heavily papillose and bear trabeculae on their inner surface. There is no other species from the Orthotrichum s.l. with such a combination of features. Therefore, Lewinsky14 also accepted this species as a single representative of one of seven subgenera of Orthotrichum s.l.
Although Podpěra34 included O. callistomum in sect. Callistoma, which was later validated by Iwatsuki and Sharp35, Lewinsky33 decided to elevate the section to subgenus as O. subg. Callistoma (Z.Iwats. & Sharp) Lewinsky. She argued that the unique morphological features of O. callistomum warranted its inclusion in its own subgenus instead of its inclusion in the section. Moreover, Lewinsky33 referred to a similar reflection by Vitt36 and used similar reasoning to propose the establishment of a separate subgenus, O. subg. Exiguifolia Vitt for Orthotrichum exiguum Sull., which was later moved to the genus Leratia Broth. & Paris based on phylogenetic analyses17. However, the results of the present study do not support the distinctiveness of this subgenus, because all of the phylogenetic analyses placed O. callistomum in a clade containing species belonging to O. subg. Pulchella (Schimp.) Vitt, effectively merging subg. Callistoma and subg. Pulchella into a single subgenus.
Regarding the position of the genus Stoneobryum, the phylogenies based on complete mitogenomes were partially incongruent with the results of a previous study17 that drew conclusions from evolutionary inferences based on four loci (two plastid, one nuclear 26S, and one mitochondrial nad5). Goffinet et al.17 resolved Stoneobryum and Sehnemobryum as sister clades to the Nyholmiella and Orthotrichum s.s. The ML analysis based on complete mitogenomes placed Stoneobryum as sister to cryptoporous Orthotrichum species (Fig. 3A), with 85% bootstrap support, whereas the BI analysis left phylogenetic relationships between Stoneobryum, Nyholmiella and Orthotrichum unresolved (Fig. 3B). The inclusion of Sehnemobryum species in this analysis would likely help to resolve this incongruence. Unfortunately, the mitogenome assembly was not available, despite several attempts, and this was likely due to highly degraded organellar DNA.
The phylogenetic position of Stoneobryum based on ML analysis could be better explained by evolutionary features proposed by Lewinsky14, who suggested that a single evolutionary event leading to the presence of cryptoporous stomata from phaneroporous ancestry could be a fundamental dichotomy within Orthotrichum s.l. The results presented in this study are compatible with that hypothesis, grouping all cryptoporous taxa into a monophyletic clade (Fig. 3A). However, the position of Stoneobryum remained unresolved based on the BI analysis.
Regarding the appearance of cryptopores, the ML analysis of mitogenomes confirmed that cryptopores appeared only once during the evolution of Orthotrichum s.l. in an event that was independent of the change of the sexual reproduction system from dioecy toward monoecy. Moreover, cryptoporous taxa of Orthotrichum and Stoneobryum share a common ancestor with Nyholmiella. However, this scenario was not confirmed (or rejected) by BI analysis. Most likely, ongoing studies of the plastomes and large sets of nuclear genes will finally resolve this problem.
In agreement with previous studies17,18,19, the phylogenetic inferences based on the data from complete mitogenomes did not support the monophyly of dioecious species of the tribe Orthotricheae. However, the A and B clades distinguished differ with respect to the patterns of sexual system evolution. According to molecular phylogenies of mosses, sexual systems are often not correlated with phylogenetic relationships36, 37, and the evolutionary mechanisms associated with the shift between dioecy and monoecy could be as complex as sex determination mechanisms in plants38, 39. Based on the present and previous studies16,17,18,19, monoecy independently evolved from dioecy at least twice in clades A and B. For instance, the basal taxa in clade A are monoecious (Ulota crispa (Hedw.) Brid. and U. hutchinsiae (Sm.) Hamm.), but dioecious Plenogemma phyllantha and Pulvigera lyellii were resolved as basal taxa for Lewinskya. A simpler picture of the evolution of sexual systems in the tribe Orthotricheae is evident in clade B, where dioecious genera Nyholmiella and Stoneobryum were resolved as basal taxa, which suggests that the shift of sexual systems in this group only occurred once.
The results presented in this study are in agreement with those of previous studies based on single plastid regions as well as mitochondrial and nuclear genomes17, 19, 40 and support recent nomenclatural decisions20, 21. However, the results of these earlier studies indicated that the common clade containing P. phyllantha and Lewinskya species was not statistically supported. Only recent studies based on three plastid genes (trnG, trnL-F and atpB-rbcL) and incomplete ITS2 loci resolved a monophyletic Ulota clade, and only in two out of four analyses41.
Resolving phylogenetic inferences among the closely related Orthotrichaceae genera requires large data sets based on complete organellar genomes or transcriptomic data, and these analyses are good examples of how the ‘omics’ era could aid in the resolution of evolutionary questions in plant systematics. The phylogenetic tree of Orthotricheae based on the mitogenome confirms the previously observed polyphyletic nature of Orthotrichum s.l. and Ulota s.l., and this tree molecularly supports recent taxonomical rearrangement20. However, since the taxon sampling is limited (especially in the case of the genera Lewinskya and Ulota), the phylogenetic inferences presented in this study have a rather preliminary character.
Since the recent reporting of complete moss mitogenomes27, 30, 32, 42, 43, only scarce data concerning potential hotspots of variability have been presented44. The comparison of two O. diaphanum mitogenomes revealed only single nonsynonymous substitutions in the rps1 and ccmFN genes and noted the latter as a potential hotspot in the Orthotrichaceae44, which also confirms the results presented above. Comparative mitogenomics between Physcomitrella patens (Hedw.) Bruch & Schimp. and Marchantia polymorpha L. revealed that nad1, nad5, and cox3 were the most polymorphic genes, and that rpl2 had the highest number of nonsynonymous mutations39. In our study, these genes were characterized by moderate variation (see Supplementary Table S2), but rpl2 was in the top three most-variable genes based on the number of nonsynonymous mutations (see Supplementary Table S2). The results also support earlier studies that reported different evolutionary patterns associated with mitochondrial gene evolution in mosses and vascular plants45. The most variable Orthotricheae genes that were responsible for the biogenesis of cytochrome c (ccmFC and ccmFN) are among the most conserved genes in vascular plant mitogenomes, especially in monocots45. However, species with ccmFC pseudogenes are also known to exist46. In the genus Silene L., atp1 was found to be the most variable among mitochondrial genes47, and it was also one of the most polymorphic genes in the present study.
The intergenic and intronic hotspots of the Orthotricheae identified in the present study do not correspond to those described in vascular plant mitogenomes. The phylogenetically informative and quickly evolving intron 4 of the nad5 gene in the genus Abies Mill48. does not exist in the known moss mitogenomes. However, most of the intergenic variation in vascular plant mitogenomes is related to repeats (≥50 bp) that are present in genomes in many copies49, which possibly lead to non-homologous, intragenomic recombination50,51,52. The lack of these repeats in moss mitogenomes is considered the main cause of their structural stability27.
A new taxonomical arrangement of the traditionally conceived genera Orthotrichum s.l. and Ulota s.l. was presented by Plášek et al.20. Taxonomic changes are graphically expressed in Fig. 5. A critical feature associated with the reclassification of this taxonomic group is the type and position of stomata. For instance, Orthotrichum s.s. is the only genus with cryptoporous stomata. Among the genera with phaneroporous stomata, an important classification character is the ploidy level. Monoecious taxa include the Ulota s.s. and Lewinskya genera, and the genera share superficial stomata and recurved leaf margins. However, Ulota species can be easily distinguished from Lewinskya by the presence of quadrate to rectangular hyaline cells that form a marginal border at the leaf base. Moreover, brood bodies are never produced in Ulota species, and asexual reproduction by propagules within Lewinskya species is extremely rare. Therefore, the dioecious genera of the Orthotricheae can be mainly characterized by the production of abundant gemmae, and this apparently compensates for the rare incidence of sexual reproduction in comparison to the monoecious species. The dioecious group consists of Nyholmiella, Stoneobryum, Pulvigera, and Plenogemma, and the latter two taxa have been recognized as new genera20. Stoneobryum, whose phylogenetic position remains ambiguous, is the only dioecious genus with cryptoporous stomata. Nyholmiella differs from Pulvigera and Plenogemma primarily based on the presence of an ovate leaf with an obtuse apex and somewhat incurved leaf margins, and the latter two genera have leaves that are linear-lanceolate or lanceolate with an acute to acuminate apex. While the gemmae in Pulvigera are scattered more or less equally on the adaxial leaf surface, in Plenogemma the conspicuous clusters of fusiform brownish gemmae are situated on the protruding costa of the upper leaves.

Materials and Methods
Material
Genomic libraries were created with the use of DNA isolated in our previous work18, 19, 53, with the exceptions of DNA samples from Stoneobryum bunyaense D.H. Norris & H. Rob., S. mirum, Lewinskya incana, and Orthotrichum callistomum, which were isolated using the Zymo Plant/Seed DNA kit (Zymo Research Corp., Irvine, CA, USA) following the manufacturer’s instructions. Specimen details and corresponding GenBank accession numbers are listed in Supplementary Table S1. Based on previous results16,17,18,19, Zygodon was selected as the outgroup. The DNA quantity was estimated with the use of a Qubit fluorimeter system and Quant-IT ds-DNA BR Assay kit (Invitrogen, Carlsbad, NM, USA).
Genomic libraries for MiSeq sequencing were developed using the Nextera XT Kit (Illumina, San Diego, CA, USA) and 1 ng of DNA; however, the normalization procedure was conducted using the Kapa Library Quantification Kit for Illumina (Kapa, Wilmington, MA, USA). The numbers of created and sequenced genomic libraries for each taxon are given in Supplementary Table S1.
The size distributions of the libraries were checked using primer sequences provided in the Sequencing Library qPCR Quantification Guide (Illumina). PCRs were performed in 20-µL reaction mixtures containing 3 µL of the genomic library, each primer at 1.0 µM, 1.5 mM MgCl2, 200 µL of dNTPs (dATP, dGTP, dCTP, and dTTP), 1X PCR buffer, and 1 U of OpenEx Taq polymerase (OpenExome, Warsaw, Poland). PCRs were performed under the following thermal conditions: (1) initial denaturation, 5 min at 94 °C; (2) denaturation, 30 s at 94 °C; (3) annealing, 30 s at 52 °C; (4) elongation, 1 min at 72 °C; and (5) final elongation, 7 min at 72 °C. Steps 2–4 were repeated 34 times. The PCR products were separated using a QIAxcel capillary electrophoresis system (Qiagen, Hilden, Germany). Electrophoresis was performed using a QIAxcel High Resolution Kit (Qiagen) with a 15–1,000-bp alignment marker and a 25–1,000-bp size marker; standard OL500 settings were used in the electrophoresis program. Validated libraries were pooled according to guidelines found in the Kapa Library Quantification Kit for Illumina. Genomic libraries were sequenced using MiSeq 500v2 and 600v3 cartridges, which supported the acquisition of 2 × 250- and 2 × 300-bp paired-end reads, respectively.
Mitogenome assembly
The obtained reads were mapped to the previously published mitogenome of Orthotrichum speciosum 31 using Geneious R6 software (Biomatters, Auckland, New Zealand) with default medium-low sensitivity settings and the previously detailed assembly workflow27. Briefly, fragments located between gaps and overlapping but incongruent sequences were extracted and moved to separate files. Raw paired-end reads with 100 iterations were mapped to every fragment using customized settings (minimum sequence overlap of 150 and 99% overlap identity). The resulting contigs underwent de novo assembly using the default settings in Geneious, and all available reads were mapped to the draft assembly of the mitogenome to determine coverage and to check for incongruence between the mapped sequences. Annotated mitogenome GenBank files were used to draw gene maps using the OrganellarGenome DRAW tool53, and the maps were examined for further comparisons of gene order and content.
Phylogenetic and variation analyses
Eleven mitogenomes obtained in the present study and six previously published genomes25, 27, 31, 44 were aligned using the Mauve genome aligner54, and two regions containing tandem repeats were adjusted manually. Unambiguously aligned DNA sequences were used in the phylogenetic analyses.
The consistency among single genes and intergenic regions was evaluated by the maximum-likelihood (ML) analysis for every locus that contained parsimony informative characters using the RAxML v7.2.3 plugin55 for Geneious 7.0.3 with the GTR + I + G model and bootstraps estimated using 500 replicates.
Potential conflicts among genes and spacers were visualized by constructing a supernetwork of 62 trees using SplitsTree456 according to the approach given by Liu et al.57, except the number of replicates was increased to 500 and the software used for creating consensus tree was Geneious 7.0.3.
PartitionFinder258 was used to determine the best partitioning schemes and corresponding nucleotide substitution models. The data-set blocks were predefined a priori based on protein-coding genes (CDS), tRNA and rRNA, introns and intergenic spacers as well as for first, second and third position for each of CDS. The Bayesian information criterion (BIC) and the ‘greedy’ algorithm with branch lengths estimated as unlinked were used to search for the best-fit scheme. The partitions with optimal substitution models are given in Supplementary Table S3. Phylogenetic reconstruction was also conducted using four partitioned data sets, which included coding regions, the 20 most variable protein-coding genes, intergenic spacers (including introns) and the 20 most variable non-coding regions (see Supplementary Figs SF1–4).
Phylogenetic analyses were conducted using ML and BI methods. The ML analysis was performed using RAxML version 8.2.455 with the partitioning scheme and nucleotide substitution blocks generated by PartitionFinder2, using default parameters. Bootstrap analyses were performed with 2,000 replicates to assess nodal support.
Bayesian analysis (BI) was conducted using MrBayes 3.259, and the MCMC algorithm was run for 20,000,000 generations (sampling every 1,000) with four incrementally heated chains (starting from random trees). The visual inspection of Tracer 1.360 plots was used to examine the parameters and to determine the number of generations needed to reach stationarity, which occurred at approximately 300,000 generations. Therefore, the first 500 trees were discarded as burn-in, and the remaining trees were used to develop a Bayesian consensus tree.
All regions, including coding regions, introns, and intergenic spacers, were extracted to identify divergent regions within the 16 mitogenomes (excluding the Zygodon outgroup) for additional phylogenetic applications. The percentage of variable characters within each region was calculated, and the number of nucleotide substitutions and indels (potentially informative characters (PIC)) among the 13 mitogenomes was determined for each region. Indels were scored in this study because they tend to be prevalent and phylogenetically informative61, 62. The preliminarily performed phylogenetic analyses based on the data set excluding indels resulted in trees with identical topology as presented here but, in some cases, with lower bootstrap support. Indels and nucleotide substitutions within indels were scored as independent, single characters to obtain the results comparable with those of previous studies45, 48. The percentage of variation (P) in each region (exons, introns, and spacers) was described using the formula of O’Donnell63, which was successfully used to identify variation in plastid regions64, 65. Mitogenome variation was visualized using Circos66 and a custom Python script.
To identify mitochondrial loci undergoing adaptation, dN/dS ratios were calculated for each gene. This measure quantifies selection pressures by comparing the rate of synonymous substitutions (dS), which are presumed neutral, to the rate of nonsynonymous substitutions (dN), which possibly experience selection67. A codon-based test of neutrality was conducted using the Nei-Gojobori method68 as implemented in MEGA7 software69, with default settings.
To estimate the level of substitution saturation, we used the approach of Xia et al.70 as implemented in DAMBE5 software71. The amount of substitutional saturation was calculated for four scenarios: the first codon position, the second codon position, the combined the first and the second codon positions, and the third codon position. Since no significant saturation was detected, all three partitions involving codon positions were used in the phylogenetic analyses.
Conclusions
The phylogenetic analyses based on complete mitogenome sequences support the new taxonomic classification within Orthotrichum s.l. (Fig. 5) as proposed by Plášek et al.20. The thirteen newly sequenced mitogenomes confirmed their stable gene order and structure in the mosses. Given the limited data availability, it is difficult to propose any universal pattern of mitochondrial gene variation in mosses. However, several ongoing studies of mitogenome variation patterns among different moss families will likely provide additional background information related to this subject.
References
Goffinet, B., Buck, W. R. & Wall, M. A. Orthotrichum freyanum (Orthotrichaceae), a new epiphytic moss from Chile. Nova Hedwigia 131, 1–11 (2007).
Fedosov, V. E. & Ignatova, E. A. Orthotrichum dagestanicum sp. nov. (Orthotrichaceae, Musci) – a new species from Dagestan (Eastern Caucasus). Arctoa 19, 69–74 (2010).
Lara, F., Garilleti, R. & Mazimpaka, V. A peculiar new Orthotrichum species (Orthotrichaceae, Bryopsida) from central Argentina. Bot. J. Linn. Soc 155, 477–482 (2007).
Lara, F., Garilleti, R. & Mazimpaka, V. Orthotrichum karoo (Orthotrichaceae), a new species with hyaline-owned leaves from southwestern Africa. Bryologist 112, 194–201 (2009).
Lara, F., Garilleti, R., Medina, R. & Mazimpaka, V. A new key to the genus Orthotrichum Hedw. in Europe and the Mediterranean Region. Cryptogam. Bryol. 30, 129–142 (2009).
Medina, R., Lara, F., Mazimpaka, V. & Garilleti, R. Orthotrichum norrisii (Orthotrichaceae), a new epiphytic Californian moss. Bryologist 111, 670–675 (2008).
Medina, R., Lara, F., Mazimpaka, V., Shevock, J. R. & Garilleti, R. Orthotrichum pilosissimum (Orthotrichaceae), a new moss from arid areas of Nevada with unique axillary hairs. Bryologist 114, 316–324 (2011).
Medina, R., Lara, F., Goffinet, B., Garilleti, R. & Mazimpaka, V. Integrative taxonomy successfully resolves the pseudocryptic complex of the disjunct epiphytic moss Orthotrichum consimile s.l. (Orthotrichaceae). Taxon 61, 1180–1198 (2012).
Medina, R. F., Lara, B., Goffinet, R., Garilleti, R. & Mazimpaka, V. Unnoticed diversity within the disjunct moss Orthotrichum tenellum s.l. validated by morphological and molecular approaches. Taxon 62, 1133–1152 (2013).
Plášek, V., Sawicki, J., Trávníčková, V. & Pasečná, M. Orthotrichum moravicum (Orthotrichaceae), a new species from the Czech Republic. Bryologist 112, 329–336 (2009).
Plášek, V., Sawicki, J. & Číhal, L. Orthotrichum pamiricum (Bryophyta), a new epiphytic moss species from Pamir Mountains in Central Asia. Turk. J. Bot. 38, 754–762 (2014).
Garilleti, R., Shevock, J. R., Norris, D. H. & Lara, F. Orthotrichum mazimpakanum sp. nov. and O. anodon (Orthotrichaceae), two similar species from California. Bryologist 114, 346–355 (2011).
Kiebacher, T. & Lüth, M. Orthotrichum dentatum T.Kiebacher & Lüth sp. nov. (Orthotrichaceae). J. Bryol., doi:10.1080/03736687.2016.1186858 (2016).
Lewinsky, J. A synopsis of the genus Orthotrichum Hedw. (Musci, Orthotrichaceae). Bryobrothera 2, 1–59 (1993).
Lewinsky-Haapasaari, J. & Hedenäs, L. A cladistic analysis of the moss genus Orthotrichum. Bryologist 101, 519–555 (1998).
Goffinet, B., Bayer, R. J. & Vitt, D. H. Circumscription and phylogeny of the Orthotrichales (Bryopsida) inferred from rbcL sequence analyses. Am. J. Bot. 85, 1324–1337 (1998).
Goffinet, B., Shaw, A. J., Cox, C. J., Wickett, N. J. & Boles, S. Phylogenetic inferences in the Orthotrichoideae (Orthotrichaceae: Bryophyta) based on variation in four loci from all genomes. Monogr. Syst. Bot. Missouri Bot. Gard 98, 270–289 (2004).
Sawicki, J., Plášek, V. & Szczecińska, M. Preliminary studies on the phylogeny of Orthotrichum inferred from nuclear ITS sequences. Ann. Bot. Fenn. 46, 507–515 (2009).
Sawicki, J., Plášek, V. & Szczecińska, M. Molecular studies resolve Nyholmiella (Orthotrichaceae) as a separate genus. J. Syst. Evol 48, 183–194 (2010).
Plášek, V., Sawicki, J., Ochyra, R., Szczecińska, M. & Kulik, T. New taxonomical arrangement of the traditionally conceived genera Orthotrichum and Ulota (Orthotrichaceae, Bryophyta). Acta Mus. Siles., Sci. Natur 64, 169–174 (2015).
Lara, F. et al. Lewinskya, a new genus to accommodate the phaneroporous and monoicous taxa of Orthotrichum (Bryophyta, Orthotrichaceae). Cryptogam., Bryol. 37(4), 361–382 (2016).
Ramsay, H. P. Australian Mosses Online. 47. Orthotrichaceae: Ulota. http://www.anbg.gov.au/abrs/Mosses_online/Orthotrichaceae_Ulota.pdf (Accessed 15 August 2016) (2012).
Medina, R., Lara, F., Goffinet, B., Garilleti, R. & Mazimpaka, V. Unnoticed diversity within the disjunct moss Orthotrichum tenellum s.l. validated by morphological and molecular approaches. Taxon 62, 1133–1152 (2013).
Doucet-Beaupre, H. et al. Mitochondrial phylogenomics of the Bivalvia (Mollusca): searching for the origin and mitogenomic correlates of doubly uniparental inheritance of mtDNA. BMC Evol. Biol. 10, 50 (2010).
Sawicki, J., Szczecińska, M., Bednarek-Ochyra, H. & Ochyra, R. Mitochondrial phylogenomics supports splitting the traditionally conceived genus Racomitrium (Bryophyta: Grimmiaceae). Nova Hedwigia 100, 293–317 (2015).
Vilstrup, J. T. et al. Mitochondrial Phylogenomics of Modern and Ancient Equids. PLoS ONE 8, e55950 (2013).
Liu, Y., Medina, R. & Goffinet, B. 350 My of mitochondrial genome stasis in mosses, an early land plant lineage. Mol. Biol. Evol 31, 2586–2591 (2014).
Szczecińska, M., Gomolińska, A., Szkudlarz, P. & Sawicki, J. Plastid and nuclear genomic resources of a relict and endangered plant species: Chamaedaphne calyculata (L.) Moench (Ericaceae). Turk. J. Bot. 38, 1229–1238 (2014).
Larraín, J., Quandt, D., Stech, M. & Muñoz, J. Lumping or splitting? The case of Racomitrium (Bryophytina: Grimmiaceae). Taxon 62, 1117–1132 (2013).
Liu, Y., Xue, J.-Y., Wang, B., Li, L. & Qiu, Y.-L. The mitochondrial genomes of the early land plants Treubia lacunosa and Anomodon rugelii: dynamic and conservative evolution. PLoS ONE 6, e25836 (2011).
Sawicki, J., Szczecińska, M., Kulik, T., Gomolińska, A. & Plášek, V. The complete mitochondrial genome of the epiphytic moss Orthotrichum speciosum. Mitochondr. DNA Part A 27, 1709–1710 (2016).
Sawicki, J. et al. The complete mitochondrial genome of the rare and endangered Orthotrichum rogeri (Orthotrichaceae, Bryophyta). Mitochondr. DNA 27, 3208–3209 (2015).
Lewinsky, J. The genus Orthotrichum Hedw. (Orthotrichaceae, Musci) in Southeast Asia. A taxonomic revision. J. Hattori Bot. Lab. 72, 1–88 (1992).
Podpěra, L. Conspectus Muscorum Europaeorum. 1–697 (ČSAV, Prague, Czech Republic, 1954).
Iwatsuki, Z. & Sharp, A. J. Interesting mosses from Formosa. J. Hattori Bot. Lab. 33, 161–170 (1970).
Villarreal, J. C. & Renner, S. S. Correlates of monoicy and dioicy in hornworts, the apparent sister group to vascular plants. BMC Evol. Biol. 13, 239 (2013).
McDaniel, S. F., Atwood, J. & Burleigh, J. G. Recurrent evolution of dioecy in bryophytes. Evolution 67, 567–572 (2013).
Tanurdzic, M. & Banks, J. A. Sex-Determining Mechanism in Land Plants. The Plant Cell 16, 61–71 (2004).
Milewicz, M. & Sawicki, J. Mechanisms of Sex Determination in Plants. Cas. Slez. Muz. Opava (A) 61, 123–129 (2012).
Tessler, M., Cunningham, S. W. & Clark, T. A. Noteworthy habitat and phylogeny updates for eastern US Ulota (Orthotrichaceae, Bryophyta). Mitochond. DNA 6, 1–5 (2016).
Caparrós, R., Lara, F., Draper, I., Mazimpaka, V. & Garilleti, R. Integrative taxonomy sheds light on an old problem: the Ulota crispa complex (Orthotrichaceae, Musci). Bot. J. Linn. Soc. 180, 427–451 (2016).
Myszczyński, K. et al. The complete mitochondrial genome of the cryptic species C of Aneura pinguis. Mitochodr. DNA 18, 1–2 (2015).
Lewis, L., Liu, Y., Rozzi, R. & Goffinet, B. Infraspecific variation within and across complete organellar genomes and nuclear ribosomal repeats in a moss. Mol. Phylogenet. Evol. 96, 195–199 (2016).
Vigalondo, B. et al. Comparing three complete mitochondrial genomes of the moss genus Orthotrichum Hedw. Mitochondr. DNA Part B: Res. 1, 168–170 (2016).
Cui, P. et al. A complete mitochondrial genome of wheat (Triticum aestivum cv. Chinese Yumai), and fast evolving mitochondrial genes in higher plants. J. Genet. 88, 299–307 (2009).
Tang, M. et al. Rapid evolutionary divergence of Gossypium barbadense and G. hirsutum mitochondrial genomes. BMC Genomics 16, 770 (2015).
Barr, C. M., Keller, S. R., Ingvarsson, P. K., Sloan, D. B. & Taylor, D. R. Variation in mutation rate and polymorphism among mitochondrial genes of Silene vulgaris. Mol. Biol. Evol. 24, 1783–1791 (2007).
Jaramillo-Correa, J. P., Aguirre-Planter, E., Eguiarte, L. E., Khasa, D. P. & Bousquet, J. Evolution of an ancient microsatellite hotspot in the conifer mitochondrial genome and comparison with other plants. J. Mol. Evol. 76, 146–57 (2013).
Chaw, S.-M., Shih, A. C.-C., Wang, D., Wu, Y.-W. & Liu, S.-M. The mitochondrial genome of the gymnosperm Cycas taitungensis contains a novel family of short interspersed elements, Bpu sequences, and abundant RNA editing sites. Mol. Biol. Evol. 25, 603–615 (2008).
André, C., Levy, A. & Walbot, V. Small repeated sequences and the structure of plant mitochondrial genomes. Trends Genet. 8, 128–132 (1992).
Maréchal, A. & Brisson, N. Recombination and the maintenance of plant organelle genome stability. New Phytol 186, 299–317 (2010).
Arrieta-Montiel, M. P., Shedge, V., Davila, J., Christensen, A. C. & Mackenzie, S. A. Diversity of the Arabidopsis mitochondrial genome occurs via nuclear-controlled recombination activity. Genetics 183, 1261–1268 (2009).
Sawicki, J., Plášek, V. & Szczecińska, M. Molecular data do not support the current division of Orthotrichum (Bryophyta) species with immersed stomata. J. Syst. Evol. 50, 12–24 (2012).
Darling, A. C. E., Mau, B., Blattner, F. R. & Perna, N. T. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403 (2014).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Huson, D. H. & Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267 (2006).
Liu, Y., Cox, C. J., Wang, W. & Goffinet, B. Mitochondrial phylogenomics of early land plants: mitigating the effects of saturation, compositional heterogeneity, and codon-usage bias. Syst. Biol. 63(6), 862–878 (2014).
Lanfear, R., Frandsen, P. B., Wright, A. M., Senfeld, T. & Calcott, B. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol., doi:10.1093/molbev/msw260 (2016).
Huelsenbeck, J. P. & Ronquist, F. R. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755 (2001).
Rambaut, A. & Drummond, A. J. Tracer v1.3. 2003. http://evolve.zoo.ox.ac.uk (Accessed 20 August 2016) (2003).
Morton, B. R. & Clegg, M. T. A chloroplast DNA mutational hotspot and gene conversion in a noncoding region near rbcL in the grass family (Poaceae). Curr. Genet. 24, 357–365 (1993).
Gilley, L., Yong-Ming, Y., Küpfer, D. & Taberlet, P. Phylogenetic use of noncoding regions in the genus Gentiana L. Chloroplast trnL (UAA) intron versus nuclear ribosomal internal transcribed spacer sequences. Mol. Phylogenet. Evol. 3, 460–466 (1996).
O’Donnell, K. Ribosomal DNA internal transcribed spacers are highly divergent in the phytopathogenic ascomycete Fusarium sambucinum (Gibberella pulicaris). Curr. Genet. 22, 213–220 (1992).
Shaw, J., Lickey, E. B., Schilling, E. E. & Small, R. L. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: The tortoise and the hare III. Am. J. Bot. 94, 275–288 (2007).
Szczecińska, M. & Sawicki, J. Genomic resources of three Pulsatilla species reveal evolutionary hotspots, species-specific sites and variable plastid structure in the family Ranunculaceae. Int. J. Mol. Sci. 16, 22258–22279 (2015).
Krzywinski, M. et al. Circos: An information Aesthetic for Comparative Genomics. Genome Res. 19, 1639–1645 (2009).
Kimura, M. Preponderance of synonymous changes as evidence for the neutral theory of molecular evolution. Nature 267, 275–276 (1977).
Nei, M. & Gojobori, T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 3, 418–426 (1986).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33(7), 1870–1874 (2016).
Xia, X., Xie, Z., Salemi, M., Chen, L. & Wang, Y. An index of substitution saturation and its application. Mol. Phylogenet. Evol. 26(1), 1–7 (2003).
Xia, X. DAMBE5: A comprehensive software package for data analysis in molecular biology and evolution. Mol. Biol. Evol. 30(7), 1720–1728 (2013).
Acknowledgements
We are very grateful to Editage company and Dr. Rod Seppelt (Tasmania) for linguistic corrections of the manuscript. The contribution by V. Plášek is part of a research project of the Institute of Environmental Technologies, reg. no. CZ.1.05/2.1.00/03.0100, Project LO1208 of the National Feasibility Programme I of the Czech Republic. This study was possible also through financial support from the Polish Ministry of Science and Higher Education (Grant Iuventus Plus IP2010-037070) for J. Sawicki. The contribution of R. Ochyra gained financial support from the Polish National Centre of Sciences through grant No. N N 303 469 338 and, in part, through the statutory fund of the Institute of Botany of the Polish Academy of Sciences.
Author information
Authors and Affiliations
Contributions
J.S. and V.P. prepared material and performed the experiments, wrote the paper; M.Ś. prepared figures and/or tables, and reviewed drafts of the paper; M.S., K.M. and T.K. analyzed data, wrote the paper; and R.O. wrote the paper and reviewed drafts of the paper.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sawicki, J., Plášek, V., Ochyra, R. et al. Mitogenomic analyses support the recent division of the genus Orthotrichum (Orthotrichaceae, Bryophyta). Sci Rep 7, 4408 (2017). https://doi.org/10.1038/s41598-017-04833-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-04833-z
This article is cited by
-
The organellar genomes of Pellidae (Marchantiophyta): the evidence of cryptic speciation, conflicting phylogenies and extraordinary reduction of mitogenomes in simple thalloid liverwort lineage
Scientific Reports (2023)
-
Breaking the limits - multichromosomal structure of an early eudicot Pulsatilla patens mitogenome reveals extensive RNA-editing, longest repeats and chloroplast derived regions among sequenced land plant mitogenomes
BMC Plant Biology (2022)
-
Insights into adaptive evolution of plastomes in Stipa L. (Poaceae)
BMC Plant Biology (2022)
-
Tea plantations and their importance as host plants and hot spots for epiphytic cryptogams
Scientific Reports (2021)
-
Molecular delimitation of European leafy liverworts of the genus Calypogeia based on plastid super-barcodes
BMC Plant Biology (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
