
Abstract
Epidermal growth factor receptor (EGFR) T790M acquired drug-resistance mutation has become a major clinical challenge for the therapy of non-small cell lung cancer. Here, we applied a structure-guided approach on the basis of the previous reported EGFR inhibitor (compound 9), and designed a series of C4-alkyl-1,4-dihydro-2H-pyrimido[4,5-d][1,3]oxazin-2-one derivatives as novel mutant-selective EGFR inhibitors. Finally, the most representative compound 20a was identified, which showed high selectivity at both enzymatic and cellular levels against EGFRL858R/T790M (H1975 cell lines) over EGFRWT (A431 cell lines). The representative compound 20a also showed promising antitumor efficiency in the in vivo antitumor efficacy study of H1975 xenograft mouse model driven by EGFRL858R/T790M. These results provide a new scaffold for the treatment of dual-mutant-driven non-small cell lung cancer.
Similar content being viewed by others


Introduction
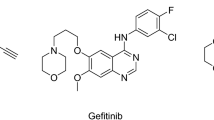
The epidermal growth factor receptor (EGFR) is a member of ErbB (HER) receptor tyrosine kinases (RTKs) family, which plays essential roles in regulation of numerous normal physiological processes, such as cell proliferation, differentiation, metabolism and apoptosis1, 2. Conversely, overexpression and abnormal activation of EGFR is associated with varieties of human epithelial malignancies, especially non-small cell lung cancer (NSCLC). Thus, targeting EGFR has provided an effective anticancer strategy, and EGFR has become a well-established critical target for the treatment of NSCLC3,4,5. Whereafter, activating mutations within the EGFR catalytic domain have been successively discovered, of which, the exon 21 single point substitution mutation (L858R) and the exon 19 deletion (del E746-A750) are the two most prevalent activating mutations. Detection of EGFR activating mutations provides a useful marker for predicting the potential of first generation EGFR inhibitors6,7,8. Thus, compounds 1 (gefitinib) and 2 (erlotinib), two of the first-generation EGFR-targeted small molecule inhibitors (Fig. 1) have been used in clinic for the treatment of advanced NSCLC patients harboring these specific activating mutations. The two agents demonstrated remarkable therapeutic responses for these NSCLC patients, however, acquired drug-resistance often emerged after treatment of 10–14 months, which has become a major clinical challenge for the therapy of NSCLC9,10,11. The emergence of point mutations in the EGFR kinase domain is also closely related to acquired resistances, among which, the gatekeeper T790M secondary mutation (threonine790 → methionine790 mutation) is the primary mechanism of the acquired resistances, as it is the most common mutation and accounts for approximately 60% of all clinically observed acquired mutants12.

Structures of first-, second- and third-generation EGFR inhibitors.
Extensive efforts have been devoted to the development of novel covalent EGFR inhibitors to overcome gefitinib- and erlotinib-resistant mutant (T790M mutation). These irreversible inhibitors are designed with electrophilic Michael-acceptor systems to covalently react with the conserved Cys797 in the EGFR active site, so as to increase inhibition potency against T790M mutant relative to reversible agents. Unfortunately, because of the dose-limiting toxicities attributed to inhibition of the wild-type (WT) EGFR, these second-generation irreversible inhibitors (Fig. 1) including 3 (afatinib)13, 4 (neratinib)14, 5 (dacomitinib)15 did not improve clinical efficacy for NSCLC patients who have developed T790M acquired resistance. Recently, the third-generation (mutant-selective) irreversible EGFR-tyrosine kinase inhibitors (TKIs) based on an amino pyrimidine scaffold, such as compounds 6 (WZ4002)16, 7 (CO-1686)17 and 8 (AZD9291)18 have demonstrated promising selectivity for EGFRL858R/T790M mutant over WT EGFR, indicating that this strategy is feasible for overcoming EGFR T790M gatekeeper mutation in NSCLC treatment (Fig. 1). Based on their clinical significant benefits for NSCLC patients with EGFR T790M acquired drug-resistance mutation, United States Food and Drug Administration (FDA) has awarded compounds 7 and 8 “Breakthrough Therapy” designations in 201419. Furthermore, 8 has been granted accelerated approval by FDA for the treatment of late-stage NSCLC patients with EGFRT790M mutation-positive who have progressed after other EGFR TKIs therapy20.
In our previous studies to develop mutant-selective EGFRL858R/T790M inhibitors, compound 9 was identified as a potent irreversible EGFR kinase inhibitor (Fig. 2A), which exhibited competitive enzymatic inhibitory activities against L858R/T790M mutant EGFR21, 22. In order to improve its cellular antiproliferative activity, meanwhile keep the selectivity profiles, we would like to describe the design and optimization of C4-alkyl-1,4-dihydro-2H-pyrimido[4,5-d][1,3]oxazin-2-one derivatives as novel mutant-selective EGFR inhibitors on the basis of compound 9.

(A) The chemical structure of compound 9. (B) The modeling binding mode of compound 9 bound to EGFRT790M (PDB code: 3IKA). The sulfur atoms are colored in dark yellow and the carbon atoms are colored in light yellow. (C) The design strategy of C4-alkyl-1,4-dihydro-2H-pyrimido[4,5-d][1,3]oxazin-2-ones scaffold. Compound 16a (orange) was aligned to the modeled conformation of compound 9 (marine) shown in panel A by SHAFTS. The EGFRWT crystal structure (shown in yellow, PDB code: 4G5J) was aligned to the EGFRT790M (shown in cyan, PDB code: 3IKA). The hinge regions of both EGFRWT and EGFRT790M were presented as stick. The carbon atoms of 9 are colored in marine and the nitrogen atoms of 9 are colored in dark blue. (D) The 2D diagram interaction of compound 16a. (E) The proposed binding mode of compound 20a bound to EGFRT790M (cyan line, PDB code: 3IKA). (F) The proposed binding mode of compound 20a bound to EGFRWT (yellow line, PDB code: 4G5J). The conformations of compound 9, 16a and 20a in the panel C,E and F are depicted as stick and highlighted in grey surface presentation.
Results
Scaffold Hopping and Binding Mode Analysis
Using a structure-based approach, we reported compound 9 as a potent irreversible inhibitor with excellent selectivity against EGFRL858R/T790M kinase over EGFRWT kinase (4.7 nM vs 474 nM, 101-fold)21. As shown in Fig. 2B, the modeling binding mode suggests that compound 9 binds to the EGFRT790M (PDB code: 3IKA) via covalently modifying the conserved Cys797 residue at the lip of the ATP-binding site. The 6,7-dioxo-6,7-dihydropteridine core is predicted to interact with the residue Met793 at the hinge region by an expected bidentate hydrogen bonding and also form a hydrophobic sandwich with the ceiling residue Ala743 and the floor residue Leu844. The N-methylpiperazine ring faces to the solvent exposed region. The two phenyl rings substituted at C2 and N8 adapt a U-shape binding mode. These interactions suggested by the docking study stabilize the basic recognition of inhibitor-kinase complex. Moreover, the methyl group at N5-position points to the mutant gatekeeper Met790 residue and strengthens nonpolar contacts with T790M mutants, which is a vital contribution proposed by the docking study to the selectivity against EGFRL858R/T790M over EGFRWT. Besides, the L858R mutation is far from the ATP-binding pocket of EGFRs and would contribute little to the interaction at the inhibitor-binding site. Given the former successful design strategy targeting EGFRL858R/T790M and aimed at improving selectivity over EGFRWT, a scaffold hopping of the 6,7-dioxo-6,7-dihydropteridine core was conducted. The piperazinediones part of compound 9 was replaced with 1,3-oxazinan-2-one in order to introduce one or two hydrophobic substituents at the C4-position, and thus it could not only occupy the lipophilic subpocket formed by the gatekeeper residue but also strengthen nonpolar contacts with Met790 residue (Fig. 2C and D). This computer-aided approach resulted in compound 16a. As expected, the superposition of compound 9 and compound 16a demonstrates that the C4-position methyl group is in van der Waals contacts (shown in little green disc, distance of 3.3 Å) with the side chain of Met790 residue in the case of the EGFRT790M while for the EGFRWT the corresponding distance is much closer (shown in large red disc, distance of 2.5 Å), resulting in a steric clash between the N5-methyl group and side chain of Thr790 residue. These different interactions between N5-position moiety and the gatekeeper residue would affect the potency and selectivity.
Chemistry
The synthesis of compounds 16a-e is outlined in Fig. 3. In summary, treatment of the ethyl 2,4-dichloropyrimidine-5-carboxylate with Boc-protected amine yielded the ethyl4-((3-((tert-butoxycarbonyl)amino)phenyl)amino)-2-chloropyrimidine-5-carboxylate (10), followed by reduction of the ethyl ester group and oxidization of the hydroxymethyl to obtain the key intermediate (12). A reaction of 12 with Grignard reagents generated compounds 13a-d. Then compounds 11, 13a-d were cyclized with 1,1′-carbonyldiimidazole to obtain the ring-closure compounds (14a-e), which were reacted with arylamine by displacement of 2-chloro in the pyrimidine to yield compounds 15a-e. Subsequently, removal of the protecting group with trifluoroacetic acid in dichloromethane and reacted with acryloyl chloride to provide the desired compounds 16a-e.

Synthesis of the Target Compounds 16a-e a. aReagents and conditions: (a) tert-butyl(3-aminophenyl)carbamate, N,N-diisopropylethylamine (DIPEA), acetonitrile (CH3CN), reflux, 6 h, 86%; (b) LiAlH4, dry tetrahydrofuran (THF), 0 °C, 4 h, 15%; (c) MnO2, CH2Cl2, r.t., overnight, 82%; (d) RBrMg, dry THF, 0 °C, 5–8 h, 74–82%; (e) 1,1′-carbonyldiimidazole, K2CO3, dry CH3CN, r.t., overnight, 69–75%; (f) arylamine, trifluoroacetic acid, isopropanol, reflux, overnight, 31–39%; (g) trifluoroacetic acid, CH2Cl2, r.t., 5 h, 77–83%; (h) acrylyl chloride, DIPEA, CH2Cl2, 0 °C to r.t., overnight, 35–49%.
Synthesis of compounds 20a-c is shown in Fig. 4. Treatment of compound 10 with appropriate Grignard reagents afforded the corresponding intermediates 17a-c, which were performed by a procedure analogous to that for synthesis of compound 16a to give the desired compounds 20a-c.

Synthesis of the target compounds 20a-c a. aReagents and conditions: (a) RBrMg, dry THF, 0 °C, 8–10 h, 33–48%; (b) 1,1′-carbonyldiimidazole, K2CO3, dry CH3CN, r.t., overnight, 65–85%; (c) arylamine, trifluoroacetic acid, isopropanol, reflux, overnight, 52–61%; (d) trifluoroacetic acid, CH2Cl2, r.t., 5 h, 78–83%; (e) acrylyl chloride, DIPEA, CH2Cl2, 0 °C to r.t., overnight, 45–51%.
In Vitro Structure-activity Relationship (SAR) and Structural Modification
Initially, a series of 1,4-dihydro-2H-pyrimido[4,5-d][1,3]oxazin-2-one derivatives (16a-d) that contain a single hydrophobic group (methyl, ethyl, propyl and isopropyl) at the C4-position of the scaffold were designed and synthesized (Fig. 3). Their in vitro enzymatic inhibitory activities against EGFRL858R/T790M and EGFRWT were evaluated by using the well-established ELISA-based assay. As shown in Fig. 5, compounds 16a and 16b indeed demonstrated different inhibitory activities for dual-mutant (DM) and WT EGFR kinases. They displayed single nanomolar inhibitory activities for EGFRL858R/T790M with IC50 values of 5.4 and 6.1 nM, respectively, while their inhibition for EGFRWT were ~4–7-fold less potent. Introduction of propyl and isopropyl groups in the 4-position of the core led to compounds 16c and 16d, which showed decreased potency for EGFR kinases and significant loss in selectivity profiles between EGFRL858R/T790M and EGFRWT (Fig. 5). The bioactivities of 16c and 16d indicated that this hydrophobic subpocket is unable to accommodate these two longer and bulkier alkyl groups, thus resulting in detrimental influence on potency and selectivity. To validate the key contribution of the introduced alkyl groups for EGFR kinases selectivity, compound 16e, a C4-unsubstituted analogue, was also prepared (Fig. 3). Compared to compound 16a, compound 16e displayed not only less potent inhibition effect for EGFRL858R/T790M (IC50 = 7.3 nM), but also slight improvement in inhibitory activity for EGFRWT (IC50 = 24 nM), leading to a 3.3-fold decrease in its selectivity between the DM and WT EGFRs. These results therefore showed that the small hydrophobic C4-substitutent, such as methyl group, occupying the lipophilic subpocket formed by the mutant gatekeeper residue was favorable to inhibitor of DM EGFR with selectivity over WT EGFR.

In Vitro Enzyme Inhibitory Activities and Cellular Antiproliferative Activities of Compounds 16a-e, 20a-ca. aKinase activities were determined through using the ELISA-based EGFR-TK assay. In vitro antiproliferation activities of the compounds were examined through using the sulforhodamine B (SRB) colourimetric assay. IC50 values are averages of at least three independent determinations and reported as the means ± standard deviations (SD). bDual-mutant (EGFRL858R/T790M). cNot determined.
On the basis of above single-substituent structural modification, we hypothesized that introducing one more suitable hydrophobic moiety at C4-position to further strengthen the nonpolar contacts with mutant gatekeeper Met790 may be beneficial for improving the mutant selectivity profile. So we applied this rationale to compound 16a through attaching a dual-alkyl group at the C4-position, and resulted in compounds 20a-c (Fig. 4). As expected, compound 20a, dual-methyl groups substituted derivative at C4-position of the 1,4-dihydro-2H-pyrimido[4,5-d][1,3]oxazin-2-one, inhibited EGFRL858R/T790M with an IC50 value of 4.5 nM, which was similar to that of compound 16a. Whereas its inhibition of WT EGFR was less potent (IC50 = 156 nM). Thus, it showed approximate 35-fold selectivity between EGFRL858R/T790M and EGFRWT, and was highly comparable to the positive control compound 7. The structure-based insights from molecular modeling are consistent with the experimental observations. As shown in the Fig. 2E, compound 20a was covalently docked into the ATP-binding site of EGFRT790M and the hinge binder part is predicted to tightly interacts with the backbone of Met793 (two H-bonds distance: 3.1 and 3.1 Å). Its upper methyl group fits the methionine gatekeeper pocket well and its lower methyl group is involved in van der Waals contacts with Met790. In the case of the proposed binding mode of EGFRWT (Fig. 2F), the branched residue Thr790 leads a steric clash to the dual methyl moiety (Fig. 2C and D) and pushed the core of compound 20a to the right side of the ATP-binding site, which attenuates the hydrogen-bonding with the hinge region (two H-bonds distance: 3.3 and 3.9 Å). These comparable docking studies also validated this selectivity profile. However, when introducing bulkier substitutions, such as dual-ethyl and dual-propyl groups at the C4-position of the core, the compounds (20b and 20c) led to a complete loss of inhibitory activities (>1000 nM) for both DM and WT EGFR kinases. These results implied that longer and bulkier substituents than dual-methyl groups are not tolerated to the hydrophobic subpocket.
In Vitro Cellular Anti-proliferation Assay
Having explored potent inhibition and obvious selectivity of EGFR kinases by introduction of alkyl groups to C4-position to the 1,4-dihydro-2H-pyrimido[4,5-d][1,3]oxazin-2-one ring, we subsequently evaluated their anti-proliferative activities in H1975 cells (expressing EGFRL858R/T790M) and A431 cells (expressing EGFRWT) in vitro by using the sulforhodamine B (SRB) colourimetric assay. The anti-proliferative activities of our designed compounds against H1975 and A431 cell lines range from 135 nM to more than 9 μM and from 554 nM to more than 7 μM, respectively (Fig. 5). Additionally, the selectivity at the cellular level is from 0.7 to 9.4-fold. Importantly, compound 20a exhibited potent growth-suppressing activities against the H1975 cell lines with IC50 values of 135 nM, and achieved high selectivity over A431 cell lines (9.4-fold, IC50 = 1274 nM), which is comparable with the positive control compound 7 (15-fold). Therefore, compound 20a, which shows best potency at both enzymatic and cellular levels, was selected as the representative compound evaluated in the following pharmacology assays.
Western Blot Analysis
In order to further validate the observed selectivity of enzymatic and cellular inhibition, we selected the representative compound 20a to examine its suppressive functions on the activation of EGFR and the downstream signaling proteins in H1975 and A431 cell lines. As illustrated in Fig. 6A, compound 20a inhibited the phosphorylation of EGFRL858R/T790M and AKT in H1975 cells at the concentration as low as 10 nM, and inhibited downstream molecule ERK phosphorylation at the concentration up to 100 nM, demonstrating similar activity with compound 8 (positive control). Meanwhile, its inhibitory activity against A431 cell lines harboring EGFRWT was obviously less potent (Fig. 6B). For example, compound 20a significantly suppressed the activation of EGFRL858R/T790M in H1975 cells at 10 nM, but it did not show obvious effect until reaching high concentration at 1 μM in A431 cells. The results demonstrated that 20a strongly inhibited EGFRL858R/T790M while EGFRWT sparing at the cellular level, further suggesting that 20a was a potent and selective EGFRL858R/T790M inhibitor.

Compound 20a inhibits phosphorylation of EGFR and downstream signaling substrates in (A) H1975 cell line harboring EGFRL858R/T790M, while less potent in (B) A431 cells harboring EGFRWT. Cells were starved in serum-free medium for 24 h before incubation of indicated concentrations of compounds 20a and 8 (positive control) and harvested after stimulation by EGF (50 ng/mL) for 10 min. Phosphorylation of EGFR and downstream signaling substrates was examined by Western Blot analysis. Blot source data is provided in Supplementary Fig. S1.
In Vivo Antitumor Efficacy Study of Compound 20a
In view of its encouraging ability of inhibition for EGFR kinases and cells, we also evaluated the preliminary in vivo antitumor activity of compound 20a in H1975 xenograft mouse model driven by EGFRL858R/T790M (Fig. 7). Compound 20a and positive control compound 7 were administered orally once per day at a dosage of 50 mg/kg for continuous two weeks (14 days). As shown in Fig. 7A, compound 20a (tumor growth inhibition (TGI) = 71%, P < 0.05) potently suppressed the proliferation and growth of the tumor, which is comparable with that of compound 7 (TGI = 72%, P < 0.05). Furthermore, the dosage of compound 20a was well tolerated, with no obvious effect on the body weight observed during the treatment period (Fig. 7B).

Preliminary in vivo antitumor efficacy of compound 20a against H1975 NSCLC xenograft mouse model. H1975 cells were implanted as described in Methods. Compounds 20a and 7 were administered orally at a concentration of 50 mg/kg/day for consecutive 14 days starting from day 0. H1975 xenograft mouse model tumor volumes (A) and body weights (B) were recorded every two or three days. All values represent mean ± SEM. The tumor growth inhibition (TGI, %) was measured on the final day of the treatment (drug-treated group versus the vehicle control), where the asterisk ∗ represents P < 0.05 (Student’s t test) compared to the vehicle group.
Discussion
Herein, we combined the ligand-based scaffold hopping with the structure-based analysis of differences of the binding pockets between EGFRL858R/T790M and EGFRWT to design a new 1,4-dihydro-2H-pyrimido[4,5-d][1,3]oxazin-2-one scaffold targeting EGFRL858R/T790M and improve selectivity over EGFRWT. The scaffold hopping method in silico helps us modify more than one hydrophobic substituent at C4-position. Compared with the compound 16e (no substitution at C4), the single methyl group substituent on the compound 16a occupies hydrophobic subpocket, leading to a slightly 1.4-fold increase of the enzymatic inhibition against EGFRL858R/T790M (7.3 nM vs 5.4 nM). With the slight potency decline against EGFRWT (16e vs 16a, 24 nM vs 38 nM), the selectivity against EGFRL858R/T790M over EGFRWT boost 2.1-fold (16e vs 16a, 3.3-fold vs 7-fold). In the second round dual-substituent optimization, the one more methyl group at C4-position stabilized the nonpolar interaction with gatekeeper residue Met790 (Fig. 2E), keeping the enzymatic inhibitory activities against EGFRL858R/T790M at the same level (16a vs 20a, 5.4 nM vs 4.5 nM). Besides, dual-methyl group introduces more steric clashes into the interaction between the inhibitor and the EGFRWT (Fig. 2F), leading to a pronouncing decrease of enzymatic inhibition against EGFRWT (16a vs 20a, 38 nM vs 156 nM). Thus, the representative compound 20a achieved the best selectivity (35-fold) in this series, which is comparable with the compound 7 (29-fold). The selectivity improvements achieved along the whole structural optimization verified the rationality of our design strategy.
According to the results from enzymatic and cellular inhibition assays and western blot analysis, this series of compounds present good targeting properties. In particular, the antiproliferation potencies against H1975 and A431 cell lines are highly correlated with the enzymatic inhibitory activities against EGFRL858R/T790M and EGFRWT, respectively (Fig. 8). The western blot analysis of representative compound 20a validated the selectivity profile observed both in the enzymatic and cellular assays (Fig. 6). Moreover, the in vivo efficacy for double-mutant tumor xenografts of 20a is highly comparable with that of the drug at clinical trials (compound 7). These good targeting and selective properties of representative compound may imply the potential clinical use for the treatment of the drug-resistance NSCLC and less side-effects caused by the EGFR inhibitor without mutant selectivity.

(A) Linear relationship between pIC50s against EGFRWT and pIC50s against A431 cell lines. (B) Linear relationship between pIC50s against EGFRDM and pIC50s against H1975 cell lines.
In summary, through structure-guided design approach based on our previous work on mutant-selective EGFR inhibitor (compound 9), we have designed and generated a novel 1,4-dihydro-2H-pyrimido[4,5-d][1,3]oxazin-2-one scaffold, followed by introduction of alkyl groups contacting the Met790 gatekeeper residue into the C4-position to keep the selectivity for the EGFRL858R/T790M over EGFRWT, leading to discovery of one of the most potent compound 20a. 20a not only showed remarkable selectivity of enzyme inhibition for EGFRL858R/T790M over EGFRWT, but also strongly inhibited the proliferation of H1975 cells, while significantly less effective on A431 cell lines. A further preliminary in vivo antitumor efficacy evaluation on 20a suggested that it significantly inhibited tumor growth in H1975 NSCLC xenograft mouse model via po dosing at 50 mg/kg/day for 14 days. Considering the encouraging enzymatic and cellular selectivity and in vivo antitumor efficiency of compound 20a, this agent might be a promising lead compound for further optimization as a selective EGFRL858R/T790M inhibitor to overcome T790M resistance mutation. The results also provide more insights for designing new classes of mutant-selective EGFR inhibitors. Further pre-clinical evaluations for the candidate compound are in progress and will be performed in due course.
Materials and Methods
Molecular Modeling
The X-ray structures (PDB code: 3IKA and 4G5J) were downloaded from the Protein Data Bank (PDB, http://www.pdb.org). The comparative docking study was conducted via three steps: First, compounds 9 and 20a were docked into the EGFRT790M (PDB code: 3IKA) and EGFRWT (PDB code: 4G5J) as reversible inhibitors using Glide23 (version 5.5) in extra precision (XP) mode with default settings; Then, the covalent bond between the Cys797 and the electrophilic group was formed by the build panel in Maestro24; Finally, to fix the bond lengths and eliminate steric clashed, the covalently modified inhibitor-kinase complexes were minimized in water solvent using MacroModel application with flexible ligand and constrained protein.
The scaffold hopping was conducted by SHAFTS25, 26. The query molecule is the docked pose of compound 9 obtained from the above docking study. The aligned pose of compound 16a (shown in Fig. 2C) was generated by fast 3D similarity calculation of SHAFTS. The score for the similarity calculation between compound 9 and 16a is 1.494. The range of the SHATFS’ similarity score is from 0 (totally dissimilar) to 2 (completely similar). The Fig. 2D was generated by LigPlot+27.
Reagents and General Methods
All chemical reagents and solvents were purchased from commercial sources, and used without further purification. The positive control compound 7 was purchased from Shanghai Future Chemical Technology Co., Ltd. Thin-layer chromatography (TLC) was used to monitor the progress of reactions. Column chromatography was performed using 300−400 mesh silica gel (Qingdao Haiyang Chemical Co.,Ltd). 1H and 13C NMR spectra were recorded on a Bruker AV-400 spectrometer at 400 MHz and 100 MHz, respectively. LC-MS and high-resolution mass spectra (HRMS) were measured at the Institute of Fine Chemistry of East China University of Science and Technology. The purity of all final compounds were analyzed by using HPLC (Hewlett-Packard 1100), with the purity of all compounds to be over than 95% (>95%). HPLC instrument: (column: Zorbax RP-18, 5 μm, 4.6 mm × 250 mm, reverse phase; detector: photodiode array detector). The mobile phases A and B were acetonitrile and 10 mM NH4OAc in water (pH 6.0), respectively. A gradient of 5−100% the mobile phase A was run at a flow rate of 1.0 mL/min.
Ethyl-4-((3-((tert-butoxycarbonyl)amino)phenyl)amino)-2-chloropyrimidine-5-carboxy late (10)
A mixture of ethyl-2,4-dichloropyrimidine-5-carboxylate (22.100 g, 100 mmol), 3-(tert-Butoxycarbonylamino)aniline (20.800 g, 100 mmol) and DIPEA (12.900 g, 100 mmol) in CH3CN (220 mL) was heated with reflux for 6 h. After cooling to room temperature, the white solid was collected by filtration and washed with CH3CN to give the product 10 (33.719 g, 86%). 1H NMR (400 MHz, DMSO-d 6) δ 10.23 (s, 1H), 9.50 (s, 1H), 8.80 (s, 1H), 7.70 (s, 1H), 7.35 (d, J = 7.6 Hz, 1H), 7.29 (t, J = 8.0 Hz, 1H), 7.24 (d, J = 8.4 Hz, 1H), 4.39 (q, J = 7.2 Hz, 2H), 1.49 (s, 9H), 1.36 (t, J = 7.2 Hz, 3H). LC-MS: m/z: 393.1 (M+H)+.
tert-Butyl(3-((2-Chloro-5-(hydroxymethyl)pyrimidin-4-yl)amino)phenyl)carbamate (11)
To a solution of compound 10 (31.360 g, 80 mmol) in dry THF (150 mL) was dropwise added LiAlH4 (2.5 M in THF, 80 mL) at 0 °C. The reaction mixture was stirred for 4 h at 0 °C and was then treated with saturated aqueous NH4Cl. The solid was filtered off, and the filtrate was extracted with ethyl acetate (3 × 100 mL). The organic layer was washed with saturated aqueous NaCl, dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by silica gel chromatography (petroleum ether/ethyl acetate = 2:1, v/v) to give the product 11 (4.209 g, 15%). 1H NMR (400 MHz, CDCl3) δ 8.38 (s, 1H), 7.87 (s, 1H), 7.70 (s, 1H), 7.34 (d, J = 8.4 Hz, 1H), 7.25 (t, J = 8.0 Hz, 1H), 7.05 (d, J = 8.0 Hz, 1H), 6.66 (s, 1H), 4.65 (s, 2H), 1.52 (s, 9H). LC-MS: m/z: 351.1 (M+H)+.
tert-Butyl(3-((2-Chloro-5-formylpyrimidin-4-yl)amino)phenyl)carbamate (12)
To a solution of compound 11 (3.500 g, 10 mmol) in dichloromethane (30 mL) was added activated manganese (IV) oxide (58%, 15.000 g, 100 mmol). The mixture was stirred overnight at room temperature. The solid was filtered off through Celite, and the filtrate was concentrated under reduced pressure. The resulting crude product was purified by silica gel chromatography (petroleum ether/ethyl acetate = 4:1, v/v) to give the product 12 (2.850 g, 82%). 1H NMR (400 MHz, CDCl3) δ 10.60 (s, 1H), 9.89 (s, 1H), 8.56 (s, 1H), 7.82 (t, J = 2.0 Hz, 1H), 7.39 (d, J = 8.8 Hz, 1H), 7.31 (t, J = 8.0 Hz, 1H), 7.20 (d, J = 8.0 Hz, 1H), 6.58 (s, 1H), 1.53 (s, 9H). LC-MS: m/z: 349.1 (M+H)+.
tert-Butyl(3-((2-Chloro-5-(1-hydroxyethyl)pyrimidin-4-yl)amino)phenyl)carbamate (13a)
To a solution of 12 (1.044 g, 3 mmol) in dry THF (15 mL) was dropwise added a solution of methylmagnesium bromide (1 M in THF, 9 mL) under the condition of argon gas at 0 °C. The mixture was stirred for 5 h at 0 °C, and was then quenched with a saturated aqueous NH4Cl solution and extracted with ethyl acetate (80 mL). The organic layer was washed with saturated aqueous NaCl, dried over anhydrous Na2SO4 and evaporated to obtain a residue. Purification by silica gel chromatography (petroleum ether/ethyl acetate = 2:1, v/v) to give the product 13a (0.896 g, 82%). 1H NMR (400 MHz, CDCl3) δ 8.83 (s, 1H), 7.77 (s, 1H), 7.70 (s, 1H), 7.29 (d, J = 8.4 Hz, 1H), 7.05 (d, J = 8.0 Hz, 1H), 6.67 (s, 1H), 4.87 (q, J = 6.4 Hz, 1H), 1.55 (d, J = 6.4 Hz, 3H), 1.52 (s, 9H). LC-MS: m/z: 365.1 (M+H)+.
tert-Butyl(3-(7-Chloro-4-methyl-2-oxo-2H-pyrimido[4,5-d][1,3]oxazin-1(4H)-yl)phenyl) carbamate (14a)
To a mixture of 13a (0.815 g, 2.2 mmol), potassium carbonate (0.455 g, 3.3 mmol) in dry CH3CN (10 mL) was added 1,1′-carbonyldiimidazole (1.069 g, 6.6 mmol). The reaction mixture was stirred overnight at room temperature. The reaction mixture was poured into water, and extracted with ethyl acetate. The organic layer was washed with saturated aqueous NaCl, dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by silica gel chromatography (petroleum ether/ethylacetate = 2:1, v/v) to give the product 14a as a white solid (0.642 g, 75%). 1H NMR (400 MHz, DMSO-d 6) δ 9.59 (s, 1H), 8.54 (s, 1H), 7.61 (s, 1H), 7.41–7.36 (m, 2H), 6.98 (d, J = 6.8 Hz, 1H), 5.86 (q, J = 6.4 Hz, 1H), 1.74 (d, J = 6.4 Hz, 3H), 1.47 (s, 9H). LC-MS: m/z: 391.1 (M+H)+.
tert-Butyl(3-(7-((2-Methoxy-4-(4-methylpiperazin-1-yl)phenyl)amino)-4-methyl-2-oxo-2H-pyrimido[4,5-d][1,3]oxazin-1(4H)-yl)phenyl)carbamate (15a)
To a mixture of 14a (0.737 g, 1.89 mmol), 2-methoxy-4-(4-methylpiperazin-1-yl)aniline (0.502 g, 2.27 mmol) in isopropanol (15 mL) was added trifluoroacetic acid (210 μL, 2.84 mmol). The reaction solution was heated with reflux for 10 h. After cooling to room temperature, the mixture was neutralized with saturated aqueous NaHCO3, and extracted with CH2Cl2. The organic layer was washed with saturated aqueous NaCl, dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by silica gel chromatography (ethylacetate/methanol = 25:1, v/v) to give the product 15a (0.403 g, 37%). 1H NMR (400 MHz, CDCl3) δ 8.46 (s, 1H), 8.18 (d, J = 8.8 Hz, 1H), 7.79 (s, 1H), 7.74 (s, 1H), 7.24–7.20 (m, 3H), 6.69 (s, 1H), 6.54 (d, J = 2.4 Hz, 1H), 6.50 (dd, J = 8.8 Hz, J = 2.4 Hz, 1H), 4.52 (q, J = 6.4 Hz, 1H), 3.85 (s, 3H), 3.17 (t, J = 4.8 Hz, 4H), 2.60 (t, J = 4.8 Hz, 4H), 2.36 (s, 3H), 1.51 (s, 9H), 1.49 (d, J = 6.4 Hz, 3H). LC-MS: m/z: 576.3 (M+H)+.
N-(3-(7-((2-Methoxy-4-(4-methylpiperazin-1-yl)phenyl)amino)-4-methyl-2-oxo-2H-pyrimido[4,5-d][1,3]oxazin-1(4H)-yl)phenyl)acrylamide (16a)
To a solution of 15a (0.277 g, 0.48 mmol) in dichloromethane (5 mL) was added trifluoroacetic acid (1 mL). The mixture was stirred for 5 h at room temperature. The reaction solution was neutralized with saturated aqueous NaHCO3, and extracted with CH2Cl2. The organic layer was washed with saturated aqueous NaCl, dried over anhydrous Na2SO4 and concentrated in vacuo to obtain a white solid (0.187 g, 82%), which was used in the next reaction without further purification.
To a solution of the above obtained compound (0.187 g, 0.39 mmol), DIPEA (0.076 g, 0.59 mmol) in CH2Cl2 (5 mL) was added acryloyl chloride (42 μL, 0.51 mmol) dissolved in CH2Cl2 (1 mL) at 0 °C. The reaction mixture was stirred overnight at room temperature. The mixture was partitioned between dichloromethane (50 mL) and saturated aqueous NaHCO3 (5 mL). The organic layer was washed with saturated aqueous NaCl, dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by silica gel chromatography (dichloromethane/methanol = 20:1, v/v) to give the product 16a (0.075 g, 36%). 1H NMR (400 MHz, DMSO-d 6) δ 10.36 (s, 1H), 8.22 (s, 1H), 7.84 (d, J = 8.0 Hz, 1H), 7.77 (s, 1H), 7.67 (s, 1H), 7.47 (t, J = 8.0 Hz, 1H), 7.25 (d, J = 8.4 Hz, 1H), 7.10 (d, J = 7.6 Hz, 1H), 6.52–6.50 (m, 1H), 6.44 (dd, J = 16.8 Hz, J = 10.0 Hz, 1H), 6.26 (dd, J = 16.8 Hz, J = 1.6 Hz, 1H), 6.08–6.06 (m, 1H), 5.77 (dd, J = 10.4 Hz, J = 1.6 Hz, 1H), 5.72 (q, J = 6.4 Hz, 1H), 3.75 (s, 3H), 3.03–3.01 (m, 4H), 2.44–2.42 (m, 4H), 2.22 (s, 3H), 1.69 (d, J = 6.4 Hz, 3H). 13C NMR (100 MHz, DMSO-d 6) δ 163.21, 159.03, 156.44, 153.15, 150.51, 139.69, 136.47, 131.68, 129.28, 127.11, 124.36, 120.18, 118.96, 106.32, 106.01, 99.73, 72.39, 55.57, 54.58, 48.61, 45.70, 19.65. HRMS(ESI) (m/z): (M+H)+ calcd for C28H32N7O4 530.2516, found, 530.2512. HPLC purity: 98.48%, retention time = 8.71 min.
The following compounds 16b-e were prepared by a method similar to that for the synthesis of compound 16a.
N-(3-(4-Ethyl-7-((2-methoxy-4-(4-methylpiperazin-1-yl)phenyl)amino)-2-oxo-2H-pyrimido[4,5-d][1,3]oxazin-1(4H)-yl)phenyl)acrylamide (16b)
1H NMR (400 MHz, DMSO-d 6) δ 10.46 (s, 1H), 8.22 (s, 1H), 7.85 (d, J = 8.4 Hz, 1H), 7.81 (s, 1H), 7.66 (s, 1H), 7.47 (t, J = 8.0 Hz, 1H), 7.26 (d, J = 8.4 Hz, 1H), 7.09 (d, J = 8.0 Hz, 1H), 6.55 (d, J = 2.0 Hz, 1H), 6.48 (dd, J = 16.8 Hz, J = 6.8 Hz, 1H), 6.27 (dd, J = 16.8 Hz, J = 1.6 Hz, 1H), 6.10–6.09 (m, 1H), 5.77 (dd, J = 10.0 Hz, J = 1.6 Hz, 1H), 5.55 (t, J = 6.8 Hz, 1H), 3.76 (s, 3H), 3.19 (t, J = 4.4 Hz, 4H), 2.92 (t, J = 4.4 Hz, 4H), 2.56 (s, 3H), 2.09–1.95 (m, 2H), 1.03 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, DMSO-d 6) δ 163.29, 156.45, 153.58, 150.39, 139.79, 136.41, 131.76, 129.26, 127.06, 124.28, 120.74, 120.08, 119.02, 106.62, 104.38, 100.11, 76.93, 55.65, 53.18, 47.17, 27.36, 8.50. HRMS(ESI) (m/z): (M+H)+ calcd for C29H34N7O4 544.2672, found, 544.2654. HPLC purity: 99.34%, retention time = 9.37 min.
N-(3-(7-((2-Methoxy-4-(4-methylpiperazin-1-yl)phenyl)amino)-2-oxo-4-propyl-2H-pyrimido[4,5-d][1,3]oxazin-1(4H)-yl)phenyl)acrylamide (16c)
1H NMR (400 MHz, DMSO-d 6) δ 10.45 (s, 1H), 8.22 (s, 1H), 7.85 (d, J = 8.0 Hz, 1H), 7.80 (s, 1H), 7.66 (s, 1H), 7.47 (t, J = 8.0 Hz, 1H), 7.25 (d, J = 8.4 Hz, 1H), 7.08 (d, J = 8.0 Hz, 1H), 6.54 (d, J = 2.0 Hz, 1H), 6.48 (dd, J = 16.8 Hz, J = 10.0 Hz, 1H), 6.26 (dd, J = 16.8 Hz, J = 1.6 Hz, 1H), 6.10–6.09 (m, 1H), 5.77 (dd, J = 9.6 Hz, J = 1.6 Hz, 1H), 5.60 (t, J = 7.2 Hz, 1H), 3.76 (s, 3H), 3.14 (t, J = 4.4 Hz, 4H), 2.79 (t, J = 4.4 Hz, 4H), 2.47(s, 3H), 2.00–1.92 (m, 2H), 1.56–1.44 (m, 2H), 0.99 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, DMSO-d 6) δ 163.27, 156.39, 153.45, 150.36, 139.78, 136.42, 131.75, 129.26, 127.04, 124.29, 120.60, 120.10, 119.01, 106.55, 104.77, 100.01, 75.68, 55.63, 53.53, 47.54, 36.18, 17.23, 13.55. HRMS(ESI) (m/z): (M+H)+ calcd for C30H36N7O4 558.2829, found, 558.2836. HPLC purity: 97.05%, retention time = 11.98 min.
N-(3-(7-((2-Methoxy-4-(4-methylpiperazin-1-yl)phenyl)amino)-2-oxo-4-isopropyl-2H-pyrimido[4,5-d][1,3]oxazin-1(4H)-yl)phenyl)acrylamide (16d)
1H NMR (400 MHz, DMSO-d 6) δ 10.52 (s, 1H), 8.22 (s, 1H), 7.87 (d, J = 8.0 Hz, 1H), 7.85 (s, 1H), 7.64 (s, 1H), 7.47 (t, J = 8.0 Hz, 1H), 7.26 (d, J = 7.6 Hz, 1H), 7.05 (d, J = 7.6 Hz, 1H), 6.57 (d, J = 1.6 Hz, 1H), 6.50 (dd, J = 16.8 Hz, J = 10.0 Hz, 1H), 6.26 (dd, J = 16.8 Hz, J = 1.6 Hz, 1H), 6.14–6.10 (m, 1H), 5.77 (dd, J = 10.0 Hz, J = 1.6 Hz, 1H), 5.40 (d, J = 4.8 Hz, 1H), 3.77 (s, 3H), 3.18 (t, J = 4.4 Hz, 4H), 2.74 (s, 3H), 2.27–2.22 (m, 1H), 1.04 (d, J = 6.8 Hz, 3H), 0.99 (d, J = 6.8 Hz, 3H). 13C NMR (100 MHz, DMSO-d 6) δ 163.33, 156.37, 154.32, 150.33, 139.87, 136.31, 131.77, 129.29, 127.08, 124.14, 120.98, 119.88, 119.03, 106.77, 103.07, 100.29, 80.57, 55.70, 52.39, 46.36, 33.25, 17.87, 15.96. HRMS(ESI) (m/z): (M+H)+ calcd for C30H36N7O4 558.2829, found, 558.2831. HPLC purity: 99.04%, retention time = 11.62 min.
N-(3-(7-((2-Methoxy-4-(4-methylpiperazin-1-yl)phenyl)amino)-2-oxo-2H-pyrimido[4,5-d][1,3]oxazin-1(4H)-yl)phenyl)acrylamide (16e)
1H NMR (400 MHz, CDCl3+CD3OD) δ 8.09 (s, 1H), 7.89 (s, 1H), 7.75 (d, J = 8.4 Hz, 1H), 7.47 (t, J = 8.0 Hz, 1H), 7.42–7.39 (m, 1H), 7.07 (d, J = 7.6 Hz, 1H), 6.42–6.38 (m, 2H), 6.15 (d, J = 7.2 Hz, 1H), 5.71 (dd, J = 9.2 Hz, J = 2.8 Hz, 1H), 5.36 (s, 2H), 3.80 (s, 3H), 3.37 (t, J = 4.8 Hz, 4H), 3.24 (t, J = 4.8 Hz, 4H), 2.82 (s, 3H). 13C NMR (100 MHz, CDCl3+CD3OD) δ 161.37, 156.64, 155.73, 149.09, 143.64, 140.07, 135.12, 135.07, 133.55, 131.59, 128.51, 126.93, 124.71, 124.63, 124.02, 112.78, 105.19, 105.14, 69.18, 59.60, 57.68, 51.93, 47.34. HRMS(ESI) (m/z): (M+H)+ calcd for C27H30N7O4 516.2359, found, 516.2364. HPLC purity: 95.07%, retention time = 10.60 min.
tert-Butyl(3-((2-Chloro-5-(2-hydroxypropan-2-yl)pyrimidin-4-yl)amino)phenyl)carbama te (17a)
To a solution of compound 10 (3.920 g, 10 mmol) in dry THF (25 mL) was dropwise added a solution of methylmagnesium bromide (1 M in THF, 30 mL) under the condition of argon gas at 0 °C. The mixture was stirred for 8 h at 0 °C, and was then quenched with a saturated aqueous NH4Cl solution and extracted with ethyl acetate (100 mL). The organic layer was washed with saturated aqueous NaCl, dried over anhydrous Na2SO4 and evaporated to obtain a residue. Purification by silica gel chromatography (petroleum ether/ethyl acetate = 2.5:1, v/v) to give the product 17a (1.811 g, 48%). 1H NMR (400 MHz, DMSO-d 6) δ 10.01 (s, 1H), 9.41 (s, 1H), 8.12 (s, 1H), 7.62 (t, J = 2.0 Hz, 1H), 7.40 (dd, J = 8.0 Hz, J = 1.2 Hz, 1H), 7.25 (t, J = 8.4 Hz, 1H), 7.13 (d, J = 8.8 Hz, 1H), 6.43 (s, 1H), 1.56 (s, 6H), 1.48 (s, 9H). LC-MS: m/z: 379.1 (M+H)+.
tert-Butyl(3-(7-Chloro-4,4-dimethyl-2-oxo-2H-pyrimido[4,5-d][1,3]oxazin-1(4H)-yl)phen yl)carbamate (18a)
To a mixture of 17a (1.512 g, 4 mmol), potassium carbonate (0.828 g, 6 mmol) in dry CH3CN (20 mL) was added 1,1′-carbonyldiimidazole (1.296 g, 8 mmol). The reaction mixture was stirred overnight at room temperature. The reaction mixture was poured into water, and extracted with ethyl acetate (3 × 25 mL). The organic layer was washed with saturated aqueous NaCl, dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by silica gel chromatography (petroleum ether/ethylacetate = 2.5:1, v/v) to give the product 18a (1.049 g, 65%). 1H NMR (400 MHz, DMSO-d 6) δ 9.57 (s, 1H), 8.64 (s, 1H), 7.57 (s, 1H), 7.41–7.36 (m, 2H), 7.01–6.99 (m, 1H), 1.79 (s, 6H), 1.47 (s, 9H). LC-MS: m/z: 405.1 (M+H)+.
tert-Butyl(3-(7-((2-Methoxy-4-(4-methylpiperazin-1-yl)phenyl)amino)-4,4-dimethyl-2-oxo-2H-pyrimido[4,5-d][1,3]oxazin-1(4H)-yl)phenyl)carbamate (19a)
To a mixture of 18a (1.010 g, 2.5 mmol), 2-methoxy-4-(4-methylpiperazin-1-yl)aniline (0.663 g, 3 mmol) in isopropanol (25 mL) was added trifluoroacetic acid (280 μL, 3.77 mmol). The reaction solution was heated with reflux for 10 h. After cooling to room temperature, the mixture was neutralized with saturated aqueous NaHCO3, and extracted with CH2Cl2 (3 × 40 mL). The organic layer was washed with saturated aqueous NaCl, dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by silica gel chromatography (ethylacetate/methanol = 25:1, v/v) to give the product 19a (0.867 g, 59%). 1H NMR (400 MHz, CDCl3) δ 8.07 (s, 1H), 7.51 (d, J = 8.4 Hz, 2H), 7.47–7.43 (m, 2H), 7.01 (d, J = 7.2 Hz, 1H), 6.45 (s, 1H), 6.44 (d, J = 2.4 Hz, 1H), 6.18–6.16 (m, 1H), 3.82 (s, 3H), 3.16 (t, J = 4.4 Hz, 4H), 2.67 (t, J = 4.4 Hz, 4H), 2.42 (s, 3H), 1.80 (s, 6H), 1.49 (s, 9H). LC-MS: m/z: 590.4 (M+H)+.
N-(3-(7-((2-Methoxy-4-(4-methylpiperazin-1-yl)phenyl)amino)-4,4-dimethyl-2-oxo-2H-pyrimido[4,5-d][1,3]oxazin-1(4H)-yl)phenyl)acrylamide (20a)
To a solution of 19a (0.500 g, 0.85 mmol) in dichloromethane (10 mL) was added trifluoroacetic acid (2 mL). The mixture was stirred for 5 h at room temperature. The reaction solution was neutralized with saturated aqueous NaHCO3, and extracted with dichloromethane (3 × 30 mL). The organic layer was washed with saturated aqueous NaCl, dried over anhydrous Na2SO4 and concentrated in vacuo to obtain a white solid (0.335 g, 81%), which was used in the next reaction without further purification.
To a solution of the above obtained compound (0.335 g, 0.68 mmol), DIPEA (0.131 g, 1.02 mmol) in CH2Cl2 (5 mL) was added acryloyl chloride (72 μL, 0.88 mmol) dissolved in CH2Cl2 (1 mL) at 0 °C. The reaction mixture was stirred overnight at room temperature. The mixture was partitioned between dichloromethane (100 mL) and saturated aqueous NaHCO3 (10 mL). The organic layer was washed with saturated aqueous NaCl, dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by silica gel chromatography (dichloromethane/methanol = 15:1, v/v) to give the product 20a (0.165 g, 45%). 1H NMR (400 MHz, CDCl3) δ 8.08 (s, 1H), 7.79 (d, J = 8.0 Hz, 1H), 7.74 (s, 1H), 7.47 (t, J = 8.0 Hz, 1H), 7.38–7.36 (m, 1H), 7.05 (d, J = 7.6 Hz, 1H), 6.41 (d, J = 2.0 Hz, 1H), 6.36–6.33 (m, 2H), 6.13 (s, 1H), 5.70 (dd, J = 9.2 Hz, J = 2.4 Hz, 1H), 3.80 (s, 3H), 3.17 (t, J = 4.4 Hz, 4H), 2.80 (t, J = 4.4 Hz, 4H), 2.50 (s, 3H), 1.80 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 155.79, 152.22, 151.27, 139.83, 139.74, 136.17, 131.23, 131.19, 129.68, 127.54, 124.41, 122.18, 122.11, 120.62, 120.54, 120.05, 119.96, 108.34, 100.54, 80.76, 55.61, 54.54, 49.21, 45.09, 28.32. HRMS(ESI) (m/z): (M+H)+ calcd for C29H34N7O4 544.2672, found, 544.2698. HPLC purity: 95.77%, retention time = 10.81 min.
The following compounds 20b and 20c were prepared by a method similar to that for the synthesis of compound 20a.
N-(3-(4,4-Diethyl-7-((2-methoxy-4-(4-methylpiperazin-1-yl)phenyl)amino)-2-oxo-2H-pyrimido[4,5-d][1,3]oxazin-1(4H)-yl)phenyl)acrylamide (20b)
1H NMR (400 MHz, CDCl3) δ 9.42 (s, 1H), 7.96 (s, 1H), 7.89 (s, 1H), 7.81 (d, J = 5.6 Hz, 1H), 7.53 (s, 1H), 7.42 (t, J = 8.0 Hz, 1H), 7.33–7.31 (m, 1H), 6.99 (d, J = 7.6 Hz, 1H), 6.55–6.49 (m, 1H), 6.36–6.32 (m, 2H), 6.12 (d, J = 7.6 Hz, 1H), 5.65 (d, J = 10.4 Hz, 1H), 3.78 (s, 3H), 3.41 (t, J = 4.4 Hz, 4H), 3.21 (t, J = 4.4 Hz, 4H), 2.80 (s, 3H), 2.11–1.96 (m, 4H), 1.00 (t, J = 7.2 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 164.29, 158.75, 156.87, 153.46, 151.27, 148.56, 144.89, 140.04, 136.51, 131.50, 129.71, 127.67, 124.29, 123.12, 120.86, 120.05, 119.46, 108.92, 105.55, 101.12, 87.13, 55.75, 53.61, 47.76, 43.41, 32.88, 7.90. HRMS(ESI) (m/z): (M+H)+ calcd for C31H38N7O4 572.2985, found, 572.2981. HPLC purity: 98.86%, retention time = 12.08 min.
N-(3-(7-((2-Methoxy-4-(4-methylpiperazin-1-yl)phenyl)amino)-2-oxo-4,4-dipropyl-2H-pyrimido[4,5-d][1,3]oxazin-1(4H)-yl)phenyl)acrylamide (20c)
1H NMR (400 MHz, CDCl3) δ 8.06 (s, 1H), 7.96 (s, 1H), 7.77 (d, J = 4.8 Hz, 1H), 7.49 (s, 1H), 7.41 (t, J = 8.0 Hz, 1H), 6.97 (d, J = 8.0 Hz, 1H), 6.40 (d, J = 2.0 Hz, 1H), 6.36 (dd, J = 16.8 Hz, J = 1.2 Hz, 1H), 6.18–6.12 (m, 2H), 5.68 (dd, J = 10.4 Hz, J = 1.2 Hz, 1H), 3.79 (s, 3H), 3.09 (t, J = 4.4 Hz, 4H), 2.58 (t, J = 4.8 Hz, 4H), 2.36 (s, 3H), 2.07–2.00 (m, 2H), 1.96–1.89 (m, 2H), 1.54–1.38 (m, 4H), 0.97 (t, J = 7.2 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 163.56, 158.89, 156.43, 153.34, 151.78, 139.74, 136.05, 131.38, 129.78, 127.26, 124.14, 121.50, 120.46, 120.21, 107.93, 99.92, 86.64, 55.58, 55.05, 49.77, 46.00, 42.86, 16.81, 14.10. HRMS(ESI) (m/z): (M+H)+ calcd for C33H42N7O4 600.3298, found, 600.3297. HPLC purity: 99.69%, retention time = 14.41 min.
In Vitro Enzymatic Activity Assay
The enzymatic activity assay was evaluated by using the well-established ELISA-based assay. Kinases domain of EGFRL858R/T790M and EGFRWT were expressed by using the Bac-to-Bac™ baculovirusexpression system (Invitrogen, US), followed by purified in Ni-NTA columns (QIAGEN Inc., US). The detailed experiment was performed according to the published literature21.
In Vitro Cellular Antiproliferation Assay
In vitro cellular antiproliferation activities of the designed compounds against H1975 and A431 cell lines were examined by using the well-established sulforhodamine B (SRB) colorimetric assay28. The detailed experiment was performed according to the published literature21. Briefly, H1975 and A431 cells were seeded in 96-well plates, and allowed to adhere overnight followed by treated with gradient concentrations of the test compounds for 72 h. The inhibition rate for cell proliferation was calculated as [1 − (A 515 treated/A 515 control)] × 100%. The IC50 values were obtained by using the Logit method and reported as the mean ± SD from three independent determinations.
Western Blot Analysis
Western blot analysis was performed with standard procedures. Briefly, H1975 and A431 cell lines were seeded in six-well plates, starved in serum-free medium for 24 h, followed by incubation for 2 h in the presence of different concentrations of compounds 20a and 8 (as positive control). Cells were stimulated by the addition of EGF (50 ng/mL, R&D Systems, US) for 10 min, then washed with PBS and lysed in SDS lysis buffer. Antibodies against p-EGFR (Y1068; #3777 S), EGFR (#4267 S), p-ERK (T202/Y204; #4370 L), ERK (#4695 S), p-AKT (Ser473), AKT and Tubulin were used (Cell Signaling Technologies, Cambridge, MA).
In Vivo Antitumor Efficacy Study
All experimental procedures involving animals were performed in accordance with the ethical guidelines from Shanghai Laboratory Animal Public Service Center and Animal Ethics Committee of the East China University of Science and Technology. The whole experimental protocol was also approved by the Animal Ethics Committee of the East China University of Science and Technology. All 4–6 weeks old nude mice were purchased from Shanghai Slac Laboratory Animal Co. Ltd (The license of experimental animal: SCXK (shanghai) 2012–0002). The mice were maintained under the 12 hour light/dark cycles condition in the specific pathogen free (SPF) cleanroom with fresh air (more than 50%), appropriate temperature (22–26 °C) and humidness (40–60%). All materials and containers were disinfected and sterilized before use. Nude mice, aged 4–6 weeks, were inoculated subcutaneously into the right flank with H1975 NSCLC cells (approximately 2 × 106 cells/mouse). When the tumor size reached a volume of ~200−400 mm3, animals were randomly assigned into control and treated groups (n = 3/group). For efficacy studies, mice in control and in treatment groups were treated with vehicle compound 7 and compound 20a (50 mg/kg/day) for continuous 14 days, respectively. The sizes of tumor volume (V) were measured every two or three days by using vernier caliper, and were calculated with the formula V = (L × W2)/2, where L and W stands for length and width, respectively.
References
Favoni, R. E. & De Cupis, A. The role of polypeptide growth factors in human carcinomas: new targets for a novel pharmacological approach. Pharmacol. Rev. 52, 179–206 (2000).
Lemmon, M. A. & Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134 (2010).
Mendelsohn, J. & Baselga, J. Status of epidermal growth factor receptor antagonists in the biology and treatment of cancer. J. Clin. Oncol. 21, 2787–2799 (2003).
Bianco, R., Melisi, D., Ciardiello, F. & Tortora, G. Key cancer cell signal transduction pathways as therapeutic targets. Eur. J. Cancer 42, 290–294 (2006).
Levitzki, A. Tyrosine kinase inhibitors: views of selectivity, sensitivity, and clinical performance. Annu. Rev. Pharmacol. Toxicol. 53, 161–185 (2013).
Lynch, T. J. et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non–small-cell lung cancer to gefitinib. N. Engl. J. Med. 350, 2129–2139 (2004).
Pao, W. et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. 101, 13306–13311 (2004).
Cheng, X. & Chen, H. Tumor heterogeneity and resistance to EGFR-targeted therapy in advanced nonsmall cell lung cancer: challenges and perspectives. Onco Targets Ther. 7 (2014).
Tiseo, M., Bartolotti, M., Gelsomino, F. & Bordi, P. Emerging role of gefitinib in the treatment of non-small-cell lung cancer (NSCLC). Drug Des. Devel. Ther. 4, 81–98 (2010).
Lin, L. & Bivona, T. G. Mechanisms of resistance to epidermal growth factor receptor inhibitors and novel therapeutic strategies to overcome resistance in NSCLC patients. Chemotherapy research and practice 2012 (2012).
Xu, Y., Liu, H., Chen, J. & Zhou, Q. Acquired resistance of lung adenocarcinoma to EGFR-tyrosine kinase inhibitors gefitinib and erlotinib. Cancer Biol. Ther. 9, 572–582 (2010).
Helena, A. Y. et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 19, 2240–2247 (2013).
Miller, V. A. et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. The lancet oncology 13, 528–538 (2012).
Giaccone, G. & Wang, Y. Strategies for overcoming resistance to EGFR family tyrosine kinase inhibitors. Cancer Treat. Rev. 37, 456–464 (2011).
Reckamp, K. L. et al. A phase 2 trial of dacomitinib (PF‐00299804), an oral, irreversible pan‐HER (human epidermal growth factor receptor) inhibitor, in patients with advanced non–small cell lung cancer after failure of prior chemotherapy and erlotinib. Cancer 120, 1145–1154 (2014).
Zhou, W. et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 462, 1070–1074 (2009).
Walter, A. O. et al. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov 3, 1404–1415 (2013).
Cross, D. A. et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov 4, 1046–1061 (2014).
Steuer, C. E., Khuri, F. R. & Ramalingam, S. S. The next generation of epidermal growth factor receptor tyrosine kinase inhibitors in the treatment of lung cancer. Cancer 121, E1–E6 (2015).
Greig, S. L. Osimertinib: First Global Approval. Drugs 76, 263–273 (2016).
Hao, Y. et al. Discovery and Structural Optimization of N5-Substituted 6, 7-dioxo-6, 7-dihydropteridines as Potent and Selective Epidermal Growth Factor Receptor (EGFR) Inhibitors Against L858R/T790M Resistance Mutation. J. Med. Chem. 59, 7111–7124 (2016).
Zhou, W. et al. Discovery of Pteridin-7 (8 H)-one-based irreversible inhibitors targeting the epidermal growth factor receptor (EGFR) Kinase T790M/L858R mutant. J. Med. Chem. 56, 7821–7837 (2013).
Glide, version 5.5 Schrödinger, LLC, New York, USA. www.schrodinger.com (2009).
Maestro, version 9.0 Schrödinger, LLC, New York, USA. www.schrodinger.com (2009).
Gong, J. et al. ChemMapper: a versatile web server for exploring pharmacology and chemical structure association based on molecular 3D similarity method. Bioinformatics 29, 1827–1829 (2013).
Liu, X., Jiang, H. & Li, H. SHAFTS: a hybrid approach for 3D molecular similarity calculation. 1. Method and assessment of virtual screening. J. Chem. Inf. Model. 51, 2372–2385 (2011).
Laskowski, R. A. & Swindells, M. B. LigPlot+: Multiple Ligand–Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 51, 2778–2786 (2011).
Li, M. et al. DW10075, a novel selective and small-molecule inhibitor of VEGFR, exhibits antitumor activities both in vitro and in vivo. Acta Pharmacologica Sinica 37, 398–407 (2016).
Acknowledgements
The research is supported in part by the National Key Research and Development Program (Grant 2016YFA0502304) (H.L.), the National Natural Science Foundation of China (Grants 21302054 and 81230076) (Z.C., H.L.), the Shanghai Committee of Science and Technology (Grant 14431902100) (Y.X.), H.L. is also sponsored by Specialized Research Fund for the Doctoral Program of Higher Education (Grant 20130074110004). H.X. is supported by “Personalized Medicines-Molecular Signature-based Drug Discovery and Development”, Strategic Priority Research Program of the Chinese Academy of Sciences (Grant XDA12020209).
Author information
Authors and Affiliations
Contributions
Y.H. and J.L. performed research and drafted the manuscript; H.L. and J.L. designed the compounds and performed the computational simulation; Y.H. and Y.X. synthesized the compounds; R.Q. performed the in vitro enzymatic and cellular inhibition assays and the western blot analysis; D.S. conducted the in vivo efficacy study of the representative compound; Z.Z., Z.C., and J.D. performed research and helped to draft the manuscript; H.X., Y.X., and H.L. designed and performed research, interpreted data and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hao, Y., Lyu, J., Qu, R. et al. Structure-Guided Design of C4-alkyl-1,4-dihydro-2H-pyrimido[4,5-d][1,3]oxazin-2-ones as Potent and Mutant-Selective Epidermal Growth Factor Receptor (EGFR) L858R/T790M Inhibitors. Sci Rep 7, 3830 (2017). https://doi.org/10.1038/s41598-017-04184-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-04184-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
