
Abstract
Epidermal growth factor receptor (EGFR) therapy using small-molecule tyrosine kinase inhibitors (TKIs) is initially efficacious in patients with EGFR-mutant lung cancer, although drug resistance eventually develops. Allosteric EGFR inhibitors, which bind to a different EGFR site than existing ATP-competitive EGFR TKIs, have been developed as a strategy to overcome therapy-resistant EGFR mutations. Here we identify and characterize JBJ-09-063, a mutant-selective allosteric EGFR inhibitor that is effective across EGFR TKI-sensitive and resistant models, including those with EGFR T790M and C797S mutations. We further uncover that EGFR homo- or heterodimerization with other ERBB family members, as well as the EGFR L747S mutation, confers resistance to JBJ-09-063, but not to ATP-competitive EGFR TKIs. Overall, our studies highlight the potential clinical utility of JBJ-09-063 as a single agent or in combination with EGFR TKIs to define more effective strategies to treat EGFR-mutant lung cancer.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout








Similar content being viewed by others

Data availability
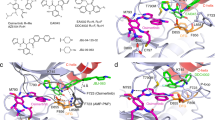
All data in this study are presented in the article and Supplementary Information. The structure in Fig. 1a has been deposited in the PDB with the accession code 7JXQ. The structure depicted in Fig. 6b was accessed via PDB accession code 4ZAU. The CRISPR sequencing data in Extended Data Fig. 1g were deposited into the NCBI Biosample database (PRJNA798765) and are publicly available. All materials are available upon request and through a material transfer agreement. Source data are provided with this paper.
References
Bray, F. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424 (2018).
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 70, 7–30 (2020).
Maemondo, M. et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N. Engl. J. Med. 362, 2380–2388 (2010).
Mitsudomi, T. et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 11, 121–128 (2010).
Zhou, C. et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 12, 735–742 (2011).
Rosell, R. et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 13, 239–246 (2012).
Park, K. et al. Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): a phase 2B, open-label, randomised controlled trial. Lancet Oncol. 17, 577–589 (2016).
Sequist, L. V. et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J. Clin. Oncol. 31, 3327–3334 (2013).
Wu, Y. L. et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): a randomised, open-label, phase 3 trial. Lancet Oncol. 18, 1454–1466 (2017).
Wu, Y. L. et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. 15, 213–222 (2014).
Soria, J. C. et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N. Engl. J. Med. 378, 113–125 (2018).
Janne, P. A. et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N. Engl. J. Med. 372, 1689–1699 (2015).
Mok, T. S. et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N. Engl. J. Med. 376, 629–640 (2017).
Cho, B. C. et al. Osimertinib versus standard of care EGFR TKI as first-line treatment in patients with EGFRm advanced NSCLC: FLAURA Asian subset. J. Thorac. Oncol. 14, 99–106 (2019).
Ramalingam, S. S. et al. Osimertinib as first-line treatment of EGFR mutation-positive advanced non-small-cell lung cancer. J. Clin. Oncol. 36, 841–849 (2018).
Soria, J. C. & Ramalingam, S. S. Osimertinib in EGFR mutation-positive advanced NSCLC. N. Engl. J. Med. 378, 1262–1263 (2018).
Mok, T. S. et al. Improvement in overall survival in a randomized study that compared dacomitinib with gefitinib in patients with advanced non-small-cell lung cancer and EGFR-activating mutations. J. Clin. Oncol. 36, 2244–2250 (2018).
Ahn, M. J. et al. Osimertinib in patients with T790M mutation-positive, advanced non-small cell lung cancer: long-term follow-up from a pooled analysis of 2 phase 2 studies. Cancer 125, 892–901 (2019).
Auliac, J. B. et al. Real-life efficacy of osimertinib in pretreated patients with advanced non-small cell lung cancer harboring EGFR T790M mutation. Lung Cancer 127, 96–102 (2019).
Igawa, S. et al. Impact of EGFR genotype on the efficacy of osimertinib in EGFR tyrosine kinase inhibitor-resistant patients with non-small cell lung cancer: a prospective observational study. Cancer Manag. Res. 11, 4883–4892 (2019).
Thress, K. S. et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med. 21, 560–562 (2015).
Rangachari, D. et al. EGFR-mutated lung cancers resistant to osimertinib through EGFR C797S respond to first-generation reversible EGFR inhibitors but eventually acquire EGFR T790M/C797S in preclinical models and clinical samples. J. Thorac. Oncol. 14, 1995–2002 (2019).
Jia, Y. et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature 534, 129–132 (2016).
To, C. et al. Single and dual targeting of mutant EGFR with an allosteric inhibitor. Cancer Discov. 9, 926–943 (2019).
De Clercq, D. J. H. et al. Discovery and optimization of dibenzodiazepinones as allosteric mutant-selective EGFR inhibitors. ACS Med. Chem. Lett. 10, 1549–1553 (2019).
Engelman, J. A. et al. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J. Clin. Invest. 116, 2695–2706 (2006).
Gallant, J.-N. et al. EGFR kinase domain duplication (EGFR-KDD) is a novel oncogenic driver in lung cancer that is clinically responsive to afatinib. Cancer Discov. 5, 1155–1163 (2015).
Gallant, J. N. et al. EGFR kinase domain duplication (EGFR-KDD) is a novel oncogenic driver in lung cancer that is clinically responsive to afatinib. Cancer Discov. 5, 1155–1163 (2015).
Alam, M. S. Proximity ligation assay (PLA). Curr. Protoc. Immunol. 123, e58 (2018).
Leonetti, A. et al. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br. J. Cancer 121, 725–737 (2019).
Lin, L. et al. Acquired rare recurrent EGFR mutations as mechanisms of resistance to Osimertinib in lung cancer and in silico structural modelling. Am. J. Cancer Res. 10, 4005–4015 (2020).
Hu, H. et al. Three subtypes of lung cancer fibroblasts define distinct therapeutic paradigms.Cancer Cell 39, 1531–1547 (2021).
Ma, S. et al. Epiregulin confers EGFR-TKI resistance via EGFR/ErbB2 heterodimer in non-small cell lung cancer. Oncogene 40, 2596–2609 (2021).
Mancini, M. et al. An oligoclonal antibody durably overcomes resistance of lung cancer to third-generation EGFR inhibitors. EMBO Mol. Med. 10, 294–308 (2018).
Cho, J. et al. Cetuximab response of lung cancer-derived EGF receptor mutants is associated with asymmetric dimerization. Cancer Res. 73, 6770–6779 (2013).
Wylie, A. A. et al. The allosteric inhibitor ABL001 enables dual targeting of BCR-ABL1. Nature 543, 733–737 (2017).
Hughes, T. P. et al. Asciminib in chronic myeloid leukemia after ABL kinase inhibitor failure. N. Engl. J. Med. 381, 2315–2326 (2019).
Eide, C. A. et al. Combining the allosteric inhibitor asciminib with ponatinib suppresses emergence of and restores efficacy against highly resistant BCR-ABL1 mutants. Cancer Cell 36, 431–443 (2019).
Yun, C. H. et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl Acad. Sci. USA 105, 2070–2075 (2008).
Winter, G. et al. DIALS: implementation and evaluation of a new integration package. Acta Crystallogr. D 74, 85–97 (2018).
Morin, A. et al. Collaboration gets the most out of software. Elife 2, e01456 (2013).
Winter, G. xia2: an expert system for macromolecular crystallography data reduction. J. Appl. Crystallogr. 43, 186–190 (2010).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010).
Zhou, W. et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 462, 1070–1074 (2009).
Kosaka, T. et al. Response heterogeneity of EGFR and HER2 exon 20 insertions to covalent EGFR and HER2 inhibitors. Cancer Res. 77, 2712–2721 (2017).
Jang, J. et al. Mutant-selective allosteric EGFR degraders are effective against a broad range of drug-resistant mutations. Angew. Chem. Int. Ed. 59, 14481–14489 (2020).
Bahcall, M. et al. Amplification of wild-type KRAS imparts resistance to crizotinib in MET exon 14 mutant non-small cell lung cancer. Clin. Cancer Res. 24, 5963–5976 (2018).
Tracy, S. et al. Gefitinib induces apoptosis in the EGFRL858R non-small-cell lung cancer cell line H3255. Cancer Res. 64, 7241–7244 (2004).
Ercan, D. et al. EGFR mutations and resistance to irreversible pyrimidine-based EGFR inhibitors. Clin. Cancer Res. 21, 3913–3923 (2015).
Acknowledgements
We thank K. Elenius (University of Turku, Finland) for generously providing the pBABE constructs, the MGH CCI DNA Core for their next-generation sequencing services, E. Cohen from the Molecular Biology Core Facilities at the DFCI for processing the DFCI52-C797S and GR-C797S cell line validation FASTA data in Integrative Genomics Viewer and all staff at the DFCI Animal Resources Facility for their mouse husbandry services. This study was supported by National Cancer Institute grants R35CA220497 (P.A.J.), RO1 CA201049 (M.J.E., N.S.G. and P.A.J) and PO1CA154303 (M.J.E., N.S.G. and P.A.J); American Cancer Society grant CRP-17-111-01-CDD (P.A.J.); the Balassiano Family Fund for Lung Cancer Research (P.A.J.); the Gohl Family Lung Cancer Research Fund (P.A.J.); and Takeda. T.S.B. is supported by a Ruth L. Kirschstein National Research Service Award (1F32CA247198-01). Y.K. is supported in part by Japan Society for the Promotion of Science Overseas Research Fellowships and Research Fellowship from Uehara Memorial Foundation. This work used NE-CAT beamlines (P30 GM124165) and an Eiger detector (S10OD021527) at the Advanced Photon Source (DE-AC02-06CH11357).
Author information
Authors and Affiliations
Contributions
C.T. and P.A.J. conceived and designed the studies, interpreted the findings and wrote the manuscript. C.T., T.S.B., W.W.F., H.M.H., B.H.S., J.K.R., D.E.H. and M.M. performed experiments and prepared figures. B.A.L., K.M.S., M.J.P. and P.C.G. conducted and coordinated the in vivo studies. J.J., T.W.G. and D.A.S. synthesized the JBJ-09-063 compound. Y.K. generated the H3255GR-C797S and DFCI52-C797S cell lines. M.B., W.W.F., K.W. and K.J.K. generated the constructs for transfection experiments. M.C. performed the pharmacokinetic studies of JBJ-09-063. A.O., C.X. and Y.Z. established the DFCI52 cell line and DFCI52 patient-derived xenograft. M.J.E. and N.S.G. provided crucial feedback on all experimental design and data interpretation. C.T. and P.A.J. supervised the studies and coordinated the efforts of all authors. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
P.A.J. has received consulting fees from AstraZeneca, Boehringer Ingelheim, Pfizer, Roche/Genentech, Takeda Oncology, ACEA Biosciences, Eli Lilly and Company, Araxes Pharma, Ignyta, Mirati Therapeutics, Novartis, LOXO Oncology, Daiichi Sankyo, Sanofi Oncology, Voronoi, SFJ Pharmaceuticals, Biocartis, Novartis Oncology, Nuvalent, Esai, Bayer, Transcenta, Silicon Therapeutics, Allorion Therapeutics, Accutar Biotech and AbbVie; receives post-marketing royalties from DFCI-owned intellectual property on EGFR mutations licensed to Lab Corp; receives or has received sponsored research funding from AstraZeneca, Astellas, Daichi-Sankyo, PUMA, Boehringer Ingelheim, Eli Lilly and Company, Revolution Medicines and Takeda and has stock ownership in Gatekeeper Pharmaceuticals. N.S.G. is a founder, science advisory board member and equity holder in Gatekeeper, Syros, Petra, C4, Allorion, Jengu, B2S, Inception, EoCys, Larkspur (board member) and Soltego (board member). The Gray lab receives or has received research funding from Novartis, Takeda, Astellas, Taiho, Jansen, Kinogen, Arbella, Deerfield and Sanofi. M.J.E. has served as a paid consultant to Novartis Institutes for Biomedical Research and H3 Biomedicine. M.J.E. receives sponsored research support from Novartis, Sanofi and Takeda. D.E.H. is a consultant for Logos Capital and the Jefferies Group. The series of compounds to which JBJ-04-125-02 and JBJ-09-063 belong is described in US patent 10,836,722 B2. All other authors declare no competing interests.
Peer review
Peer review information
Nature Cancer thanks Rafael Rosell, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Structure, potency, pharmacodynamic studies of JBJ-09-063 and the development and validation of H3255GR-C797S and DFIC52-C797S model in vitro.
a, Positive Fo-Fc electron density at 3 sigma for JBJ-09-063. b, Cell viability assay and c, Western Blot analyses of EGFRL858R/C797S Ba/F3 cells treated with increasing concentrations of JBJ-09-063, osimertinib and gefitinib. Data shown in panel b is a representative figure from N=3 independent experiments with 3 technical replicates in each experiment. d, Pharmacodynamic study of JBJ-09-063 in lung tumor tissues of H1975 xenograft mice dosed with 50 mg/kg of the compound. Samples were collected at 0 or 2, 8, 16 and 24h after dosing and samples were processed for Western Blotting analyses. e, Treatment history of patient who was given the first-generation TKI erlotinib (pink bar). Pleural effusion was subsequently drawn (orange arrow with PDX) to establish patient-derived xenograft (PDX) model. DFCI52 cell line was derived from PDX tumors (mouse #135 derived from the effusion). f, Workflow of EGFRL858R/T790M/C797S cell line generation: H3255GR and DFCI52 cells, which already harbor the EGFRL858R/T790M mutation, were transfected by nucleofection with a sgRNA designed to incorporate the C797S mutation in cis with EGFRT790M. 100 nM osimertinib was used for selection for 1 week and samples were sent for next-generation sequencing to determine the presence and frequency of C797S mutation. g, Visualization of the presence and frequency of T790M and C797S allele variants in cis in H3255GR-C797S and DFCI52-C797S cells using Integrated Genome Viewer (IGV). h, Cell viability assays and i, Western Blot analyses of H3255GR vs. H3255GR-C797S and DFCI52 vs. DFCI52-C797S cells treated with increasing concentrations of osimertinib. Data shown in panel h is a representative figure from N=3 independent experiments with 3 technical replicates in each experiment. All cell viability assays shown in this figure were graphed as a percentage of activity relative to DMSO control (mean ± SD) over indicated concentrations.
Extended Data Fig. 2 JBJ-09-063 efficacy is reduced in vitro unless combined with gefitinib in H3255GR and DFCI52 cells or with osimertinib in H3255GR-C797S and DFCI52-C797S cells.
a, Cell viability and b, Western blot analyses of H3255GR cells treated with indicated concentrations of osimertinib, JBJ-09-063 and JBJ-04-125-02. c, Cell viability, d, apoptosis and e, Western Blot analyses of DFCI52 cells treated with indicated concentrations of gefitinib, JBJ-09-063 or the combination of both agents. Cell viability of H3255GR-C797S cells in panel f, and DFIC52-C797S cells in panel g treated with indicated concentrations of JBJ-09-063 and osimertinib as a single agent or in combination of both compounds. h, Western Blot analyses of H3255GR-C797S and DFIC52-C797S cells treated with indicated concentrations of JBJ-09-063 and osimertinib as a single agent or in combination of both compounds. All cell viability assays shown in this figure were graphed as a percentage of activity relative to DMSO control (mean ± SD) over indicated concentrations. Data shown in panels a, c, f, g are representative figures from N=3 independent experiments with 3 technical replicates in each experiment. The apoptosis experiments were graphed as normalized caspase-3/7 activity (in average arbitrary units ± SEM)) over time. Data shown in panel f is a representative figure from N=3 independent experiments with 12 technical replicates in each experiment.
Supplementary information
Supplementary Tables
Supplementary Table 1. Data collection and refinement statistics (molecular replacement) of JBJ-09-063. Supplementary Table 2. IC50 of JBJ-04-125-02 and JBJ-09-063 in enzymatic assays, Ba/F3 cells, H1975 cells, and human cancer cells. Supplementary Table 3. Pharmacokinetic profile of JBJ-09-063. Supplementary Table 4. Sequences and primers for plasmids and cell lines generation.
Source data
Source Data Fig. 1
Unprocessed western blots with molecular markers.
Source Data Fig. 1
Quantitation of statistical data.
Source Data Fig. 2
Unprocessed western blots with molecular markers.
Source Data Fig. 2
Quantitation for statistical data.
Source Data Fig. 3
Unprocessed western blots with molecular markers.
Source Data Fig. 3
Quantitation of statistical data.
Source Data Fig. 4
Unprocessed western blots with molecular markers.
Source Data Fig. 4
Quantitation for statistical data
Source Data Fig. 5
Unprocessed western blots with molecular markers.
Source Data Fig. 5
Quantitation of statistical data.
Source Data Fig. 6
Unprocessed western blots with molecular markers.
Source Data Fig. 6
Quantitation of statistical data.
Source Data Fig. 7
Unprocessed western blots with molecular markers.
Source Data Fig. 7
Quantitation for statistical data.
Source Data Fig. 8
Unprocessed western blots with molecular markers.
Source Data Fig. 8
Quantitation for statistical data.
Source Data Extended Data Fig. 1
Unprocessed western blots with molecular markers.
Source Data Extended Data Fig. 1
Quantitation of statistical data
Source Data Extended Data Fig. 2
Unprocessed western blots with molecular markers..
Source Data Extended Data Fig. 2
Quantitation of statistical data.
Rights and permissions
About this article

Cite this article
To, C., Beyett, T.S., Jang, J. et al. An allosteric inhibitor against the therapy-resistant mutant forms of EGFR in non-small cell lung cancer. Nat Cancer 3, 402–417 (2022). https://doi.org/10.1038/s43018-022-00351-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s43018-022-00351-8
This article is cited by
-
Unveiling the mechanisms and challenges of cancer drug resistance
Cell Communication and Signaling (2024)
-
Nobiletin alleviates atherosclerosis by inhibiting lipid uptake via the PPARG/CD36 pathway
Lipids in Health and Disease (2024)
-
A macrocyclic kinase inhibitor overcomes triple resistant mutations in EGFR-positive lung cancer
npj Precision Oncology (2024)
-
Linking ATP and allosteric sites to achieve superadditive binding with bivalent EGFR kinase inhibitors
Communications Chemistry (2024)
-
Recent advances on high-efficiency of microRNAs in different types of lung cancer: a comprehensive review
Cancer Cell International (2023)
