
Abstract
Polyploidy or genome doubling (i.e., the presence of two or more diploid parental genome sets within an organism) are very important in higher plants. Of particular interest are the mechanisms in the new microenvironment of the common nucleus, where doubled regulatory networks interact to generate a viable genetic system capable of regulating growth, development and responses to the environment. To determine the effects of whole genome merging and doubling on the global gene expression architecture of a new polyploid, derived from protoplast fusion of the A1A1 genome of Gossypium arboreum and the E1E1 genome of Gossypium stocksii, we monitored gene expression through cDNA-AFLP in the somatic hybrids (G. arboreum + G. stocksii). The genomic expression patterns of the somatic hybrids revealed that changes in expression levels mainly involved regulatory genes (31.8% of the gene expression profiles), and the AA and EE genomes contributed equally to genome-wide expression in the newly formed AAEE genome from additivity and dominance effects. These results provide a novel perspective on polyploid gene regulation and hint at the underlying genetic basis of allopolyploid adaption in the new microenvironmental nucleus.
Similar content being viewed by others


Introduction
The genus Gossypium (cotton) currently consists of approximately 45 diploid species that are divided into eight monophyletic groups, each designated by a single letter (“A-G” and “K genome”) and 6 polyploid species1. Ancient hybridization between A and D diploids resulted in a new allopolyploid (AD) lineage in the New World approximately 1 million–2 million years ago2, 3. Two of the descendant allopolyploid species: Gossypium hirsutum (A1A1D1D1) and Gossypium barbadense (A2A2D2D2), as well as two African-Asian A diploids-Gossypium herbaceum (A1A1) and Gossypium arboreum (A2A2), were each independently domesticated for their long, spinnable, epidermal seed trichomes. These four species collectively account for the world’s cotton fiber production, more than 90% of which is provided by upland cotton G. hirsutum 1. While these polyploid cotton species are currently geographically separated, their monophyletic origin makes the Gossypium genus ideal for investigating emergent consequences of polyploidy. Understanding the cotton genome is important for facilitating advances in crop variety development and utilization. Furthermore, the mechanism of polyploid evolution in cotton can be used as an example to understand other polyploid crops4.
Polyploidization causes a simultaneous duplication of all nuclear DNA, and some of the genomic consequences of polyploidization could be dramatic1, 4,5,6,7. In plants, polyploidy is often associated with novel and presumably advantageous ecological attributes, such as range expansion8, novel secondary chemistry and morphology9, and increased pathogen resistance10, although the underlying genetic basis for these novel adaptations remains obscure. The reunion of two diverged genomes in a common nucleus during allopolyploid speciation entails a suite of genomic accommodations11,12,13, including non-additivity of gene expression14, 15 and expression partitioning among tissues and organs16,17,18,19. The most important point for us is the mechanisms by which doubled regulatory networks interact to generate a viable genetic system capable of regulating growth, development, and responses to the environment20.
One consequence of polyploidization is the unequal expression of homoeologous loci which was firstly described in cotton1, 21,22,23,24. Homoeologous gene expression levels were quantified in diploid and tetraploid flower petals of Gossypium using the Gossypium raimondii genome sequence as a reference. In the polyploid, most homoeologous genes were expressed at equal levels, although a subset had an expression bias for AT and DT copies. The direction of gene expression bias is conserved in natural and recent polyploids of cotton. Conservation of the direction of bias and additional comparisons between diploids and tetraploids suggest that the different regulation mechanisms of gene expression are different4. Regardless of the growth stage, tissue, or stress, the degree of bias between duplicated gene pairs is distributed across a spectrum of different expression ratios including the 50:50 ratio of most homoeologous gene pairs18, 25, 26.
Another consequence of polyploidization is expression level dominance. Expression level dominance has been characterized by the abundance of a transcript rather than the transcript origin by comparing expression levels in Gossypium tetraploids to those in related diploids for a given gene22. Expression level dominance of one of the two genomes has been found in leaf20, 26 and petal25, 26 tissue of interspecific hybrids and natural Gossypium polyploids. Expression level dominance has also been observed in other polyploid species such as Coffea27, Spartina6, and wheat7.
Previously, the transcript contributions of the two co-resident cotton genomes have been quantified using custom microarrays24, 25, 28, RNA-seq and EST assemblies26, and transcriptome RNA-seq4, 29, 30. Here, we used the cDNA-AFLP method to measure global genomic expression in the diploid parents (G. arboreum and G. stocksii) and their somatic hybrids (G. arboreum + G. stocksii). A modification of the method using complementary DNA (cDNA) samples in the analysis, known as AFLP-cDNA or cDNA-AFLP display, allows the characterization of tissue-specific gene expression patterns31 and the detection of gene expression differences in allopolyploids32,33,34. The cDNA-AFLP method is an extremely efficient and sensitive mRNA fingerprinting technique for identifying both common and rare or unknown transcripts35,36,37 and is an open architecture technology for global transcriptional analysis in a non-model plant species14, 38, 39. This technique is a robust and high-throughput tool for the analysis of genome-wide gene expression, and it can be used to identify genes that are differentially expressed in allopolyploids. Quantitative cDNA-AFLP was used to monitor variation in the expression levels of cotton fiber transcripts among a population of inter-specific Gossypium hirsutum × G. barbadense recombinant inbred lines (RILs), proving to be a cost-effective and highly transferable platform for genome-wide and population-wide gene expression profiling39. cDNA-AFLP has been used previously in cotton to compare the transcriptomes of two cotton lines (one fertile and the other male sterile)40, to identify genes involved in somatic embryogenesis41 and to study gene silencing42, and to construct genetic maps39, 43.
Results
Differential transcript profiling through cDNA-AFLP was used to investigate and compare transcript changes in the new tetraploid somatic hybrid (G. arboreum + G. stocksii, a new genotype of A1A1E1E1) relative to its diploid parents (G. arboreum and wild species G. stocksii). The morphology of the somatic hybrid was significantly different to that of the parental plants (Fig. 1A): the leaves of the somatic hybrid were thicker and darker green in color and the plants were more vigorous than the parental plants (Fig. 1B). Leaves at different developmental stages were collected from each of the three species for cDNA-AFLP analysis to investigate the global gene expression changes in the allopolyploid relative to the diploid parental genomes.

Comparison of the morphology of parental plants and their somatic hybrids.
cDNA-AFLP analysis and TDF detection
Each of the selective AFLP primer combinations amplified between 100 and 600 fragments, most of which were between 100 and 300 base pairs long. cDNA-AFLP analysis using 64 primer combinations resulted in the identification of more than 6800 clear and unambiguous differentially expressed transcript-derived fragments (TDFs) (Fig. 2).

cDNA-AFLP fingerprints of G. arboreum (Ga), G.stocksii (Gs) and their somatic hybrids (AS). Mse + CAC means Mse I primer plus 3 primer_2 (5′-GAC GAT GAG TCC TGA GTA A – CAC-3′, which are listed in Table 2).
More than 4000 differentially expressed fragments based on presence/absence or differences in intensity were eluted from the gels, re-amplified and sequenced. The DNA sequence of each TDF was assigned a putative biological function by checking against the cotton EST in the GenBank database (BLASTN/BLASTX). From the sequence alignment, some different TDFs were aligned to the same transcript at different fragments, representing one gene. The sequences of the 1627 sequenced differentially expressed TDFs were aligned. Among the differentially expressed TDFs, the functions of some genes were annotated and not studied in cotton previously. Some of them were cloned with full-length cDNA or genomic DNA sequence and listed in Table 1. TDF113 (Cotton_A_21823) was annotated as Acyl-CoA N-acyltransferase, the same gene was found in the male sterility mutant of cotton described in our previous paper44.
TDFs representing differentially expressed genes were classified into different categories on the basis of their presence/absence (qualitative variation) or differences in expression levels (quantitative variation) among G. arboreum, G stocksii and their somatic hybrids (Table 3). Different TDFs representing genes controlling biological processes were classified as follows: regulation (31.8%), general and secondary metabolism (18.8%), signal transduction (15.8%), transportation (9.9%), cellular organization (11.8%), defense and response to stimuli (5.8%), photosynthesis & energy (4.1%), transposable elements (1%) and unknown (1%) (Fig. 3).

Functional classification of transcript derived fragments: transcript derived fragment categories.
TDFs representing genes were implicated in the biosynthesis of proteins & amino acids (9%), fatty acids (6%) and carbohydrates (4%) in general and secondary metabolism. TDFs (11.8%) were involved in cellular function and organization, and some TDFs (9.9%) were transporters. Genes involved in signal transduction (15.8%) and regulation (31.8%), including Zn finger binding proteins and ARF guanyl-nucleotide exchange factors, were also detected.
Differential expression patterns in the two parents and their somatic hybrid
To detect additivity, transgressive expression, and expression level dominance, we counted 2240 units (one amplification in G. arboreum, G. stocksii and the somatic hybrid with one combination of primers) of differentially expressed bands for the three individual lines (G. arboreum, G stocksii and the somatic hybrid) and then grouped genes that showed a change in expression level in the somatic hybrid relative to the expression level in their parents into 12 different categories. These categories were described as additivity (I and XII), E-expression level dominance (II and XI), A-expression level dominance (IV and IX), transgressive expression lower than either parent (III, VII and X), or transgressive expression higher than either parent (V, VI and VIII). The additivity categories (I and XII in Fig. 4) made up 10.7% (239/2240 units), and equivalent expression (approximately 1000 ‘no change’ units were excluded from the total count; these were considered to be mid-parent expression values) of the differentially expressed genes in the somatic hybrid and the two parents, representing the initial stage of the merging genomes, displayed additivity in the allopolyploid somatic hybrid of 5.1% and 5.5% from the AA genome and the EE genome, respectively.

The 12 possible differential expression states in the somatic hybrid relative to its diploid parents. Roman numerals indicate the same categorization as used in Rapp et al. (2009), with figures schematizing their respective gene expression pattern for diploid parent G. arboreum (Ga: A-genome), tetraploid somatic hybrid (G.arboreum + G.stocksii: AS, AE genome) and diploid parent G. stocksii (Gs: E-genome). Additivity (I and XII), E-expression level dominance (II and XI), A-expression level dominance (IV and IX), transgressive expression lower than either parent (III, VII and X) or transgressive expression higher than either parent (V, VI and VIII).
The two effects of dominance (23.8% from the AA genome, 21.2% from the EE genome) and transgressive regulation (19.7% of genes were downregulated, 24.6% were upregulated) contributed to global gene expression in the somatic hybrid at levels of 46% and 44.3%, respectively (Fig. 4).
Approximately 45.2% of genes showed expression level dominance (Fig. 4). Analogous to the expression levels of the two diploid parents and their tetraploid somatic hybrid, the genome-wide expression level dominance resulted from the A genome (23.8%) and the E genome (21.2%), while the direction of expression level dominance showed that gene silencing (15.7% from the A genome; 16.6% from the E genome) occurred simultaneously in the somatic hybrid, and one parent was severe, two – three times for acquired dominant expression (8.1% from the A genome; 4.6% from the E genome) in the somatic hybrid. The degree of biased expression level dominance was the most severe in the somatic hybrid, where the expression levels of 533 genes (23.8% of all genes, categories IV and IX) were statistically equivalent to the A genome parent, compared with 474 genes (21.2%, categories II and XI) for the E genome parent. Thus, gene pairs from the A and E genomes (533 vs 474) exhibited expression level dominance from the A parent and the E parent at the same level.
More genes were transgressively upregulated (24.6%, 552/2240; categories V, VI, and VIII in Fig. 4) than downregulated (19.7%, 442/2240; categories III, VII and X in Fig. 4) in allopolyploids.
Among the transgressively upregulated genes, the percentage of acquired expression in somatic hybrids reached 15.3% (category VIII in Fig. 5), significantly more than the other two types of transgressive upregulation (categories V and VI in Fig. 4, Fig. 5), and significantly more than the transgressive downregulated genes. With regard to the dominance effects of the AA and EE genomes, the proportion of silent expression (category IX for the AA genome and category XI for the EE genome) was significantly greater than dominant expression (category IV for the AA genome and category II for the EE genome) (Fig. 5).

Percentages of differential expression patterns in somatic hybrids and thediploid parents.
Relationship between homoeolog-specific expression and expression level dominance
To evaluate how the expression of individual homoeologs relates to joint homoeolog expression, we examined homoeolog expression in each of the 12 categories of differential expression. The results showed that the number of genes showing homoeolog expression bias varied depending on the origin of the parent. In G. arboreum, novel expression of the AA genome was 8.1%, and silenced expression was 15.7%; in G. stocksii, novel expression of the EE genome was 4.6%, and silenced expression was 16.6%; in the somatic hybrid, novel expression reached 28.6%, and silenced expression was 40.3% from the AA genome and 44.6% from the EE genome (Table 4). For the parental plants, the proportion of silenced expression was the same, whereas the proportion of novel expression in G. arboreum was significantly higher than in G. stocksii. In the somatic hybrids, the proportion of genes showing novel expression or silenced expression was significantly higher than in the two parental plants. The proportion of homoeologous genes that were silenced depended on the parent of origin in the somatic hybrid.
Transcript quantification of selected TDFs in five different cotton species
The 12 TDFs were located in the G. arboreum, G. ramondii or G. hirsutum genomes; the CDS sequences derived from the TDFs were obtained, and primers were designed according to the sequences for gene expression analysis in different cotton species, including G. arboreum (Ga), G. stocksii (Gs), their somatic hybrid (G. arboreum + G. stocksii, AS), G. hirsutum (Gh) and G. barbadense (Gb).
The 12 TDFs representing genes for pectinesterase (TDF211), disease resistance protein RPS2 (TDF507), nucleobase:cation symporter-1, NCS1 (TDF557), Heat shock protein Hsp20 (TDF275), Cytochrome P450 (TDF293), Auxin responsive SAUR protein (TDF20), Acyl-CoA N-acyltransferase (TDF113), ARF guanyl-nucleotide exchange factor (TDF1), gibberellin receptor GID1 (TDF10), bZIP transcription factor (TDF177), MADS-box transcription factor (TDF497), RPS2 (RESISTANT TO P. SYRINGAE 2), and disease resistance protein RPS2 (TDF57) (Table 1), showed significantly changed transcript abundance in five different cotton species.
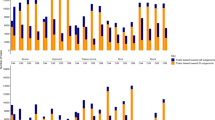
A relatively greater abundance of TDFs was observed for TDF557 (nucleobase:cation symporter-1, NCS1), TDF20 (Auxin responsive SAUR protein), TDF113 (Acyl-CoA N-acyltransferase), and TDF1 (ARF guanyl-nucleotide exchange factor) genes in the somatic hybrid, G. hirsutum and G. barbadense, which are tetraploid cotton species (Fig. 6). For pectinesterase (TDF211) and Cytochrome P450 (TDF293) genes, a relatively lower expression level was observed in all tested tetraploid somatic hybrid cotton species, G. hirsutum and G. barbadense, significantly greater transcript abundance was observed in diploid wild species of G. stocksii, and the two genes were silenced in the somatic hybrid (Fig. 6). Acyl-CoA N-acyltransferase (TDF113) showed acquired expression in the somatic hybrid. NCS1 (TDF557), ARF guanyl-nucleotide exchange factor (TDF1), gibberellin receptor GID1 (TDF10), MADS-box transcription factor (TDF497), and disease resistance protein RPS2 (TDF57) were overexpressed in the somatic hybrid (Fig. 6). In the two parental plants G. arboreum and G. stocksii, gene silence or very low expression was observed for NCS1 (TDF557), Acyl-CoA N-acyltransferase (TDF113), ARF guanyl-nucleotide exchange factor (TDF1), gibberellin receptor GID1 (TDF10), and MADS-box transcription factor (TDF497) in G. stocksii (Fig. 6). MADS-box transcription factor (TDF497) showed significantly higher expression levels in somatic hybrids, G. barbadense, G. arboreum, and G. hirsutum, which have long fibers, and was silent in G. stocksii, which only has fuzz fibers (Fig. 6).

Expression of genes from TDFs in different cotton species. Ga: Gossypium arboreum, Gs: G. stocksii, AS: somatic hybrid of G. arboreum and G. stocksii, Gh: G. hirsutum, Gb: G. barbadense.
The RPS2 gene from TDF507 encoding resistance to Pseudomonas syringae protein 2 specifically recognizes the AvrRpt2 type III effector avirulence protein from Pseudomonas syringae to guard the plant against pathogens. The RPS2 gene was expressed in all tested cotton species with high levels of expression in wild species G. stocksii and G. barbadense. Acyl-CoA N-acyltransferase (TDF113) is involved in the metabolism of fatty acids and enters the citric acid cycle, eventually forming several molecules of ATP; it is also involved in the metabolism of carbon sugars as the starting point for the citric acid cycle and in fatty acid metabolism as a balance between carbohydrate metabolism and fat metabolism. Furthermore, acyl-CoA N-acyltransferase is required for the synthesis of flavonoids and related polyketides for elongation of fatty acids. Acyl-CoA N-acyltransferase showed high levels of expression in G. barbadense and G. hirsutum, and acquired expression in the somatic hybrid. The NCS1gene from TDF557 showed high levels of expression in natural tetraploid cotton of G. hirsutum and G. barbadense, and in the somatic hybrid of the new synthetic tetraploid cotton.
Discussion
Here, we report a cotton transcriptome study of a genomic polyploid analyzed by cDNA-AFLP in a somatic hybrid (G. arboreum + G. stocksii) and two parental plants (G. arboreum and G. stocksii); our results provide a demonstration of a genomic expression profile after polyploidy in cotton. Changes in gene expression after polyploidy were mainly focused on genes involved in regulation, followed by genes involved in general and secondary metabolism and signal transduction. The contribution effects derived from the parental genomes of AA and EE were equivalent for the double genome of AAEE.
Changes in gene expression associated with polyploidy
Here, the main changes in gene expression were to genes involved in regulating polyploidy. The differentially expressed genes mainly included genes involved in cell division, the SAUR family, zinc fingers, brassinosteroid insensitive 1-associated receptor kinases, ubiquitin hydrolase and ubiquitin ligase, telomerase activating protein, transcription elongation factors, RNA recognition motifs, DNA-binding motifs, and transcription factors (e.g., MADS-box, TGA, Auxin responsive protein, Zinc finger, bZIP, and K-box). Some differentially expressed genes included seed maturation proteins, pentatricopeptide repeats, tetratricopeptide repeats, and the DDT domain superfamily. These genes are involved in the process of DNA replication, transcription, translation and protein metabolism, and the biosynthesis and interaction of endogenous growth regulators during plant growth and development.
AA and EE subgenomes have the same contribution to the genome-wide expression of the somatic hybrid
To explore and categorize the expression alterations accompanying polyploid formation, we grouped the differentially expressed genes into the 12 possible patterns of differential expression through clear and unambiguous differentially expressed band patterns. The 2240 units of differentially expressed bands clearly displayed additivity of 5.1% from the A genome and 5.5% from the E genome for genome-wide expression in the newly formed somatic hybrids. The exhibited genome-wide expression level dominance resulted from the A genome (23.8%) and the E genome (21.2%) at the same level. The two effects of additivity and dominance contributed 55.6% to the genome-wide expression of somatic hybrids of two duplicated genomes.
For the two effects, the AA genome contributed 28.9%, and the EE genome contributed 26.7% to the expression of the somatic hybrid genome. The somatic hybrid of the AAEE genome contained two parental genomes of AA and EE, which had the same level of contribution to the new synthetic AAEE genome, different from the bias toward the A genome in a diploid hybrid and natural allopolyploids as described by Yoo et al.26.
Gene expression patterns in interspecific hybrid F1 and leaf transcriptomes from synthetic and natural allopolyploid cotton indicated that genome-wide expression level dominance was biased toward the A genome in the diploid hybrid and natural allopolyploids, whereas the direction was reversed in the synthetic allopolyploid, mainly caused by up- or downregulation of the homoeolog from the ‘non-dominant’ parent26.
Expression effects from the AA and EE genomes
For the dominance effect, the AA and EE genomes almost contributed to genome-wide expression of the AAEE genome at the same level (23% from the AA genome and 21.2% from the EE genome). In the two donor genomes of AA and EE, the silent expression pattern in the dominance effect held the major position in the genome-wide expression of the AAEE genome.
For the transgressive effect in the genome-wide expression of somatic hybrids, the number of genes upregulated (24.6%, 552/2240; categories V, VI and VIII in Fig. 4) was more than those downregulated (19.7%, 442/2240; categories III, VII and X in Fig. 4) in allopolyploids. The result of the polyploidization of the somatic hybrid was an increase in genes transgressively upregulated over those transgressively downregulated (Figs 4, 5). Among the transgressively upregulated genes, acquired expression in somatic hybrids reached 15.3% (categories VIII in Fig. 4), significantly more than the other two types of transgressive upregulation (categories V and VI in Fig. 4). Another remarkable result of polyploidization was an increase of transgressive upregulated genes compared to downregulated genes, especially the activation of the acquired expression of new genes that were silent or not present in both of the parental plants.
For the homoeolog expression in somatic hybrid plants, the proportion of novel acquired expression (15.3%; category VIII in Fig. 4) was roughly equivalent to the proportion of silent expression (13.3%; categories III and VII) while simultaneously expressed in G. arboreum and G. stocksii. While in the somatic hybrid, the proportion of silenced expression was over 40% from the AA and EE genomes. The phenomena of gene silencing resulting from polyploidization were severe and universal.
In allopolyploid cotton including two natural allopolyploids and an interspecific diploid F1 hybrid (G. arboreum (A2A2) × G. raimondii (D5D5)), higher rates of transgressive and novel gene expression patterns as well as homoeolog silencing were observed in natural allopolyploids compared to the F1 hybrid or synthetic allopolyploid cotton. Extensive alterations in homoeolog expression bias and expression level dominance accompany the initial merger of two diverged diploid genomes, suggesting a combination of regulatory (cis or trans) and epigenetic interactions that may arise and propagate through the transcriptome network26.
Validation of TDFs
Because of the large number of differentially expressed gene identified, 12 cloned TDFs were tested by quantitative RT-PCR across the five different cotton species. TDFs with differential expression patterns belonged to genes involved in regulation, general and secondary metabolism, signal transduction, transportation, cellular organization, defense and response to stimuli (Fig. 6, Table 1).
The genes for regulation, general and secondary metabolism, signal transduction, transportation, cellular organization, and defense and response to stimuli were expressed at different levels in the diploid parents and their somatic hybrids; the expression levels of some genes were the same in tetraploid species including the somatic hybrids, G. hirsutum and G. barbadense, suggesting polyploidization enhanced gene expression; however, some genes became silent after polyploidization. These genes are all candidate sequences for validation of the cDNA-AFLP technique. The identified unigenes could be screened by qRT-PCR for verification of quantitative changes in transcript abundance (gene expression) for the cDNA-AFLP. AFLP based TDFs could represent the identification of differentially expressed genes in genome-wide-expression analysis.
The RPS2 gene was expressed in all tested cotton species, with high abundance in wild species of G. stocksii and G. barbadense. The disease resistance (R) protein specifically recognizes the AvrRpt2 type III effector avirulence protein from Pseudomonas syringae, interacts with RIN4, and probably triggers plant resistance when RIN4 is degraded by AvrRpt245. In this experiment, RPS2 had high expression levels in the five cotton species. Serine/Threonine Kinase receptors play a role in the regulation of cell proliferation, programmed cell death (apoptosis), cell differentiation, and embryonic development. The NCS1 gene (nucleobase:cation symporter-1) showed high levels of expression in the natural tetraploid cotton species G. hirsutum and G. barbadense and high levels of expression in the somatic hybrid of new synthetic tetraploid cotton, but showed very low expression levels in the diploid parental plants, perhaps related to polyploidization. The expression level changes in these genes occurring in the diploid parental species and their hybrids were validated in the global expression pattern change of the whole genome of the newly synthesized polyploid hybrid.
Genes duplicated by polyploidy (homoeologs) may be differentially expressed in the synthesized hybrid by protoplast fusion of G. arboreum and G. stocksii compared with their parental species. Compared to previous studies, a surprising level of expression homeostasis was observed in the expression patterns of polyploid genomes; in the new microenvironmental nucleus of somatic hybrids, the main functional classes of changed gene expression were attributed to regulation; the AA and EE genomes showed equal contributions to genome-wide expression of the newly formed AAEE genome from additivity and dominance effects. Mechanisms of gene regulation in the cotton genome warrant further investigation.
Materials and Methods
The diploid species G. arboreum (A1A1 genome) and the wild species G. stocksii (E1E1 genome) and their somatic hybrids (G. arboreum + G. stocksii, A1A1E1E1 genome) via protoplast fusion were planted in the greenhouse of our campus and used in this experiment. The somatic hybrids of G. arboreum + G. stocksii were confirmed by cytological examination, molecular markers and ploidy analysis by DNA content with flow cytometry. More than ten plants of each taxon were grown in growth chambers. Plants were grown at 26 °C with a photoperiod of 14 h of light and 10 h of dark and watered as necessary. Samples of fresh young leaves in different developmental stages (germination, seedling, bud, following and boll) were collected (from May to the end of August). Samples were harvested between 9 and 10 AM, immediately frozen in liquid nitrogen and stored at −80 °C before total RNA extraction.
RNA extraction and cDNA synthesis
Young leaves at different developmental stages were mixed equivalently and ground in liquid nitrogen. Total RNA was isolated from leaves using the RNAprep Pure Plant Kit (Qiagen, GmbH, German) according to the manufacturer’s instructions.
RNA quality was verified on a 1.4% denaturing agarose gel. Total nucleic acids were quantified using a Nanodrop 2000 °C spectrophotometer (Thermo Scientific, USA), and DNA contamination was quantified using a DNA-free Kit (Ambion, USA).
Double-stranded cDNA was synthesized using an iScriptTM cDNA Synthesis Kit (Bio-rad, USA) according to a standard double-stranded cDNA synthesis protocol.
cDNA-AFLP analysis
A total of 200 ng of double stranded cDNA was subjected to standard AFLP template production according to Vuylsteke et al.46 with little modification. cDNA was digested with restriction enzymes MseI and EcoR I (NEB, England). Digested products were then ligated to adapters with the following sequences: MseI adapter 5′-GACGATGAGTCCTGAG-3′, 3′-TACTCAGGACTCAT-5′; EcoR I adaptors 5′-CTCGTATACTGCGTACC-3′, 3′-AATTGGTACGCAGTA-5′. Adapter ligated DNA served as a template for pre-amplification, with PCR parameters of 30 cycles of 30 s at 94 °C, 60 s at 56 °C, and 60 s at 72 °C. The diluted (30-fold) amplified products were used as the template for selective amplification. Equal amounts of pre-amplified products were amplified with primers having selective nucleotides at the 3′ end in a total volume of 20 μl. The primers were listed in Table 2. First selective amplification cycle consisted of 30 s at 94 °C, 30 s at 65 °C, and 60 s at 72 °C; annealing temperature was lowered by 0.7 °C per cycle during the next 12 cycles, followed by 23 cycles at 94 °C for 30 s, 56 °C for 30 s, and 72 °C for 60 s. All PCR reactions were carried out in Applied Biosystem model 9902 Veriti thermal cycler. To each PCR product 7.5 μl of formamide dye (98% formamide, 10 mM EDTA, 0.005% xylene cyanol FF, and 0.005% bromophenol blue) was added, and 7 μl of each sample was loaded onto a pre-warmed 6% polyacrylamide gel using 1x Tris–borate–EDTA (TBE) buffer. Electrophoresis was then run for 2.5 h at 65 W and the gels were silver stained using a silver staining kit (Promega cat. #Q4132, Madison, WI), following the manufacturer’s instructions.
Transcript-derived fragment (TDF) isolation and re-amplification
Differentially expressed TDFs based on presence, absence or differences in intensity were carefully excised from the gel with a sharp blade to avoid any contaminating fragment(s), eluted in 50 μl of sterile double distilled water, incubated at 95 °C for 15 min and then hydrated overnight at 4 °C. An aliquot of 2 μl was used for re-amplification in a total volume of 25 μl, using the same set of corresponding selective primers and PCR conditions as used for the selective amplification, except that an annealing temperature of 56 °C for 35 cycles was used. PCR products were resolved in a 2% agarose gel; each single band was isolated and eluted using the QIA quick DNA gel extraction kit (Qiagen, USA). The reproducibility of cDNA-AFLP was verified by repeating two times.
Cloning and sequencing of TDFs
Eluted TDFs were cloned into the plasmid pGEM-T easy® vector (Promega, Madison, USA) and transformed into E. coli DH5α following the manufacturer’s protocol and then sequenced. For each TDF, three individual clones were isolated and sequenced. The nucleotide sequences were compared with publicly available cotton EST databases using BLAST sequence alignments. Lists of cloned TDFs, primers and other features are summarized in Table 2.
Sequences of TDFs (with vector sequences trimmed off, where the plasmid was used as the template) were then analyzed for their homology against the publicly available nonredundant genes/ESTs/transcripts in databases of Gossypium arboreum L., Gossypium raimondii Ulbr., and Gossypium hirsutum L. (https://www.cottongen.org/;http://www.ncbi.nlm.nih.gov/BLAST, http://cgp.genomics.org.cn/page/species/blast.jsp, http://www.arabidopsis.org/Blast) using BLASTN and BLASTX algorithms, according to Gupta et al.38. Then, all the unigenes of 1627 were annotated using a BLASTx search of the UniProt database (http://www.ebi.ac.uk/uniprot/). GO-KEGG-EC annotation was performed based on the Annot8r platform46, 47. TDFs were also checked for putative function against the Arabidopsis database and the cotton genome database (Institute of Cotton Research of CAAS) using the FASTA tool (http://www.arabidopsis.org/cgi-bin/fasta/nph-TAIRfasta.pl, http://cgp.genomics.org.cn/).
From the aligned and annotated differentially expressed genes, analysis of expression level dominance and homoeolog expression bias, we first explored the data for novel expression (new expression of a gene in a tissue) and homoeolog silencing patterns (no expression of one homoeolog) in the somatic hybrid and parents. Novel expression was inferred when both parental species had no bands for a gene, yet allopolyploids displayed clear bands in all three replicates. If both parental species had clear bands for a homoeolog, but somatic hybrids had no band for the same homoeolog, this was considered silencing. These two cases were eliminated from further analysis, focusing on genes that are expressed in at least one parent and where both homoeologs are expressed in the somatic hybrids. Genes identified as differentially expressed in the somatic hybrid relative to their diploid parents were grouped into 12 possible classes of differential expression (see Fig. 2), that is, expression level dominance, additivity and transgression (outside the range of either parent), according to Rapp et al.20. Briefly, genes were parsed into these 12 categories (using Roman numerals; see Fig. 2), depending on relative expression levels between the two parents and those of the somatic hybrids. Examined in this manner, genes may display additivity (I and XII), E-expression level dominance (II and XI), A-expression level dominance (IV and IX), transgressive expression lower than either parent (III, VII and X) or transgressive expression higher than either parent (V, VI and VIII).
For each of the 12 categories above (which are based on joint expression levels for both homoeologs), we tabulated homoeolog-specific bands to examine how homoeolog usage for each gene pair was related to total gene expression for each homoeolog pair for each of the 12 categories.
Quantitative Real Time-PCR
As cDNA-AFLP bands are anonymous, 12 selected TDF fragments were isolated from gels and sequenced to tentatively confirm by qPCR the population-wide quantitative cDNA-AFLP profile. The sequences of these 12 TDFs were aligned against the cotton sequence databases using a Blast algorithm.
The 12 TDFs were subsequently tested by QRT-PCR. Sequence homology of the TDFs with cotton EST sequences allowed the design of gene-specific primers. Expression profiling over the tested cotton species (two natural tetraploid cotton species G. hirsutum and G. barbadense, somatic hybrids of G. arboreum + G. stocksii, two parental plants of G. arboreum and G. stocksii) was carried out using qRT-PCR. For three TDFs, for which the sequence blast result could not discriminate between several possible database accessions, primer pairs were designed for each accession and tested independently by qPCR.
Total RNA was extracted from fresh leaves according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA, USA) and treated extensively with RNase-free DNase I. Double-stranded cDNA was synthesized from 100 ng RNA using iScriptTM cDNA Synthesis Kit (Quanta Quantscript RT kit) according to a standard double-stranded cDNA synthesis protocol. Real-time PCR assays were performed using the SYBR Green Real-Time PCR Master Mix (Promega Gotaq@ qPCR master mix, Madison, USA) and the qRT-PCR reaction was performed using the Eppendorf real-time PCR instrument (Mastercycler ep realplex, Hamburg, Germany). Specificity of the amplified PCR product was determined based on melting curve analysis. Primers for target genes were designed using Premier5 software (Premier Biosoft, Palo Alto, CA). The cotton Ubiquitin7 gene (GhUBQ7, Gen Bank accession number: DQ116441, GhUBQ7F: 5′-GAAGGCATTCCACCTGACCAAC-3′, GhUBQ7R: 5′-CTTGACCTTCTTCTTCTTGTGCTTG-3′) was used as an internal control for the assays. The expression levels of endogenous genes in cotton were obtained and standardized to the constitutive GhUBQ7 gene expression level. In each study, three independent experiments were conducted. The relative expression were calculated by the 2−ΔΔCT method48. Analysis of variance (ANOVA) and means were performed via the statistical software SPSS10.0.
References
Wendel, J. F. & Cronn, R. C. Polyploidy and the evolutionary history of cotton. Adv. Agron. 78, 139–186 (2003).
Wendel, J. F. New world tetraploid cottons contain old world cytoplasm. Proc. Natl.Acad. Sci. USA 86, 4132–4136 (1989).
Li, F. G. et al. Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat. Biot. 33, 524–530 (2015).
Rambani, A., Page, J. T. & Udall, J. A. Polyploidy and the petal transcriptome of Gossypium. BMC Plant Biol. 14, 3 (2014).
Salmon, A., Flagel, L., Ying, B., Udall, J. A. & Wendel, J. F. Homoeologous nonreciprocal recombination in polyploid cotton. New Phytol. 186, 123–134 (2010).
Chelaifa, H., Monnier, A. & Ainouche, M. Transcriptomic changes following recent natural hybridization and allopolyploidy in the salt marsh species Spartina × townsendii and Spartina anglica (Poaceae). New Phytol. 186, 161–174 (2010).
Qi, B. et al. Global transgenerational gene expression dynamics in two newly synthesized allohexaploid wheat (Triticum aestivum) lines. BMC Biol. 10, 3 (2012).
Hijmans, R. J. et al. Geographical and environmental range expansion through polyploidy in wild potatoes(Solanum section Petota). Global Ecol.Biogeogr. 16, 485–495 (2007).
Leitch, A. R. & Leitch, I. J. Genomic plasticity and the diversity of polyploid plants. Science 320, 481–483 (2008).
Nuismer, S. L. & Thompson, J. N. Plant polyploidy and non-uniform effects on insect herbivores. Proc. R. Soc. Lond. B. Biol. Sci. 268, 1937–1940 (2001).
Osborn, T. et al. Understanding mechanisms of novel gene expression in polyploids. Trends Genet. 19, 141–147 (2003).
Adams, K. L. & Wendel, J. F. Polyploidy and genome evolution in plants. Curr. Opin. Plant Biol. 8, 135–141 (2005).
Chen, Z. J. Genetic and epigenetic mechanisms for gene expression and phenotypic variation in plant polyploids. Annu. Rev. Plant Biol. 58, 377–406 (2007).
Wang, J. L. et al. Genome wide nonadditive gene regulation in Arabidopsis allotetraploids. Genetics 172, 507–517 (2006).
Hegarty, M. J. et al. Transcriptome shock after interspecific hybridization in Senecio is ameliorated by genome duplication. Curr. Biol. 16, 1652–1659 (2006).
Adams, K. L. & Liu, Z. Expression partitioning between genes duplicated by polyploidy under abiotic stress and during organ development. Curr. Biol. 17, 1669–1774 (2007).
Adams, K. L. & Wendel, J. F. Allele-specific, bi-directional silencing of an alcohol dehydrogenase gene in different organs of interspecific cotton hybrids. Genetics 17, 2139–2142 (2006).
Hovav, R. et al. Partitioned expression of duplicated genes during development of a single cell in a polyploid plant. Proc. Natl. Acad. Sci. USA 105, 6191–6195 (2008).
Flagel, L. E., Udall, J. A., Nettleton, D. & Wendel, J. F. Duplicate gene expression in allopolyploid Gossypium reveals two temporally distinct modes of expression evolution. BMC Biol. 6, 16 (2008).
Rapp, R. A., Udall, J. A. & Wendel, J. F. Genomic expression dominance in allopolyploids. BMC Biol. 7, 18 (2009).
Krapovickas, A. & Seijo, G. Gossypium ekmanianum (Malvaceae), algodón silvestre de la República Dominicana. Bonplandia 17, 55–63 (2008).
Grover, C. E. et al. Homoeolog expression bias and expression level dominance in allopolyploids. Plant J. 196, 966–971 (2012).
Grover, C. E., Grupp, K. K., Wanzek, R. J. & Wendel, J. F. Assessing the monophyly of polyploid Gossypium species. Plant Syst. Evol. 298, 1177–1183 (2012).
Udall, J. A. A novel approach for characterizing expression levels of genes duplicated by polyploidy. Genetics 173, 1823–1827 (2006).
Flagel, L. E. & Wendel, J. F. Evolutionary rate variation, genomic dominance and duplicate gene expression evolution during allotetraploid cotton speciation. New Phytol. 186, 184–93 (2010).
Yoo, M. J., Szadkowski, E. & Wendel, J. F. Homoeolog expression bias and expression level dominance in allopolyploid cotton. Heredity 110, 171–180 (2013).
Bardil, A., De Almeida, J. D., Combes, M. C., Lashermes, P. & Bertrand, B. Genomic expression dominance in the natural allopolyploid Coffea arabica is massively affected by growth temperature. New Phytol. 192, 760–774 (2011).
Hovav, R. et al. A majority of cotton genes are expressed in single-celled fiber. Planta 227, 319–329 (2007).
Costa, V., Angelini, C., De Feis., I. & Ciccodicola, A. Uncovering the complexity of transcriptomes with RNA-Seq. J. Biomed. Biotechnol. 2010, 853916 (2010).
Wang, Z., Gerstein, M. & Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63 (2009).
Bachem, C. W. et al. Visualization of differential gene expression using a novel method of RNA fingerprinting based on AFLP: analysis of gene expression during potato tuber development. Plant J. 9, 745–753 (1996).
Comai, L. Genetic and epigenetic interactions in allopolyploid plants. Plant Mol. Biol. 43, 387–399 (2000).
Lee, H. S. & Chen, Z. J. Protein‐coding genes are epigenetically regulated in Arabidopsis polyploids. Proc. Natl. Acad. Sci. USA 98, 6753–6758 (2001).
Madlung, A. et al. Remodeling of DNA methylation and phenotypic and transcriptional changes in synthetic Arabidopsis allotetraploids. Plant Physiol. 129, 733–746 (2002).
Xiao, X. H., Li, H. P. & Tang, C. R. A silver-staining cDNA-AFLP protocol suitable for transcript profiling in the latex of Hevea brasiliensis (para rubber tree). Mol Biotechnol. 42, 91–99 (2009).
Fukumura, R. et al. A sensitive transcriptome analysis method that can detect unknown transcripts. Nucleic Acids Res. 31, e94 (2003).
Song, Y. et al. Transcriptional profiling by cDNA-AFLP analysis showed differential transcript abundance in response to water stress in Populus hopeiensis. BMC Genomics 13, 286 (2012).
Gupta, N., Naik, P. K. & Chauhan, R. S. Differential transcript profiling through cDNA-AFLP showed complexity of rutin biosynthesis and accumulation in seeds of a nutraceutical food crop (Fagopyrum spp.). BMC Genomics 13, 231 (2012).
Claverie, M. et al. cDNA-AFLP-based genetical genomics in cotton fibres. Theor. Appl. Genet. 124, 665–683 (2012).
Ma., X. et al. Analysis of differentially expressed genes in genic male sterility cotton (Gossypium hirsutum L.) using cDNA-AFLP. J. Genet. Genom. 34, 536–543 (2007).
Leng, C. X., Li, F. G., Chen, G. Y. & Liu, C. L. cDNA-AFLP analysis of somatic embryogenesis at early stage in TM-1 (Gossypium hirsutum L.). Acta Botanica Boreali-Occidentalia Sinica 27, 233–237 (2007).
Adams, K. L., Cronn, R., Percifield, R. & Wendel, J. F. Genes duplicated by polyploidy show unequal contributions to the transcriptome and organ-specific reciprocal silencing. Proc. Natl. Acad. Sci. USA. 100, 4649–4654 (2003).
Liu, R., Wang, B., Guo, W., Wang, L. & Zhang, T. Differential gene expression and associated QTL mapping for cotton yield based on a cDNA-AFLP transcriptome map in an immortalized F2. Theor. Appl. Genet. 123, 439–454 (2011).
Fu, W. F. et al. Acyl-CoA N-acyltransferase influences fertility by regulating lipid metabolism and jasmonic acid biogenesis in cotton. Scientific Reports 5, 11790 (2015).
Mackey, D., Belkhadir, Y., Alonso, J. M., Ecker, J. R. & Dangl, J. L. Arabidopsis RIN4 is a target of the type III virulence effector AvrRpt2 and modulates RPS2-mediated resistance. Cell 112, 379–389 (2003).
Vuylsteke, M., Peleman, J. D. & van Eijk, M. J. AFLP-based transcript profiling (cDNA-AFLP) for genome-wide expression analysis. Nature Protocol 2, 1399–1413 (2007).
Schmid, R. & Blaxter, M. L. annot8r: GO, EC and KEGG annotation of EST datasets. BMC Bioinforma. 9, 180 (2008).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408 (2001).
Acknowledgements
The authors would like to thank Professor Zhenming Pei of Duke University for numerous helpful discussions and comments. We acknowledge the support of The National Key Research and Development Program of China (2016YFD0100203-7), Zhejiang Provincial Natural Science Foundation under Grant No. LR14C130001 and General Program of National Natural Science Foundation of China (31671738), State Key Laboratory of Cotton Biology Open Fund (CB2017A01).
Author information
Authors and Affiliations
Contributions
Y.S. and L.K. conceived and designed the experiments. X.Y., B.L., Q.L. and M.Z. performed the research. L.K. participated in data analysis. Y.S and J.S. wrote and corrected the article. All of the authors discussed the results and commented on the article. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ke, L., Luo, B., Zhang, L. et al. Differential transcript profiling alters regulatory gene expression during the development of Gossypium arboreum, G.stocksii and somatic hybrids. Sci Rep 7, 3120 (2017). https://doi.org/10.1038/s41598-017-03431-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-03431-3
This article is cited by
-
CleanBSequences: an efficient curator of biological sequences in R
Molecular Genetics and Genomics (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
