
Abstract
We present the formation possibility for Pd-hydrides and Pd-Rh hydrides system by density functional theory (DFT) in high pressure upto 50 GPa. Calculation confirmed that PdH2 in face-centered cubic (fcc) structure is not stable under compression that will decomposition to fcc-PdH and H2. But it can be formed under high pressure while the palladium is involved in the reaction. We also indicate a probably reason why PdH2 can not be synthesised in experiment due to PdH is most favourite to be formed in Pd and H2 environment from ambient to higher pressure. With Rh doped, the Pd-Rh dihydrides are stabilized in fcc structure for 25% and 75% doping and in tetragonal structure for 50% doping, and can be formed from Pd, Rh and H2 at high pressure. The electronic structural study on fcc type Pd x Rh1−xH2 indicates the electronic and structural transition from metallic to semi-metallic as Pd increased from x = 0 to 1.
Similar content being viewed by others
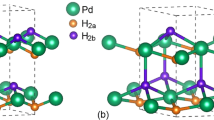

Introduction
As it is well known that metal hydrides are very interesting systems because of their favourable characteristics including hydrogen-storage capacity, kinetics, toxicity, cyclic behaviour, pressure and thermal response1. Especially, the hydrides of platinum group metals are highly attractive due to a number of favourable properties. For instance, the hydrogen absorption by palladium is reversible and therefore has been investigated for hydrogen storage2, the catalytic properties, kinetic reversibility3 and superconductivity4, 5 of palladium hydrides also have been investigated.
It has been reported that hydrogen atoms randomly occupy the octahedral interstices in the Pd-metal lattice with neutron diffraction studies. The limit of absorption at normal pressures is PdH0.7, indicating that approximately 70% of the octahedral holes are occupied6. Hydrogen absorption in Rh requires extremely high hydrogen pressures (of the order of GPa)7 and under normal conditions this metal can only adsorb hydrogens on the surface. Recently, rhodium dihydride was discovered as a first dihydride compound in the platinum group metals by compressing rhodium in fluid hydrogen8. The mechanical stability, thermodynamic and elastic properties of RhH2 were also studied9. With the discovery of RhH2, the dihydride of platinum group metals with tetrahedral sites occupied structure was considered to construct the dihydrides of palladium and Pd-Rh-H system alloys.
It is known that the addition of a second metal to palladium changes hydrogen absorption properties of system. It is a consequence of the alteration of crystal lattice structure, elastic and electronic properties10,11,12,13. Among various Pd alloys, the Pd-Rh system is an exceptional system because the amount of absorbed hydrogen in Pd-rich Pd-Rh alloys is larger than in case of pure Pd14. This is in contrast to the general rule that Pd alloys with a non-absorbing metal (e.g., Au, Ag and Pt) are characterised by a decrease in the maximum amount of absorbed hydrogen15. An Pd-Rh alloy containing 92.6 at.% Pd has been characterised by the highest hydrogen absorption capacity with H/M ratio exceeding 0.8016 using cyclic voltammetry and chronoamperometry in acidic solution.
In this work, we have calculated the formation enthalpy of the hydrides (mono-, di- and tri-) of palladium and rhodium and also Pd-Rh dihydride alloys using DFT approach under high pressure to study the formation possibility. The electronic structure of Pd-Rh dihydride alloys are also analysed by total and partial density of states calculation. The concentration of Pd in Pd-Rh dihydride system alloys is 25%, 50% and 75%, respectively.
Results and Discussion
Crystal Structure
The total energy of Pd-Rh-H compounds as a function of volume are shown in Fig. 1. The results show that the PdH2 compound in fcc phase is energetically more stable than in hcp phase for the volume range from 7.5 Å 3/atom to 6.1 Å 3/atom. PdH3 in hcp phase is more stable than fcc phase for the volume range of 7.1 Å 3/atom to 5.6 Å 3/atom.

Total energy as a function of volume of fcc and hcp phases for both PdH2 and PdH3.
In our calculations, the metal hydride are stabilized in fcc and hcp structures. For monohydride compound PdH, the crystal structure is fcc in space group Fm3m and the hydrogen atom resides in a octahedral sites. For dihydride compounds PdH2 and RhH2, the crystal structures are same with monohydrides, but the hydrogen atom resides the tetrahedral sites. The trihydride compound RhH3 is stabllized in fcc structure, in which the hydrogen atoms are occupied in both tetrahedral and octahedral sites. but PdH3 is stabllized in hcp structure in space group of P63/mmc.
Table 1 shows the lattice parameters and bulk modulus of all the compounds compared with experimental data. The bulk modulus are obtained by fitting B-M equation of state with fixed B0′ = 4. The bulk modulus B0 for Rh-hydrides are larger than Pd-hydrides. As the insertion of H in the octahedral sites of Pd leads to an expansion of the lattice constant from Pd to PdH, the bulk modulus are increased as hydrogen concentration increased. With Rh doped, the bulk modulus of Pd x Rh1−x H2 are increased with various Rh concentration from 0% to 100%.
Formation possibility driven by high pressure
The enthalpy deferences for Pd-H and Rh-H systems are shown in Fig. 2(a). For PdH and RhH, the enthalpy energy differences is regarding to the chemical reaction equations:

(a) The formation enthalpy difference for Pd-H and Rh-H systems. (b) The formation enthalpy for Pd x Rh1−x H2 alloy system. The combined enthalpy of the stable constituent elements establish the reference line corresponding to each compound expressed with dash. ReferenceLine = ∑ [xH(Pd) + (1 − x)H(Rh) + 2H(0.5H 2)].
The PdH has the negative enthalpy of formation, which is consistent with the knowledge that the formation of PdH should be favourable under pressure17. The PdH2 also should be formed under pressure higher than 2.1 GPa due to the negative enthalpy of formation. The formation properties for RhH and RhH2, which both compounds can be formed under pressure, are in good aggreement with the recent work of rhodium dihydride8. Whereas for the trihydirde compounds, the positive enthalpy of formation for PdH3 and RhH3 suggest that they are unfavourable to be formed even with compressing upto 10 GPa.
The formation enthalpy of Pd x Rh1−x H2 system as a function of pressure is shown in Fig. 2(b). As is shown, the negative enthalpy of formation for Pd0.5Rh0.5H2 in the range of pressure suggests it can be formed even at ambient pressure. While for Pd0.25Rh0.75H2 and Pd0.75Rh0.25H2, the formation enthalpy convert to negative at 0.1 GPa and 0.4 GPa, respectively. Therefore they are more favour to be formed when pressure respectively above 0.1 GPa and 0.4 GPa. Besides, with pressure increasing, the decrease trend of negative formation enthalpy for Pd x Rh1−x H2 suggests they are more likely to be formed with compressing.
The enthalpy difference of Pd-Rh-H were carried out in total enthalpy between production compound Pd x Rh1−x H2 and the sum enthalpy of reaction compounds Pd, Rh, and H2:
Consider the reaction of Pd and H2, PdH as a product of reaction, will compete with PdH2 in all range of pressure. To make a further investigation, three reaction paths of PdH2 are figured out which respectively is
Figure 3 shows the reaction enthalpy of PdH2 with compression upto 50 GPa. The enthalpy of reaction 5 keeps positive in the range of pressure, which suggests PdH and H2 is more favourable competing with PdH2. Whereas the reaction 6 suggests a decrease trend on the reaction enthalpy with pressure increase, and the reaction enthalpy convert to negative at 5.5 GPa. In this case, the PdH2 is more likely to be formed than PdH, Pd and H2 when pressure above 5.5 GPa. Therefore, summarising the three reactions above, we conclude that PdH2 is metastable and will directly dissociate into PdH and H2.

Reaction enthalpy of PdH2 according to the three reactions paths: (a) PdH2 = Pd + H 2; (b) 2PdH 2 = 2Pd + H 2; (c) \(4Pd{H}_{2}=2PdH+2Pd+3{H}_{2}.\)
Electronic Structure Properties
The density of state (DOS) of Pd-Rh-H compounds are calculated at equilibrium volume, as shown in Fig. 4(a). The electronic structure indicated a mixture of metallic and covalent bonding. Below Fermi level there are only occupied states by metal Rh or Pd, and hydrogen electron located in a deeply lower valence band and above fermi level states. In the doped hydrides Pd x Rh1−x H2, d-electron of Palladium shows a strong itinerant electronic properties than Rhodium. By replace 25%, 50%, 75% and 100% Rh atoms with Pd in fcc RhH2, the fermi surface shift to lower energy from 8.24 eV for RhH2 to 8.06 eV, 7.78 eV, 7.62 eV and 8.03 eV for fcc type PdH2, respectively. The DOS of PdH2 shows a semimetallic property in which the electronic states around fermi level is less than 0.06.

(a) Density of state of Pd-Rh-H system at ambient pressure. (b) Density of state of palladium hydrides at ambient pressure.
Figure 4(b) shows the electronic density of states as a function of hydrogen concentration on PdH x series while x goes from 0 to 3. In PdH due to the number of H-filled Pd octahedra, the hybridized band H s /Pd d appeared and partially filled on the range of −6.2 ∼ −7.9 eV. In additional, the valence/conduction band on the fermi level is dominated by the 4d orbitals of the palladium atoms in PdH, but the fermi level shifted from 8.94 eV for pure Pd to 8.86 eV for PdH. The electronic states on fermi level are decreased from 3.32 to 0.51 states/eV/f.u.
When hydrogen number increases to 2, two structures fcc and hcp PdH2 are considered. The dispersion of the density of states for fcc PdH2 mainly followed the curve on fcc PdH, in which fermi level shift from 8.86 eV for PdH to 8.03 eV for PdH2. The observed difference is the electronic property that changed from metallic to “near insulator” owing to the density of states on fermi level tends to zero. For hcp PdH2 that electron dispersion is following the curve on hcp PdH3, in which hydrogen s orbital contributes to the valence/conduction band on the fermi level.
Conclusion
In conclusion, three different types of palladium and rhodium hydrides and Pd-Rh-H dihydride alloys have been investigated by first principle calculation. We have found that PdH2 is not stable and dissociate to PdH and H2 at ambient and even higher pressure. While when palladium is involved in the reaction, PdH2 can be easy formed from lower pressure around 10 GPa. With Rh doping alloy hydrides Pd x Rh1−x H2 is formed from fcc metal Pd and Rh in H2 atmosphere at even lower pressure. The electronic density of states investigations show that the Pd x Rh1−x H2 has metallic properties whereas PdH2 semimetallic property.
Methods
The DFT calculations are carried out by employing the Vienna Ab-initio Simulation Package(VASP)18 implementing the Projector Augmented Wave method. The generalized gradient approximation19 was used for the correlation energy function20 with the Perdew Burke Ernzerhof parameterisation. The valence electron configurations for Pd and Rh were 4p65 s14d9 and 4p65 s14d8, respectively. The relaxation convergence for ions and electrons are 10−2 and 10−6 eV, respectively. The electronic wave function was expanded in a plane wave with an energy cut-off 800 eV. For energy formation calculation, the 24 × 24 × 24 Monkorst-Pack(MP) K mesh for Brillouin zone integration was used for the structure optimisation and static calculation. For DOS calculations, the K mesh was increased to 32 × 32 × 32 for fcc compounds, and 15 × 15 × 7 mesh for hcp PdH3, respectively.
References
Sakintuna, B., Lamari-Darkrim, F. & Hirscher, M. Metal hydride materials for solid hydrogen storage: A review. Int. J. Hydrogen. Energ. 32, 1121–1140 (2007).
Grochala, W. & Edwards, P. P. Thermal Decomposition of the Non-Interstitial Hydrides for the Storage and Production of Hydrogen. Chem. Rev. 104, 1283–1316 (2004).
Adams, B. D. & Chen, A. The role of palladium in a hydrogen economy. Mater. Today. 14, 282–289 (2011).
Tripodi, P., Gioacchino, D. D., Borelli, R. & Vinko, J. D. Possibility of high temperature superconducting phases in PdH. Physica. C 388–389, 571–572 (2003).
Tripodi, P., Di Gioacchino, D. & Vinko, J. D. Superconductivity in PdH: phenomenological explanation. Physica. C: Superconductivity 408–410, 350–352 (2004).
Klotz, E. & Mattson, B. Hydrogen and Palladium Foil: Two Classroom Demonstrations. J. Chem. Educ. 86, 465 (2009).
Tkacz, M. High pressure studies of the rhodium–hydrogen system in diamond anvil cell. J. Chem. Phys. 108, 2084 (1998).
Li, B. et al. Rhodium dihydride (RhH2) with high volumetric hydrogen density. Proc. Natl. Acad. Sci. USA 108, 18618–18621 (2011).
Pan, G. et al. Ab initio study of mechanical stability, thermodynamic and elastic properties of Rh, RhH, and RhH2 under high temperature and pressure. J. Mater. Res. 29, 1334–1343 (2014).
Burch, R. Theoretical aspects of the absorption of hydrogen by palladium and its alloys. Part 1.-A reassessment and comparison of the various proton models. Trans. Faraday Soc. 66, 736–748 (1970).
Sakamoto, Y., Yuwasa, K. & Hirayama, K. X-ray investigation of the absorption of hydrogen by several palladium and nickel solid solution alloys. J. Less. Common. Met. 88, 115–124 (1982).
Sakamoto, Y., Baba, K. & Flanagan, T. B. The Effect of Alloying of Palladium on the Hydrogen-Palladium Miscibility Gap. Zeitschrift für. Physikalische. Chemie 158, 223–235 (1988).
Wicke, E. & Frölich, K. Electronic and Elastic Effects in the Phase Diagrams of Binary Pd Alloy Hydrides. Zeitschrift für. Physikalische. Chemie 163, 173706 (1989).
Koss, U., Łukaszewski, M., Hubkowska, K. & Czerwiński, A. Influence of rhodium additive on hydrogen electrosorption in palladium-rich Pd–Rh alloys. J. Solid. State. Electr. 15, 2477 (2011).
Brodowsky, H. The Palladium Hydrogen System. Angew. Chem-ger Edit 80, 498–498 (1968).
Koss, U., Hubkowska, K., Łukaszewski, M. & Czerwiński, A. Influence of temperature on hydrogen electrosorption into palladium-noble metal alloys. Part 3: Palladium–rhodium alloys. Electrochim. Acta. 107, 269–275 (2013).
Houari, A., Matar, S. F. & Eyert, V. Electronic structure and crystal phase stability of palladium hydrides. J. Appl. Phys 116, (2014).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci 6, 15–50 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Kohn, W. & Sham, L. J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev 140, A1133–A1138 (1965).
Andersen, O. K. Electronic Structure of the fcc Transition Metals Ir, Rh, Pt and Pd. Phys. Rev. B 2, 883–906 (1970).
Kittel, C. Introduction to Solid State Physics. 23, 59 (John Wiley & Sons, Inc, 1996).
Antonov, V. E., Belash, I. T., Malyshev, V. Y. & Ponyatovsky, E. G. The solubility of hydrogen in the platinum metals under high pressure. Platinum Met Rev 28, 158–163 (1984).
Schirber, J. E. & Morosin, B. Lattice constants of PdHx and PdDx with x near 1.0. Phys. Rev. B 12, 117–118 (1975).
Baranowski, B., Tkacz, M. & Majchrzak, S. Pressure dependence of hydrogen volume in some metallic hydrides in Molecular Systems Under High Pressure. (eds Pucci, R. & G, Piccitto.) 139–156 (Elsevier, 1991).
Acknowledgements
Thanks to SI (Sweden Institute) for VISBY collaboration research funding, SSF-Sweden and NRF-Korea for joint collaborative research funding. W. Luo and R. Ahuja acknowledge the Swedish Research Council (VR) for the financial support. X. Yang and H. Li acknowledges the China Scholarship Council for funding and the financial support from graduate innovation funding project of Hebei (00302-6370012). This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (No. 2014R1A2A1A12066298) The Swedish National Infrastructure for Computing (SNIC) at NSC (Matter and Triolith clusters) and at Umeå (Abisko and Akka clusters) provided the computing facilities for this project.
Author information
Authors and Affiliations
Contributions
W.L. and R.A. designed the project, analysed the results and review the manuscript. X.Y. did the calculation and wrote the manuscript. H.L. and T.K. reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, X., Li, H., Ahuja, R. et al. Formation and electronic properties of palladium hydrides and palladium-rhodium dihydride alloys under pressure. Sci Rep 7, 3520 (2017). https://doi.org/10.1038/s41598-017-02617-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-02617-z
This article is cited by
-
Superconductivity Versus Magnetism in the Amorphous Palladium “Ides”: Pd1−c(H/D/T)c
Journal of Low Temperature Physics (2022)
-
Metastable hexagonal close-packed palladium hydride in liquid cell TEM
Nature (2022)
-
In-situ observation of plasmon-controlled photocatalytic dehydrogenation of individual palladium nanoparticles
Nature Communications (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
