
Abstract
Differentiation of Brucella canis from other Brucella species are mainly performed through PCR-based methods and multilocus variable-number tandem-repeat (VNTR) analysis (MLVA) procedures. Both PCR-based and MLVA methods are limited in discriminating B. canis strains. A new MLVA-13Bc method for B. canis genotyping was established by combining eight newly-developed VNTRs with five published ones. During 2010 and 2016, 377 B. canis PCR-positives were identified from 6,844 canine blood samples from 22 U.S. states, resulting in 229 B. canis isolates. The MLVA-13Bc method was able to differentiate each of these 229 isolates. The Hunter-Gaston Discriminatory Index of the individual VNTR loci ranged from 0.516 to 0.934 and the combined discriminatory index reached 1.000. Three major clusters (A, B and C) and 10 genotype groups were identified from the 229 B. canis isolates. Cluster A mainly contains genotype groups 1 and 2, and a few group 3 isolates; nearly all Cluster B isolates were from group 6; other genotype groups were classified into Cluster C. Our newly developed MLVA-13Bc assay is a highly discriminatory assay for B. canis genotyping, and can serve as a useful molecular epidemiological tool, especially for tracing the source of contamination in an event of a B. canis outbreak.
Similar content being viewed by others

Introduction
Brucella canis is a gram-negative, facultative intracellular pathogen mainly responsible for causing canine brucellosis. B. canis infections can lead to abortion in females and epididymitis and prostatitis in males1. Main routes of transmission are through direct contact with aborted fetus, placenta, fetal fluids, or vaginal discharge2. The organism stays in the animal system longer than other Brucella spp., making intervention more challenging3. An aborting bitch can discharge the bacterium at very high concentrations for 4–6 weeks after a single abortion2, 4. The disease has been widely reported in different continents2, 5,6,7,8, and it has been a major problem in canine breeding facilities1. Infection rate of canine brucellosis is on the rise. The Wisconsin Veterinary Diagnostic Laboratory tested 510 samples in 1995 and 1996, and only resulted in 10 positives (1.96%); Testing on 174 samples during 2003 and 2004 by the same lab resulted in 4.6% positivity9. The positive rate can reach up to 26.8% during an outbreak9. Although it is uncommon, human B. canis infections have been reported10 with symptoms from mild flu-like illnesses to more severe complications11,12,13,14. Thus, its zoonotic potential has become a public health concern.
Variable number of tandem repeat (VNTR) and multilocus VNTR analysis (MLVA) have been widely used for genotyping strains of different bacterial species15,16,17,18,19,20. Several MLVA systems have been developed and described for the genotyping of Brucella species and biovars21,22,23. Bricker et al.21 used a MLVA method named HOOF-Prints with eight tandem repeat (TR) loci (MLVA-8) and was able to differentiate Brucella isolates at both the species and biovar levels. A rather comprehensive screening of 107 TRs identified 15 TRs (MLVA-15) that were more informative and were able to differentiate most Brucella species and for some strains even at the biovar level22. Al Dahouk et al.24 added another TR into the MLVA-15 method to form a new assay called MLVA-16 and was used to study genetic diversity of 128 human B. melitensis strains. The study identified 110 genotypes that differentiated most of the 128 strains, yet the MLVA-16 method provided much lower discriminatory power against the eight B. canis and 18 B. ovis strains in the study. Using MLVA-16, B. suis biovar 1, B. suis biovar 2, B. abortus, B. melitensis and B. ovis can be clearly identified. B. suis biovar 5, B. neotomae and the marine mammal strains are closely related strains, and they can be differentiated by this method. However, this method appeared to be insufficient in differentiating strains within the species of B. canis in different studies, especially for those that were from closely related geographic regions22, 24, 25. Whatmore et al.23 grouped 121 Brucella isolates into 119 genotypes based on 21 VNTR loci. The approach based on 21 VNTR loci provided better strain genotyping information for B. abortus, B. melitensis and B. suis, but was less informative in differentiating strains of B. canis, B. ovis and B. neotomae. Therefore, the goal of this study were: 1) to develop and validate a MLVA genotyping method for the differentiation of B. canis strains; and 2) to study genetic diversity of a collection of 229 B. canis isolates collected from the US in recent years using the newly developed MLVA-13Bc method.
Results
PCR identification and isolation of B. canis strains from canine blood samples
From a total of 6,844 canine blood samples, the duplex diagnostic PCR identified 377 Brucella positives, and all Brucella positives were confirmed to be B. canis strains. The MLVA-13Bc PCR amplifications for 10 genotype groups visualized by QIAxcel were shown in Figure S1. Among the 377 PCR-positive samples, 229 B. canis isolates were obtained. Selected positive samples (n = 20) were further verified by sequencing a region flanking a 976 bp fragment that is deleted only from the B. canis genome, and is intact in all other Brucella species26, 27. Positive samples were observed in 10 states, mainly from the Midwest region, of the US with an average positive rate of 5.5% (377/6,844). The positive rate for the 6,844 samples ranged from 3.8% (58/1,533 IN) to 23.1% (3/13 MS) when sorted by sampling state, and varied from 2.9% (13/445 2015) to 9.1% (24/264 2013) if sorted by sampling year. The remaining 99 samples collected from 12 other states were negative for B. canis (Table 1).
Development and optimization of the MLVA genotyping method for B. canis
We particularly analyzed the polymorphism generated by each of the 16 VNTR loci in the MLVA-16 method22, 24 using published data. Our analysis indicated that five of the 16 loci published earlier should generate genetic polymorphism among different B. canis strains (Table 2), yet may not be enough to differentiate the 229 isolates in our collection. Additional 8 loci (starts with “BCTR”) that can potentially aid to differentiate strains within the B. canis species were selected for the design of new VNTRs. Results of the 13 individual VNTRs on 20 isolates generated large genetic polymorphism, indicating they are useful loci for genotyping strains of B. canis (Figure S2). Based on the average size range and primer annealing temperatures of each VNTR locus, 12 of them were grouped into 6 duplex PCR reactions, namely Reaction 1 (for loci BCTR09 and BCTR06), 2 (BCTR12 and Bruce07), 3 (BCTR03 and Bruce16), 4 (BCTR02 and Bruce09), 5 (BCTR01 and BCTR08), and 6 (BCTR11 and Bruce04), respectively. Reaction 7 is a singular one for locus Bruce18 (Table 2). All primers were specific to targeted regions and did not amplify other regions of the B. canis genome. The results were compared to individual PCR reactions. No interference was observed while comparing the singular and duplex PCR reactions. There were no overlapping amplicons observed between the 2 loci within each duplex reaction. The result of Sanger sequencing (data not shown) was consistent with the size obtained by the QIAxcel Advanced System.
Genotyping 229 B. canis isolates using MLVA-13Bc method
The newly established method (Table 2) was then used for genotyping the remaining 229 isolates. Copy numbers of the tandem repeats for each locus were compiled to indicate the genotype for each isolate, and then used to further group the isolate into one of the 10 genotype groups (Figure S2). Each of the 229 isolates can be differentiated by this newly-developed MLVA-13Bc (MLVA-13 for B. canis) method. To evaluate the discriminatory power of the selected loci, the Hunter and Gaston28 discrimination index (HGDI) was calculated for the MLVA-13Bc method as a combined method, and for each of the 13 loci used in this study. Among these 229 isolates, the newly developed MLVA assay allowed the classification of all isolates to unequivocal genotypes (HGDI = 1.000). The HGDI value ranged from 0.516 to 0.934 among the 13 individual VNTR loci (Table 3).
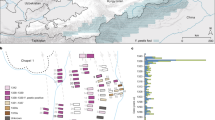
Genotyping of the 229 B. canis isolates by the newly developed MLVA-13Bc method identified 10 genotype groups (group 1 - group 10), with the similarity cutoff at 20% (Figure S2). The US strain 2010009751 was classified into group 1; the Korean strain HSK A52141 was in group 2; US strains ATCC 23365, RM6/66 and 2009004498 were classified into group 3; US strain 2009013648 and the South Africa strain F7/05 A were in group 4; the Argentina strain CNGB 1342 was in group 6; and the Sweden strain SVA13 was in group 9. Group 3 is the largest group with 76 isolates widely spread in 8 states, and it is the predominant group in Colorado (100%) and Kansas (71%). The second largest group, group 6, had strains only from Ohio (71%), Indiana (58%) and Missouri (3%). The 33 isolates in group 2 were mostly obtained from Oklahoma (30); only two from Kansas and one from Missouri. The 22 isolates in group 1 were from Iowa, Missouri, Indiana and Ohio, while the 21 isolates in group 7 were from Idaho, Kansas, Minnesota, Missouri and Indiana, which were widely spread in the US. There were no more than 10 isolates in groups 4, 5, 8, 9 and 10, and each of these groups was observed in only one or two states (Figure S2). Distribution of the 10 genotype groups were also illustrated in a cluster analysis (Insert in Fig. 1).

Geographical and temporal distribution of 229 B. canis isolates in 10 U.S. states. Different colors indicate different states in the main chart, or one of the 10 genotype groups of B. canis isolates in the inserted chart. Red: Colorado in the main chart, or group 1 in the insert chart; dark teal: Iowa or group 2; pink: Idaho or group 3; gray: Indiana or group 4; lavender: Kansas or group 5; light teal: Minnesota or group 6; orange: Missouri or group 7; maroon: Mississippi or group 8; light green: Ohio or group 9; and light orange: Oklahoma or group 10.
Cluster analysis and geographical and temporal distributions of B. canis strains
A cluster analysis of the MLVA allele profiles of the 229 B. canis isolates with nine reference sequences by BioNumerics generated three large clusters, Cluster A, Cluster B and Cluster C, as well as 3 sub-clusters, C1, C2 and C3 (Fig. 1). Distribution the 229 isolates in different states is shown in Fig. 2. Cluster A mainly contained isolates from Oklahoma in 2010 (8/9) and 2011 (22/32), as well as all the isolates from Iowa in 2013 (6/6). Cluster B mainly consisted of isolates from Indiana (19/25) and Ohio in 2013 (18/33), as well as from Ohio in 2012 (6/8). Most isolates in Cluster C were from Kansas (43/45) and Idaho (7/8). All isolates from Colorado (14/14) and Missouri (3/3) were also classified into Cluster C. Isolates from Indiana in 2013 were in Cluster B, while isolates in other years (2012, 2015 and 2016) were all in Cluster C. Isolates in different years were spread in different clusters except samples collected in 2015 and 2016 (Fig. 1). Most isolates in group 1 (81.8%, 18/22) and 2 (94.1%, 32/34) belonged to Cluster A, and most isolates in group 6 (83.6%, 41/49) belonged to Cluster B. All isolates in group 4 (10), 7 (21) and 10 (5) as well as most isolates in group 3 (64/76) and 5 (8/9) belonged to Cluster C. The five isolates each in groups 8 and 9 were located in Cluster A and C, respectively (Insert in Fig. 1).

Distribution of B. canis genotype groups in different states. Number in brackets after the state abbreviation indicate total positives in that state; pie chart indicates percentages of each genotype group in that state. Pie size corresponds to the number of isolates. Those with state abbreviations but without a number indicate samples that were collected but all tested negative (number of submission are shown in Table 1). No sample was collected from those that do not have the state abbreviation. Different colors indicate different genotype groups. Red: genotype group 1; dark teal: group 2; pink: group 3; gray: group 4; lavender: group 5; light teal: group 6; orange: group 7; maroon: group 8; light green: group 9; and light orange: group 10. The map template was downloaded from www.vectortemplates.com (Graphics Factory CC, Western Cape, Sourth Africa) with permission.
Distributions of B. canis isolates in different dog breeds and genders
Breed information was available for 118 B. canis-positive dogs, which included 24 different dog breeds. Isolates from German Shepherd, French Bulldog, Siberian Husky and Bichon Frise were mostly grouped in Cluster A. All four isolates from Cocker Spaniel and five out of 20 isolates from Shih Tzu were in Cluster B. Isolates collected from more breeds were in Cluster C (Supplementary Table S1). These Cluster C isolates include 15 of 20 isolates from Shih Tzu, 11 of 12 from Poodle, 8 of 9 from Pomeranian, all 6, 5, and 4 isolates from Miniature Schnauzer, Chihuahua and Golden Retriever, respectively, and a few each from other breeds. Gender information was available for 133 positive dogs (Supplementary Table S1) including 87 females and 46 males. No correlation between animal gender and B. canis genotype was observed.
Stability of the 13 VNTR loci on B. canis genomes
The original isolates from samples D11-106637 #69 that were collected in 2011, D13-123417 #42 in 2013, and D16-008400 #3 in 2016, and their 10th and 20th subcultures were used for DNA extractions and MLVA-13Bc PCR. For each isolate, identical MLVA-13Bc profiles were generated for the original isolates, and their 10th and 20th subcultures, indicating the 13 VNTRs were stable during propagation. The data of 13 VNTR loci on the original isolate, its 10th and 20th subcultures of sample D13-123417 #42 is shown in Fig. 3.

Electrophoresis images of B. canis isolate D13-123417 #42 amplified with 13 VNTR locus primers after in vitro propagations. Lane A1–A3: original, 10th and 20th subcultures by BCTR09 (upper) and BCTR06 (lower); Lane B1–B3: original, 10th and 20th subcultures by BCTR12 (upper) and Bruce07 (lower); Lane C1–C3: original, 10th and 20th subcultures by BCTR03 (upper) and Bruce16 (lower); Lane D1–D3: original, 10th and 20th subcultures by BCTR02 (upper) and Bruce09 (lower); Lane E1–E3: original, 10th and 20th subcultures by BCTR01 (upper) and BCTR08 (lower); Lane F1–F3: original, 10th and 20th subcultures by BCTR11 (upper) and Bruce04 (lower); Lane G1–G3: original, 10th and 20th subcultures by Bruce18. Lane M1: QX Size Marker 50–800 bp. Lane M2: QX Size Marker FX174/HeaIII.
Discussion
A highly discriminatory MLVA assay was developed and validated with a relatively large number of B. canis isolates. With the combination of the 13 VNTRs, each of the 229 isolates were differentiated. This MLVA-13Bc assay was specifically designed for genotyping of B. canis strains with high discriminatory power. The MLVA-13Bc assay established and validated in this study can be a useful tool in epidemiological studies, especially during B. canis outbreaks.
The MLVA-16 assay24 is probably the most used method for genotyping different Brucella spp., and for strains within certain Brucella spp. including B. canis. As the method was not designed specifically to differentiate strains within B. canis, it provided limited discriminatory power against B. canis strains. For example, MLVA-16 differentiated the 29 Chinese B. canis strains into 21 genotypes, as reported by Di et al.25. Kang et al.29 classified 77 Korean B. canis strains into 30 genotypes using the same procedure. As we are using a different procedure, those strains could not be in silico analyzed except for the nine reference strains that have full or near full genome sequences. Our MLVA-13Bc assay differentiated each of the 229 strains used in this study. In comparison, the five most informative loci from the MLVA-16 assay classified the 229 strains into 131 genotypes. To further confirm that the remaining 11 VNTRs in the MLVA-16 assay would not generate useful information for B. canis strains, we have tested the 11 primer pairs on each of the genotype groups, and resulted identical amplicon sizes (Figure S3). Our data from this study together with other published data suggested that the remaining 11 VNTRs from the MLVA-16 assay do not provide useful discriminatory power for B. canis genotyping.
A survey on canine infection with B. canis was carried out in 22 U.S. states between 2010 and 2016, and the result indicated that the total average positive rate is 5.5% (377/6,844; Table 1). Although B. canis can cause human infections, information of the zoonotic potential of these 229 strains are not available. Due to not all positive samples in years 2010 and 2015, and none in 2014 were subjected to bacterial isolation, the more realistic B. canis isolation rate is reflected by data in years 2011, 2012, 2013 and 2016, which was 79.5% (194/244). This data demonstrated a good correlation between the culture and the molecular detection methods, yet about 20% of PCR positives would not be detected by the traditional culture method, indicating PCR can be more sensitive in B. canis detection.
Dog breed information was available for about half of our strain collection. We identified 24 different dog breeds from 118 out of 229 B. canis isolates. Although we did not see a clear-cut distribution, isolates from certain dog breeds were primarily located in a given cluster (Table S1). Clusters A and B are smaller clusters. Isolates from German Shepherd, French Bulldog, Siberian Husky and Bichon Frise were mostly grouped in Cluster A, and all four isolates from Cocker Spaniel and five out of 20 isolates from Shih Tzu were in Cluster B. We also have dog age information for 60 samples, but there was no correlation between dog age and isolate genotypes observed (Table S1).
There were 133 positive samples (Supplementary Table S1) that were indicated with animal genders. Although we received more samples from females (n = 87) than males (n = 46), it is worth mentioning that we received a significant portion from B. canis-barring male animals that has not been extensively documented, as most of the reports were on abortion cases9, 30. Our data indicated that the organism can circulate in blood streams of both male and female animals. Thus for B. canis screening, especially for breeding facilities, it is important to screen both female and male breeding dogs.
Our MLVA genotyping data on 229 B. canis strains may be the largest genotyping study for B. canis isolates in the US. Although the distribution of different genotypes in each state was not clear-cut, some interesting information was revealed. Genotype group 6 was the predominant genotype for both Indiana and Ohio, which are neighbor states. Group 1 isolates were mainly distributed in Missouri, Iowa and Indiana, states that are geographically related. Group 2 isolates were mainly found in Oklahoma and some in Missouri, which are also bordering states. Groups 3 and 7 though were widely distributed in different states.
In a temporal distribution analysis for these 229 B. canis isolates, all 18 samples collected in 2016 were in Cluster C. Most isolates in 2015 (12/13) were in Cluster C, only one was in Cluster A. Most isolates collected from 2011 (28/38) were in Cluster A, some (9/38) were in Cluster C and only one in Cluster B.
In conclusion, most currently-used MLVA assays (MLVA-8, MLVA-10, MLVA-11, MLVA-15 and MLVA-16) were not specifically designed for B. canis at the strain level, although they are good molecular genotyping methods of Brucella at the species, biovar or at strain level for certain Brucella species. Our newly developed MLVA-13Bc method was specifically designed to discriminate strains from within the species of B. canis, and had higher discriminatory power than previous assays25, 31, 32. Although our data is not associated with pathogenesis as such information is not available, the new MLVA-13Bc method genotyped each of the 229 isolates in our collection, indicating that it is a useful molecular epidemiological tool to trace the source of strain introduction in an event of a B. canis outbreak.
Methods
B. canis sample preparation
From January 2010 to May 2016, canine whole blood samples (n = 6,844) from 22 states were submitted to the Kansas State Veterinary Diagnostic Laboratory (KSVDL) for B. canis identifications (Table 1). All samples were diagnostic samples submitted by clients and no experimental samples were used. All procedures were carried out strictly following institutional biosafety procedures. Samples were freshly collected in sodium citrate (blue-top) or EDTA (purple-top) blood collection tubes, and delivered to KSVDL on ice for next-day delivery. Upon receiving, samples were accessioned and 1 ml blood was transferred into 5 mL Brain Heart Infusion (BHI) broth (HARDY Diagnostics, Santa Maria, CA, USA), mixed, and kept at −80 °C for 3 h (for samples received in the mornings) or at −20 °C for overnight (for those received in the afternoons), then incubated at 37 °C aerobically in a shaker incubator for 40–48 h prior to DNA extraction. A duplex PCR assay was used for B. canis identification. Isolates used in this study were confirmed by culture isolation followed by PCR confirmation on single colonies, and by sequencing on selected isolates (detailed below).
DNA extraction, PCR identification and sequencing confirmation
After the 40–48 h incubation, 150 µL of culture was used for DNA extraction with the DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA, USA) following the manufacturer’s protocol. Extracted DNA was subjected to a duplex PCR assay that was composed of two molecular targets: a 308 bp fragment of the rRNA gene that is present in all Brucella spp. (forward primer: BruCom-F4, CCGCCTTCGTTTCTCTTTCT; reverse primer: BruCom-R4, GGGATCGAACCGACGAC); and a 185 bp B. canis-specific region (1,161 bp for non-canis Brucella spp.) flanking the 976 bp deletion27 that occurs only in B. canis strains (forward primer: BcanisF, GGCTGTCAAGGCGATAAAAC; reverse primer: BcanisR, CAGCTTTACTGCCGGGTTAG). The 20 µl PCR reaction contained 1 µl of the primer mix (0.4 µM for each primer), 10 µL of BioRad (Hercules, CA) iQ Multiplex Powermix, 2 µL of extracted DNA, and 7 µL nuclease-free water. The PCR amplification program included a 5 min denaturation at 95 °C, followed by 35 cycles of 95 °C for 20 sec, and 60 °C for 50 sec. The PCR products were separated and visualized on a Qiagen QIAxcel capillary electrophoresis system. The 185 bp PCR product from selected isolates was outsource sequenced for confirmation.
Isolation of B. canis from PCR positive samples
Culture isolation of B. canis was achieved by streaking the BHI culture on 5% blood agar plates (HARDY Diagnostics). The inoculated plates were incubated at 37 °C aerobically for 72–120 h. Single colonies morphologically consistent with Brucella were sub-cultured in BHI broth for 24 h, and confirmed by PCR. All isolates confirmed as B. canis were stored with 15% glycerol at −80 °C for long-term use.
Development of the Multiple Locus Variable-Number Tandem Repeat Analysis method for B. canis genotyping
Published data using MLVA-1522 or MLVA-1624, 29 methods were analyzed with an emphasis on their ability to differentiate B. canis strains. The purpose of analysis on published data was to select those VNTRs that generated polymorphism on B. canis strains and to compile them with new VNTRs (described below) to form a new MLVA method that can be used for genotyping of B canis strains with increased discriminatory power. An effort of identifying additional VNTR loci was performed by searching B. canis whole genome sequences (chromosome I: NC_010103, NC_016778, NZ_CP007758, NZ_CP007629; chromosome II: NC_010104, NC_016796, NZ_CP007759, NZ_CP007630) using Tandem Repeats Finder described by Benson33. Additional VNTR loci were selected based on following criteria recommended by Nadon et al.34: all loci had no insertions and deletions in the tandem repeats; each tandem repeat was equal or greater than 8 bp; and the loci had conserved flanking sequences from which the PCR primers can be identified. Primers flanking each locus were designed using the online primer design software, Primer 3 (http://bioinfo.ut.ee/primer3-0.4.0/). Twenty B. canis isolates selected from different dog breeds and different colleting states spanning different years were used to evaluate and optimize the method. All individual VNTR were run on the 20 isolates first, and the optimized protocol was used for the remaining isolates.
Multiple Locus Variable-Number Tandem Repeat Analysis procedure
B. canis stocks stored at −80 °C were streaked on blood agar plates, and cultured at 37 °C for 72–120 h. A single colony from each plate was inoculated into 5 ml BHI broth and incubated at 37 °C with shaking for 48–72 h. The resulting culture was used for genomic DNA extraction with Qiagen DNeasy Blood & Tissue Kit. Takara Premix Taq™ DNA Polymerase Hot-Start Version kit (Clontech Laboratories, Mountain View, CA, USA) was used for PCR amplifications. The 20 μL PCR reaction contains 11.9 μL (13.4 μL for Reaction 7) double distilled water, 2 μL 10 × buffer, 1.5 μL each primer pair (final concentration of 375 pM for each primer), 1 μL dNTP Mix (2.5 mM each), 0.1 μL rTaq enzyme, and 2 μL of extracted DNA. The thermal cycling profile consisted of an initial denaturation at 95 °C for 5 min, followed by 35 cycles of 95 °C for 30 sec, 58 °C for Reactions 1, 2 and 3, 60 °C for Reactions 4 and 7, or 62 °C for Reactions 5 and 6 for 30 sec and 72 °C for 45 sec, plus a final extension step at 72 °C for 5 min. VNTR PCR amplicons were separated and visualized on a Qiagen QIAxcel Advanced System with QX DNA Size Marker FX174/III and QX Size Marker 50–800 bp. Fragment analysis was performed with QIAxcel Biocalculator software. Selected amplicons of different sizes obtained by QIAxcel Advanced System were verified by Sanger DNA sequencing.
MLVA data analysis
The genotype of each isolate was identified by compiling the genotype generated by each VNTR locus. The phylogenetic tree was generated using an unweighted pair group method with arithmetic mean (UPGMA) method35 that is embedded in BioNumerics version 7.6 (Applied Maths, Austin, TX). The cluster analysis was performed using the UPGMA with a minimum spanning tree (MST) and distance matrices for categorical data by BioNumerics version 7.6. The Geographic distribution map template was downloaded from www.vectortemplates.com (Graphics Factory CC, Western Cape, Sourth Africa) with permission, and the added pie charts were constructed using BioNumerics version 7.6. Hunter and Gaston discrimination index (HGDI) was calculated using Ridom EpiCompare software version 1.0 (www.ridom.de/epicompare/) to elucidate the discriminatory power of the genotyping methods, which explained the probability of two unrelated and different isolates sampled from the test population grouping as different subtypes by a specific typing method28. The 95% confidence intervals (CI) were calculated according to the method previously described36.
VNTR loci stability of the B. canis genome
In order to investigate the stability of the 13 VNTR loci in the B. canis genome, three B. canis isolates collected from 2011 (D11-106637 #69), 2013 (D13-123417 #42), and 2016 (D16-008400 #3) were subcultured by inoculating 50 μL of stock culture into 5 ml BHI medium and incubated at 37 °C for 2 days. A new 5 ml BHI broth was inoculated with 50 μL of fresh culture, and incubated at 37 °C for 2 days. A total of 20 consecutive subcultures were made for each strain. Portions of each subculture were frozen at −20 °C till use. When 20 subcultures were completed, the original culture, subculture number 10 and number 20 were used for DNA extraction and PCR amplification with the newly developed genotyping method.
References
Wanke, M. M. Canine brucellosis. Anim Reprod Sci 82–83, 195–207 (2004).
Hollett, R. B. Canine brucellosis: outbreaks and compliance. Theriogenology 66, 575–87 (2006).
Xavier, M. N., Paixão, T. A., Hartigh, A. B., Tsolis, R. M. & Santos, R. L. Pathogenesis of Brucella spp. The Open Veterinary Science Journal 4, 109–118 (2010).
Olsen, S. C. & Palmer, M. V. Advancement of knowledge of Brucella over the past 50 years. Vet Pathol 51, 1076–89 (2014).
Corbel, M. J. Brucellosis: an overview. Emerg Infect Dis 3, 213–21 (1997).
Sanchez-Jimenez, M. M., Isaza, J. P., Alzate, J. F. & Olivera-Angel, M. Comparison of Brucella canis genomes isolated from different countries shows multiple variable regions. Genomics 106, 43–51 (2015).
Holst, B. S. et al. The first case of Brucella canis in Sweden: background, case report and recommendations from a northern European perspective. Acta Vet Scand 54, 18 (2012).
Hofer, E. et al. First detection of Brucella canis infections in a breeding kennel in Austria. New Microbiol 35, 507–10 (2012).
Brower, A. et al. Investigation of the spread of Brucella canis via the U.S. interstate dog trade. Int J Infect Dis 11, 454–8 (2007).
Lucero, N. E., Escobar, G. I., Ayala, S. M. & Jacob, N. Diagnosis of human brucellosis caused by Brucella canis. J Med Microbiol 54, 457–61 (2005).
Lucero, N. E. et al. Brucella canis causing infection in an HIV-infected patient. Vector Borne Zoonotic Dis 10, 527–9 (2010).
Marzetti, S., Carranza, C., Roncallo, M., Escobar, G. I. & Lucero, N. E. Recent trends in human Brucella canis infection. Comp Immunol Microbiol Infect Dis 36, 55–61 (2013).
Krueger, W. S., Lucero, N. E., Brower, A., Heil, G. L. & Gray, G. C. Evidence for unapparent Brucella canis infections among adults with occupational exposure to dogs. Zoonoses Public Health 61, 509–18 (2014).
de la Cuesta-Zuluaga, J. J., Sanchez-Jimenez, M. M., Martinez-Garro, J. & Olivera-Angel, M. Identification of the virB operon genes encoding the type IV secretion system, in Colombian Brucella canis isolates. Vet Microbiol 163, 196–9 (2013).
Timmons, C. et al. Multiple-locus variable-number tandem repeat analysis for strain discrimination of non-O157 Shiga toxin-producing Escherichia coli. J Microbiol Methods 125, 70–80 (2016).
Ranjbar, R., Ahmadi, M. & Memariani, M. Multiple-locus variable-number tandem repeat analysis (MLVA) for genotyping of Salmonella enterica subspecies enterica serotype Infantis isolated from human sources. Microb Pathog 100, 299–304 (2016).
Du, Q. et al. Direct molecular typing of Bordetella pertussis from nasopharyngeal specimens in China in 2012–2013. Eur J Clin Microbiol Infect Dis 35, 1211–4 (2016).
Anniballi, F. et al. Multiple-locus variable number of tandem repeat analysis as a tool for molecular epidemiology of botulism: The Italian experience. Infect Genet Evol 46, 28–32 (2016).
Chalmers, R. M. et al. Suitability of loci for multiple-locus variable-number of tandem-repeats analysis of Cryptosporidium parvum for inter-laboratory surveillance and outbreak investigations. Parasitology, 1–11 (2016).
Hu, Y. et al. Identification of Variable-Number Tandem-Repeat (VNTR) Sequences in Acinetobacter pittii and Development of an Optimized Multiple-Locus VNTR Analysis Typing Scheme. Biomed Environ Sci 28, 855–63 (2015).
Bricker, B. J., Ewalt, D. R. & Halling, S. M. Brucella ‘HOOF-Prints’: strain typing by multi-locus analysis of variable number tandem repeats (VNTRs). BMC Microbiol 3, 15 (2003).
Le Fleche, P. et al. Evaluation and selection of tandem repeat loci for a Brucella MLVA typing assay. BMC Microbiol 6, 9 (2006).
Whatmore, A. M. et al. Identification and characterization of variable-number tandem-repeat markers for typing of Brucella spp. J Clin Microbiol 44, 1982–93 (2006).
Al Dahouk, S. et al. Evaluation of Brucella MLVA typing for human brucellosis. J Microbiol Methods 69, 137–45 (2007).
Di, D. et al. Genetic polymorphism characteristics of Brucella canis isolated in China. PLoS One 9, e84862 (2014).
Rajashekara, G., Glasner, J. D., Glover, D. A. & Splitter, G. A. Comparative whole-genome hybridization reveals genomic islands in Brucella species. J Bacteriol 186, 5040–51 (2004).
Garcia-Yoldi, D. et al. Multiplex PCR assay for the identification and differentiation of all Brucella species and the vaccine strains Brucella abortus S19 and RB51 and Brucella melitensis Rev1. Clin Chem 52, 779–81 (2006).
Hunter, P. R. & Gaston, M. A. Numerical index of the discriminatory ability of typing systems: an application of Simpson’s index of diversity. J Clin Microbiol 26, 2465–6 (1988).
Kang, S. I. et al. Genetic comparison of Brucella canis isolates by the MLVA assay in South Korea. J Vet Med Sci 73, 779–86 (2011).
Gyuranecz, M. et al. Detection of Brucella canis-induced reproductive diseases in a kennel. J Vet Diagn Invest 23, 143–7 (2011).
Duvnjak, S. et al. Characterisation of Brucella suis isolates from Southeast Europe by multi-locus variable-number tandem repeat analysis. Vet Microbiol 180, 146–50 (2015).
Gyuranecz, M. et al. Genotyping of Brucella melitensis strains from dromedary camels (Camelus dromedarius) from the United Arab Emirates with multiple-locus variable-number tandem repeat analysis. Vet Microbiol 186, 8–12 (2016).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27, 573–80 (1999).
Nadon, C. A. et al. Development and application of MLVA methods as a tool for inter-laboratory surveillance. Euro Surveill 18, 20565 (2013).
Zhang, X. et al. MLVA distribution characteristics of Yersinia pestis in China and the correlation analysis. BMC Microbiol 9, 205 (2009).
Grundmann, H., Hori, S. & Tanner, G. Determining confidence intervals when measuring genetic diversity and the discriminatory abilities of typing methods for microorganisms. J Clin Microbiol 39, 4190–2 (2001).
Acknowledgements
This project was supported by KSVDL revenue, the National Natural Science Foundation of China grant 31272575, the Scientific Innovation Research of College Graduate in Jiangsu province grant KYZZ16_0496, the Priority Academic Program Development of Jiangsu Higher Education Institutions, and China Scholarship Council.
Author information
Authors and Affiliations
Contributions
Y.Y., Y.W., R.R., X.L., E.P. and B.A. performed sample processing, bacteria isolation and identification. Y.Y. performed DNA extraction, PCR, genetic sequencing and sequence analysis. J.B., C.W., Y.Y. and N.L. performed the data analysis. J.B., C.W. and Y.Y. designed the study and wrote the manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, Y., Wang, Y., Poulsen, E. et al. Genotyping Brucella canis isolates using a highly discriminatory multilocus variable-number tandem-repeat analysis (MLVA) assay. Sci Rep 7, 1067 (2017). https://doi.org/10.1038/s41598-017-01114-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-01114-7
This article is cited by
-
MALDI-TOF MS and genomic analysis can make the difference in the clarification of canine brucellosis outbreaks
Scientific Reports (2020)
-
Molecular typing of Mycobacterium kansasii using pulsed-field gel electrophoresis and a newly designed variable-number tandem repeat analysis
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
