
Abstract
Clonal clusters and gene repertoires of Staphylococcus aureus are essential to understand disease and are well characterized in industrialized countries but poorly analysed in developing regions. The objective of this study was to compare the molecular-epidemiologic profiles of S. aureus isolates from Sub-Saharan Africa and Germany. S. aureus isolates from 600 staphylococcal carriers and 600 patients with community-associated staphylococcal disease were characterized by DNA hybridization, clonal complex (CC) attribution, and principal component (PCA)-based gene repertoire analysis. 73% of all CCs identified representing 77% of the isolates contained in these CCs were predominant in either African or German region. Significant differences between African versus German isolates were found for alleles encoding the accessory gene regulator type, enterotoxins, the Panton-Valentine leukocidin, immune evasion gene cluster, and adhesins. PCA in conjunction with silhouette analysis distinguished nine separable PCA clusters, with five clusters primarily comprising of African and two clusters of German isolates. Significant differences between S. aureus lineages in Africa and Germany may be a clue to explain the apparent difference in disease between tropical/(so-called) developing and temperate/industrialized regions. In low-resource countries further clinical-epidemiologic research is warranted not only for neglected tropical diseases but also for major bacterial infections.
Similar content being viewed by others


Introduction
According to WHO, neglected tropical diseases (NTD) are caused by defined, often rare, pathogens, and multiple research programs aim at investigating the epidemiology, causative pathogens, and treatment of these NTDs. Conversely, this may suggest that even in resource-limited regions, prevalent pathogens such as Staphylococcus aureus are not ‘neglected’, and that the epidemiology and microbiology is rather well known or can be inferred from data from developed countries. Literature and practical experience from these regions, however, tell a different story: diagnosis and treatment of infections caused by common pathogens is hampered by a lack of microbiological facilities and a paucity of epidemiologic data1.
S. aureus is a major public health threat and economic burden to health care systems worldwide causing important morbidity and high attributable mortality. Doubtlessly, S. aureus is a major pathogen also in (so-called) developing, tropical areas such as Sub-Saharan Africa, frequently causing invasive disease (for review: refs 2,3,4). In developed regions, epidemiologic registries provide data on the clonal structure of the prevalent isolates5,6,7,8 yielding insight into the association of clonality, gene repertoire, and disease course. In Sub-Saharan Africa, numerous studies exist describing various S. aureus strain collections with phenotypic and/or genotypic methods9,10,11,12,13,14,15,16,17,18,19,20, yet, they have been collected from retrospective strain collections, lack accompanying clinical data, are not controlled for hospital acquisition of the isolate/disease, and have not been performed strictly comparing the genotype (as clonal complex [CC] attribution and putative virulence gene content). In other words, cross-sectional molecular epidemiologic studies on both methicillin-sensitive and methicillin-resistant S. aureus are largely lacking.
Hence, the goal of this study was to investigate the hypothesis that prevalent clones of clinically defined, prospectively collected, non-nosocomial, i.e. community-associated, human S. aureus isolated in a temperate climate/developed region (Germany) differ with respect to their genetic repertoire and clonal lineage when compared to clones isolated in a tropical/developing region (Sub-Saharan Africa).
Results
Healthy participants’ and patients’ characteristics are summarized in Tables 1 and 2, respectively. The median age of asymptomatic carriers (volunteers) was 18 (0–61) years and 23 (0–89) years in the African and German study sites, respectively. Patients in Africa had a median age (range) of 3 (0–71) years, in Germany 53 (0–98) years. German patients had a higher rate of previous hospital care, or overall healthcare. German patients more frequently showed risk factors for invasive S. aureus infection (as reflected by elevated rates of Charlson comorbidity score), and in Germany a larger proportion of clinical isolates was obtained from blood cultures when compared to clinical isolates from Africa. African patients had a higher rate of skin and soft tissue infections, while deep invasive infections of the bone/joint, or respiratory tract were more frequently reported among German patients. Patients with a history of HIV infection were only found in the African group.
1,190 isolates of the 1,200 S. aureus isolates could be assigned to 32 CC and three singleton STs. For seven isolates, the CC could not be deduced because they belonged to new MLSTs not covered by known array profiles. These isolates were either from Africa (n = 4, ST2734, ST2744, ST2370) or Germany (n = 3 ST2733, ST2678, ST2735). Three isolates (1.3%) that were not CC attributable by Iconoclust were attributed to CCs by affinity propagation (based on their MA profiles).
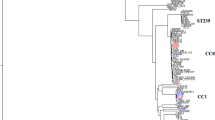
Figure 1 displays the distribution of CCs of isolates from African and German study sites. Except for four CCs (CC80 and CC88 in Africa, CC50 and CC398 in Germany), all CCs with a number of at least six isolates were found in Africa as well as in Germany. For 17 of the 40 detected CCs and STs, significant geographic distribution differences were found. 16/22 (73%) of the most frequently encountered CCs, and the vast majority of isolates contained within these CCs (896/1168, 77%) were significantly (p < 0.05) predominant either in Africa or in Germany. In the subgroup of clinical isolates, CCs were again significantly (p < 0.05) predominant in Africa or Germany, respectively (with the exception of clusters of low abundance, i.e. CC6 and CC50), while of the CCs contained in the subgroup of commensal isolates, among African isolates only CC88, CC121 and CC152, and among German isolates only CC 7 and CC30 were significantly predominant. In addition, we used already published whole genome sequences of a randomly selected subset of isolates (n = 154) of this study to construct a neighbor-joining tree based on the allelic profiles of 1861 S. aureus core genome features (cgMLST, Figure S1)21. On visual inspection, this analysis also shows that the majority of clusters are based on the geographical region. Clusters of isolates from infection or colonization were not detected.

Distribution of the 22 most prevalent clonal complexes (CC) in Africa and Germany among isolates from colonization and infection. CCs of low prevalence (<6 isolates) where grouped together (others). The CCs were sorted in ascending order according to the total number of isolates in the respective CC. The proportions of clinical (red) and nasal (green) isolates in the African and German group are shown. Differences in the distribution of CCs between Africa and Germany were calculated with Fisher’s exact test; *p < 0.05, **p < 0.001.
From the MA repertoire, all genes with a known or presumed regulator, virulence, and/or pathogenicity role were extracted, and compared between isolates from Africa and Germany (Supplementary Table 1). African isolates contain accessory gene regulators agr type I through type IV (with apparently some cross-hybridization between agrI and agrIV as the total number adds up to >100%); in contrast, within German isolates the majority is of agrI while agrIV was rarely found. Overall, enterotoxin gene recognition was low; yet, seb hybridized positively with DNA from African isolates, while sec, sed, sel, and the enterotoxin gene cluster egc was preferentially detected in isolates from Germany. A major difference was observed with leukocidins: the genes encoding for the Panton-Valentine leukocidin (PVL) lukF-PV and lukS-PV were recognized in almost one half of African clinical strains and were virtually absent in German isolates. The edinA and edinB immune evasion genes encoding the epithelial differentiation inhibitors were more frequently found amidst African isolates as was the gene isaB encoding the immunodominant antigen B and the protease gene splB. Only fragments and not the full map gene encoding the extracellular adhesive protein Eap were detected among African isolates; the gene sasG encoding for the biofilm associated surface protein G was more frequently found among African isolates (Table S1).
The majority of resistance genes were equally distributed among isolates from Africa and Germany. In general, methicillin resistance (mecA) was low in isolates from Africa (7/300 nasal [2.3%] vs 10/300 clinical [3.3%]) and Germany (2/300 nasal [0.7%] vs 22/300 clinical [7.3%]). Only blaZ was more frequently detected in African (560/600, 93%) vs. German isolates (400/600, 67%). Similar results were found for ermC (43/600 [7%] vs 91/600 [15%], p < 0.0001) and tetK (15/600 [3%] vs 211/600 [35%], p < 0.0001). merA, ermA, and tetM also displayed a significant difference between German and African isolates, yet, at an overall rate of target recognition of less than 10%. These findings correspond well to the phenotypic resistance profiles (Supplementary Table 2); here, striking differences in phenotypic resistance could be observed for tetracycline and trimethoprim-sulfamethoxazole with a larger proportion of resistant isolates in the African population, and clindamycin, with resistance more prevalent among German isolates.
The combined PCA/Silhouette analysis allowed to identify nine PCA clusters (labelled #1–9, Fig. 2). Overall, the CC attribution of the isolates corresponded to these PCA clusters, i.e. the isolates confined to a PCA/silhouette cluster could be attributed to a specific CC. Clusters with preferential composition of ‘African’ isolates are primarily found on the left side of the PCA plot (#2 [CC15], #3 [CC121], #4 [CC152]), whereas the clusters on the right side of the plot were preferentially of ‘German’ provenance (#6 [CC398], and #9 [CC30]). CC45 can be separated into two clusters (cluster #7 and cluster #8) of different geographic origin. In addition to these well-defined clusters (#1–9), there are additional clusters of isolates (Fig. 2, dashed line) which are associated with various CCs (i.e. CC1, CC5, CC6, CC7, CC9, CC12, CC20, CC25, CC49, CC50, CC59, CC80, CC88, CC97, CC101, CC188, CC395, CC509, CC707, CC913, CC1021, CC1290) or STs (i.e. ST580, ST1093, ST2370, ST2733, ST2734, ST2735, ST2744, ST2678).

Characteristic genotypic patterns of isolate subgroups detected by DNA microarray. The cluster analysis of 1200 S. aureus isolates was performed using the principal component analysis (PCA). Each dot represents one isolate. Dots are colour coded according to the study sites in Africa (Ifakara, Tanzania (IT), Lambarene, Gabon (LG), Manhiça, Mozambique (MM)) and Germany (Münster (MW), Freiburg (FR), Homburg (HS)). Major clusters that correspond to multilocus sequence typing clonal complexes (CC) are highlighted. Genes that were significantly (p < 0.01) associated with the respective CC are mentioned. Virulence factors that were significantly associated with ≥4 CCs are not displayed. Isolates encircled with a dashed line belong to CC1, CC5, CC6, CC7, CC9, CC12, CC20, CC25, CC49, CC50, CC59, CC80, CC88, CC97, CC101, CC188, CC395, CC509, CC707, CC913, CC1021, CC1290 or ST580, ST1093, ST2370, ST2733, ST2734, ST2735, ST2744, and ST2678.
With the Kolmogorov-Smirnoff test we identified the MA hybridization targets, which distinguished the isolates in the respective clusters #1–9 out of all MA hybridization signals for each isolate of the collection; these genes are denoted in Fig. 2.
Discussion
Here we present a prospective, cross-sectional geographic comparative study on strictly community-associated S. aureus isolates recovered under controlled, identical conditions in Germany and Sub-Saharan Africa and demonstrate that the cluster repartition among African and German isolates is profoundly inhomogeneous.
Studies from Europe revealed CC45, CC5, CC15, CC30, CC8 to be the most frequently encountered clusters5, 6, 8, 22,23,24. The overall smaller studies from Sub-Saharan Africa indicate that CC5, CC15, and CC30 are prevalent14, 20, that MSSA-CC8 has been primarily reported from North Africa (whereas MRSA-CC8 has been found in Central and South Africa), and that CC121 was more frequently reported from Sub-Saharan countries compared to Europe2. Methicillin-sensitive CC80 has been more frequently reported from North Africa, and may be related to the community-associated MRSA clone ST80 prevalent in Europe25. CC88 isolates are also typically methicillin-resistant; because this cluster has almost uniquely been recovered from African regions, it was attributed the acronym ‘African clone’2. The PVL positive clonal complex CC152 may also have originated from Africa14, expanded through central Europe, then acquired the methicillin resistance26. From these literature reports we conclude: first, ‘typical’ S. aureus clusters such as CC5, CC15, and CC30 appear to be prevalent in both Europe and Africa. Secondly, another set of ‘typical’ clones (such as CC80, CC88, or CC152) is reported from Africa rather than from Europe, yet, these clones do not seem to make up the bulk of isolates recovered in a non-endemic setting. Third, and probably most importantly, clear-cut studies allowing for frequency comparison between European and African clusters are lacking. The underlying mechanism of different population structures of S. aureus from Africa and Germany is unclear. The conservation of genomic patterns (e.g. gene clusters) and a subsequent clonal expansion could account for these differences. For instance, the ΦSa 2 prophage which carries lukF-PV and lukS-PV was integrated in the CC80 lineage at few occasions and subsequently clonally expanded in Africa and Europe25. Factors that favor the expansion of one clone in one geographic area might be associated with the bacterium itself (e.g. competition between different clones and species). However, host and environmental factors certainly play as well an important role which should be addressed in future studies.
Our cross sectional, comparative study now proves certain CCs of isolates from Africa to be indeed significantly prevalent (CC15, CC121, CC152) or even unique (CC88, CC80) compared to Germany. On the other hand, among isolates from Germany, other CCs are either significantly prevalent (such as CC45, CC30, CC7, CC22) or unique (CC398, CC50). PCA, avoiding multiple comparisons of single target recognition, confirmed this analysis allowing clear separation of the predominant ‘African’ from the ‘German’ clusters.
Does this clonal repartition imbalance contribute to a difference in disease spectrum? In Africa, higher rates of S. aureus-related pyomyositis are reported, frequently with bone, skin, and soft tissue involvement15, 27, at times presenting with multifocal lesions28. Moreover, S. aureus is particularly frequent in skin and soft tissue infections in Africa2. Molecular epidemiologic studies (from US and Europe) describe CC15 (in our study, ‘African’), CC30 (‘German’), and CC5, CC8, CC25 (in our study, ‘balanced’) as associated with invasive disease7, 23, 29, yet, they do not provide clear clues towards as to a different disease presentation as a function of predominant CCs in tropical/temperate geographic areas. Our study now provides such indication as the two CCs in our study significantly linked with clinical (as opposed to commensal) origin were the ‘African’ clones CC121 and CC152 (while the two CCs associated with nasal provenience were the ‘German’ clones CC45 and CC101).
In addition to the clonal repartition difference between Sub-Saharan Africa and Germany, does the gene repertoire composition contained in the respective, imbalanced CCs contribute to different disease presentation? In our analysis, agrIV was identified with an over-representation in African and agrI in German isolates, respectively, consistent with previous studies demonstrating agrIV to be prevalent in African CC12130. Moreover, the previously reported difference in the positivity rate for lukF-PV and lukS-PV was clearly confirmed also in this study14, 15, 31. The enterotoxin gene seb was also found to be predominant in African isolates, in line with results from studies performed in isolates from remote pygmy populations31, particularly among isolates of CC121. Of note is the difference in recognition of isaB target encoding a gene only expressed in vivo 32 inhibiting autophagic flux, thus allowing S. aureus to evade host degradation33. The proteases are also considered of importance to virulence34, and splB was significantly more often detected in African isolates (while splE was predominant in German isolates). Among adhesion factors, significant differences were found for map, the gene conferring extracellular adherence protein (Eap) expression35 and for the surface protein gene sasG. For map/eap this difference was largely attributable to a lacking recognition of the eap variant in isolates of CC152 (an African isolate whose genome did also fail to hybridize with the sdrC target) while for sasG the difference was mainly attributable to CC121. These observations allow to conclude that not only the clonal attribution but also certain regulatory, pathogenicity and virulence genes are differently distributed when comparing African and German S. aureus isolates obtained from patients with community associated infection.
The MRSA prevalence in our study was very low (nasal isolates: 2%, clinical isolates: 3%) compared to many other studies from Sub-Saharan Africa (23–55%)36. However, these studies should be interpreted with caution as, in contrast to our study, species of S. aureus and methicillin resistance were not confirmed. It is therefore likely that methicillin resistance is over reported in these studies.
The low rates of methicillin resistance could be also the result of strict exclusion of nosocomial, hospital-associated cases of infection. In accordance to the phenotypic data of many African studies showing a high resistance to penicillin30 and tetracycline (21.8–92%)37, we found a significant predominance of the beta lactamase operon and of the tetracycline resistance determinants tetK and tetM in the African isolates. Moreover, the erythromycin resistance genes ermA and ermC were more frequently found in German and African isolates, respectively (in line with a recent study38). In part, these findings were also confirmed by the phenotypic resistance profile demonstrating significant differences in susceptibility to tetracycline (but not to erythromycin).
This study has a number of limitations. First, the discrepancy in population age and comorbidities between the German and African cohort potentially biases the ‘true’ distribution of clones and genes between isolates from the different geographic regions (although application of a multiple linear regression model for the detection rate of Panton-Valentine leucocidin genes failed to provide evidence that age acts as a confounding variable [not shown]). In line, the imbalance in the type of infection (as shown in Table 2) between patients from Germany and Africa may also be a confounder with respect to the CC and virulence gene profile. Ex ante we deliberately did not attempt to match patients from Germany and Africa for age, comorbidity profiles, or type of clinical disease; instead, it was our goal to compare the patient characteristics and S. aureus isolates of a typical patient population presenting for primary medical care at German and African Medical Centers, and to avoid a potential bias incurred by imbalanced strata sizes. Secondly, the MA technique does not allow to distinguish between allelic variants not recognized by hybridization, and complete absence of alleles or genes (this issue has been investigated recently by our group comparing whole genome sequencing (WGS) and MA of exemplar isolates demonstrating that both techniques are highly but not fully reliable with respect to the gene/allele identification [with 1.7% WGS errors and 1.8% MA errors]39). Furthermore, the amount of gene transcripts or gene products was not assayed; thus, no correlation between transcript levels and geographic isolate provenience can be inferred. Thirdly, it was not possible to quality control the reliability of clinical case ascertainment beyond the instruction of the clinical personnel on following the written detailed instruction provided together with the structured questionnaires, and attribution of clinical characteristics may therefore lack scrutiny. Fourthly, we did not engage additional study sites from Europe; therefore, our comparison is limited to German isolates. However, in contrast to MRSA, MSSA have a similar population structure across Europe5. As the majority of our isolates were MSSA, results from Germany could be used as a surrogate for Europe.
In conclusion, prospectively collected, community-associated S. aureus isolates obtained from asymptomatic carriers and patients demonstrate profound and significant differences between Germany and various Sub-Saharan African regions, both with respect of clonal cluster attribution and gene repertoire, and for many genes the difference between the cohorts appears to be even more pronounced when only clinical isolates from both regions are analyzed. Thus, based on the overall clonal attribution and allele repertoire, our data provide first clues to explain the purported difference in clinical presentation and course of diseases caused by Staphylococcus aureus, a pathogen of major significance both in developing and developed regions.
Methods
Study design and participants
This is a cross sectional, geographical correlation study. Wherever applicable, described definitions and items on molecular epidemiology for infectious diseases study designs were applied40. Between years 2010 and 2012, a total of 1200 community-associated isolates was collected in three African (Lambaréné, Gabon; Bagamoyo, Tanzania; Manhiça, Mozambique) and three German study sites (Homburg, Freiburg, Münster). Every study site collected 100 non-duplicate isolates of healthy asymptomatic carriers. Exclusion criteria were (i) hospitalization within the past four weeks, (ii) antibacterial treatment within the past four weeks, and (iii) antituberculous treatment in the past four weeks. In addition, 100 clinical non-duplicate isolates were collected from human infection at each study site. The inclusion criteria were (i) clinical suspicion of infection by the treating physician, and (ii) community-onset of disease (outpatient clinic, or <48 h after admission). Clinical data were systematically recorded, electronically transmitted to the Freiburg study site, and checked for data consistency.
Ethical approval was obtained from the Ministry of Health and Social Welfare of Tanzania (A 81–2009), Institutional Ethics Committee of the Medical Research Unit of the International Foundation of the Albert Schweitzer Hospital (CERIL 15/09), National Committee of Bioethics for the Health System Mozambique (325/CNBS/12), Ethics Committee of the University of Münster (2009-227-b-S), Ethics Committee of Freiburg (248/09_120491) and the Ethics Committee of the Chamber of Physicians of Saarland (19/09). A written informed consent was obtained from all study subjects or their legal guardians. All experiments were performed in accordance with relevant guidelines and regulations.
Isolate collection and microbiological methods
Nasal swabs from asymptomatic carriers and appropriate specimens from infection sites were collected, and species identification performed by standard methods and confirmed at the Homburg site by MALDI-TOF (BRUKER Daltonics GmbH, Bremen, Germany). Antimicrobial susceptibility testing was performed at the various study centers using standard techniques (Clinical and Laboratory Standards Institute, M100).
All isolates were transferred into storage tubes, and shipped on dried ice to a central sample repository (Fraunhofer IBMT, Sulzbach, Germany) for long-term storage at −140 °C.
DNA microarray-based genotyping and MLST
All isolates were genotyped using the IdentiBAC® DNA microarray (MA, Alere Technologies GmbH, Jena, Germany). DNA extraction (Qiagen, Hilden, Germany) and hybridization were performed according to the manufacturer’s instructions. Spot signals were analyzed using ArrayMate® reader and corresponding Iconoclust® software (Alere Technologies GmbH, Jena, Germany) attributing specific multilocus sequence typing (MLST) clonal complex (CC) and sequence type (ST) designations. MLST was carried out for samples that were not assigned by the MA41.
CC assignment confirmation and statistics
Correctness of the CC identification by MA was confirmed by WGS of 154 exemplars39 defined by affinity propagation42. Principal component analysis (PCA) was performed to represent the isolate genotype in a two-dimensional projection. Given a large set of data, PCA identifies a small number of uncorrelated variables (termed principal components) that explain the maximum amount of variance in the data. In particular, the first two variables termed PCA1 and PCA2 describe the largest and second-largest variance in the data (Fig. 2).
Statistical analysis (Kolmogorov-Smirnoff test, Hommel p-value adjustment) was used to determine genotypic differences of isolate clusters defined by PCA. All comparisons were statistically analyzed by Chi-Square adjusted for multiple testing (Hommel p-value adjustment). Chi-Square, multivariate and principal component analysis were performed with the software “R”, version 3.2.0. Silhouette analysis was carried out to determine the number of different isolate clusters in the PCA, and was performed with “R”, version 3.2.2, function silhouette and package “cluster” version 2.0.3 on default parameters.
References
Herrmann, M. et al. Staphylococcal disease in Africa: another neglected ‘tropical’ disease. Future Microbiol. 8, 17–26 (2013).
Schaumburg, F., Alabi, A. S., Peters, G. & Becker, K. New epidemiology of Staphylococcus aureus infection in Africa. Clin. Microbiol. Infect. 20, 589–596 (2014).
Abdulgader, S. M., Shittu, A. O., Nicol, M. P. & Kaba, M. Molecular epidemiology of Methicillin-resistant Staphylococcus aureus in Africa: a systematic review. Front. Microbiol 6, 348 (2015).
Huson, M. A. et al. Methicillin-resistant Staphylococcus aureus as a cause of invasive infections in Central Africa: a case report and review of the literature. Infection 42, 451–457 (2014).
Grundmann, H. et al. Geographic distribution of Staphylococcus aureus causing invasive infections in Europe: a molecular-epidemiological analysis. PLoS Medicine 7, e1000215 (2010).
Feil, E. J. et al. How Clonal Is Staphylococcus aureus? J. Bacteriol. 185, 3307–3316 (2003).
Fowler, V. G. Jr. et al. Potential associations between hematogenous complications and bacterial genotype in Staphylococcus aureus infection. J. Infect. Dis. 196, 738–747 (2007).
Wertheim, H. F. et al. Associations between Staphylococcus aureus Genotype, Infection, and In-Hospital Mortality: A Nested Case-Control Study. J. Infect. Dis. 192, 1196–1200 (2005).
Ako-Nai, A. K., Ogunniyi, A. D., Lamikanra, A. & Torimiro, S. E. The characterisation of clinical isolates of Staphylococcus aureus in Ile-Ife, Nigeria. J. Med. Microbiol. 34, 109–112 (1991).
Ghebremedhin, B. et al. Emergence of a community-associated methicillin-resistant Staphylococcus aureus strain with a unique resistance profile in Southwest Nigeria. J. Clin. Microbiol. 47, 2975–2980 (2009).
Shittu, A. et al. Characterization of methicillin-susceptible and -resistant staphylococci in the clinical setting: a multicentre study in Nigeria. BMC Infect. Dis. 12, 286 (2012).
Anguzu, J. R. & Olila, D. Drug sensitivity patterns of bacterial isolates from septic post-operative wounds in a regional referral hospital in Uganda. African Health Sciences 7, 148–154 (2007).
Ramdani-Bouguessa, N. et al. Detection of methicillin-resistant Staphylococcus aureus strains resistant to multiple antibiotics and carrying the Panton-Valentine leukocidin genes in an Algiers hospital. Antimicrob. Agents Chemother. 50, 1083–1085 (2006).
Ruimy, R. et al. The carriage population of Staphylococcus aureus from Mali is composed of a combination of pandemic clones and the divergent Panton-Valentine leukocidin-positive genotype ST152. J. Bacteriol. 190, 3962–3968 (2008).
Breurec, S. et al. Epidemiology of methicillin-resistant Staphylococcus aureus lineages in five major African towns: emergence and spread of atypical clones. Clin. Microbiol. Infect. 17, 160–165 (2011).
Okon, K. O. et al. Cooccurrence of predominant Panton-Valentine leukocidin-positive sequence type (ST) 152 and multidrug-resistant ST 241 Staphylococcus aureus clones in Nigerian hospitals. J. Clin. Microbiol. 47, 3000–3003 (2009).
Moodley, A., Oosthuysen, W. F., Duse, A. G., Marais, E. & South African, M. S. G. Molecular characterization of clinical methicillin-resistant Staphylococcus aureus isolates in South Africa. J. Clin. Microbiol. 48, 4608–4611 (2010).
Egyir, B. et al. Molecular epidemiology and antimicrobial susceptibility of clinical Staphylococcus aureus from healthcare institutions in Ghana. PloS One 9, e89716 (2014).
Egyir, B. et al. Insights into nasal carriage of Staphylococcus aureus in an urban and a rural community in Ghana. PLoS One 9, e96119 (2014).
Conceicao, T., Coelho, C., Santos Silva, I., de Lencastre, H. & Aires-de-Sousa, M. Staphylococcus aureus in Portuguese former colonies from Africa and the far East: missing data to help fill the world map. Clin. Microbiol. Infect. 21, 842.e1–842.e10 (2015).
Leopold, S. R., Goering, R. V., Witten, A., Harmsen, D. & Mellmann, A. Bacterial whole-genome sequencing revisited: portable, scalable, and standardized analysis for typing and detection of virulence and antibiotic resistance genes. J. Clin. Microbiol. 52, 2365–2370 (2014).
Melles, D. C. et al. Natural population dynamics and expansion of pathogenic clones of Staphylococcus aureus. J. Clin. Invest. 114, 1732–1740 (2004).
Rasmussen, G., Monecke, S., Ehricht, R. & Soderquist, B. Prevalence of clonal complexes and virulence genes among commensal and invasive Staphylococcus aureus isolates in Sweden. PloS One 8, e77477 (2013).
Mehraj, J. et al. Methicillin-sensitive and methicillin-resistant Staphylococcus aureus nasal carriage in a random sample of non-hospitalized adult population in northern Germany. PLoS One 9, e107937 (2014).
Stegger, M. et al. Origin and evolution of European community-acquired methicillin-resistant Staphylococcus aureus. MBio 5, e01044–01014 (2014).
Otter, J. A. & French, G. L. Molecular epidemiology of community-associated meticillin-resistant Staphylococcus aureus in Europe. Lancet Infect. Dis. 10, 227–239 (2010).
Sina, H. et al. Variability of antibiotic susceptibility and toxin production of Staphylococcus aureus strains isolated from skin, soft tissue, and bone related infections. BMC Microbiol. 13, 188 (2013).
Ntusi, N. B. & Khaki, A. Primary multifocal pyomyositis due to Staphylococcus aureus. QJM 104, 163–165 (2011).
Rieg, S. et al. Microarray-based genotyping and clinical outcomes of Staphylococcus aureus bloodstream infection: an exploratory study. PloS One 8, e71259 (2013).
Kolawole, D. O. et al. Characterization of colonizing Staphylococcus aureus isolated from surgical wards’ patients in a Nigerian university hospital. PloS One 8, e68721 (2013).
Schaumburg, F. et al. Population structure of Staphylococcus aureus from remote African Babongo Pygmies. PLoS Neglect. Trop. Dis 5, e1150 (2011).
Mackey-Lawrence, N. M. & Jefferson, K. K. Regulation of Staphylococcus aureus immunodominant antigen B (IsaB). Microbiol. Res. 168, 113–118 (2013).
Liu, P. F. et al. IsaB Inhibits Autophagic Flux to Promote Host Transmission of Methicillin-Resistant Staphylococcus aureus. J. Invest. Dermatol. 135, 2714–2722 (2015).
Zdzalik, M. et al. Prevalence of genes encoding extracellular proteases in Staphylococcus aureus - important targets triggering immune response in vivo. FEMS Immunol. Med. Microbiol. 66, 220–229 (2012).
Stapels, D. A. C. et al. Staphylococcus aureus secretes a unique class of neutrophil serine protease inhibitors. Proc Natl Acad Sci USA 111, 13187–13192 (2014).
Falagas, M. E., Karageorgopoulos, D. E., Leptidis, J. & Korbila, I. P. MRSA in Africa: filling the global map of antimicrobial resistance. PloS One 8, e68024 (2013).
Djoudi, F. et al. Panton-Valentine leukocidin positive sequence type 80 methicillin-resistant Staphylococcus aureus carrying a staphylococcal cassette chromosome mec type IVc is dominant in neonates and children in an Algiers hospital. New Microbiol. 36, 49–55 (2013).
Phaku, P. et al. Unveiling the molecular basis of antimicrobial resistance in Staphylococcus aureus from the Democratic Republic of the Congo using whole genome sequencing. Clin. Microbiol. Infect. (2016).
Strauss, L. et al. Detecting Staphylococcus aureus Virulence and Resistance Genes - a Comparison of Whole Genome Sequencing and DNA Microarray Technology. J. Clin. Microbiol. (2016).
Field, N. et al. Strengthening the Reporting of Molecular Epidemiology for Infectious Diseases (STROME-ID): an extension of the STROBE statement. Lancet Infect. Dis. 14, 341–352 (2014).
Enright, M. C., Day, N. P., Davies, C. E., Peacock, S. J. & Spratt, B. G. Multilocus sequence typing for characterization of methicillin-resistant and methicillin-susceptible clones of Staphylococcus aureus. J. Clin. Microbiol. 38, 1008–1015 (2000).
Frey, B. J. & Dueck, D. Clustering by passing messages between data points. Science 315, 972–976 (2007).
Acknowledgements
The authors acknowledge Arnaud Flamen, Alexander Friedrich, Günther Fuhr, Karin Hilgert, Saadou Issifou, Harry Kaba, Michael Kaeser, Eloise Müller-Schulte, Anna Nimmesgern, Stefan Monecke, Gabriele Peyerl-Hoffmann, Michaela Steib-Bauert, Jasmin Wüsthoff, and Christof von Eiff for contributions during study design and implementation, for technical support, and/or for valuable advice. This study has been supported by grants of the Deutsche Forschungsgemeinschaft (BR 1564/7-1 to H.v.B., HE 1850/11-1 to M.H., HE 3875/12-1 to V.H., KE 700/3-1 to W.V.K., ME 3205/4-1 to A.M., and PE 296/6-1 to G.P.). The funder of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Author information
Authors and Affiliations
Contributions
I.M., S.A., P.A., M.P.G., V.H., W.V.K., P.G.K., A.M., G.P., F.S., M.T., H.v.B., L.v.M. and M.H. designed the study; U.R., A.A., T.K., D.C.V., R.A., A.G., J.H., S.S., L.S., L.W., F.S. and L.v.M. did the study; and U.R., R.A., M.B., A.G., M.P.G., V.H., W.V.K., P.G.K., A.M., G.P., F.S., M.T., L.v.M. and M.H. were involved in data acquisition, analysis, and interpretation. U.R. drafted the manuscript, M.H. wrote the final version, and all authors reviewed, commented on, and approved the final manuscript for submission.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Ruffing, U., Alabi, A., Kazimoto, T. et al. Community-Associated Staphylococcus aureus from Sub-Saharan Africa and Germany: A Cross-Sectional Geographic Correlation Study. Sci Rep 7, 154 (2017). https://doi.org/10.1038/s41598-017-00214-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-00214-8
This article is cited by
-
Temporal trends of skin and soft tissue infections caused by methicillin-resistant Staphylococcus aureus in Gabon
Antimicrobial Resistance & Infection Control (2024)
-
Exploring the virulence potential of Staphylococcus aureus CC121 and CC152 lineages related to paediatric community-acquired bacteraemia in Manhiça, Mozambique
Scientific Reports (2024)
-
Prevalence, antimicrobial susceptibility and genotypic characteristics of Staphylococcus aureus in Tanzania: a systematic review
Bulletin of the National Research Centre (2021)
-
An in vitro study on Staphylococcus schweitzeri virulence
Scientific Reports (2021)
-
Antimicrobial resistant and enteropathogenic bacteria in ‘filth flies’: a cross-sectional study from Nigeria
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
