
Abstract
The Legume family (Leguminosae or Fabaceae), is one of the largest and economically important flowering plants. Heartwood, the core of a tree trunk or branch, is a valuable and renewable resource employed for centuries in constructing sturdy and sustainable structures. Hongmu refers to a category of precious timber trees in China, encompassing 29 woody species, primarily from the legume genus. Due to the lack of genome data, detailed studies on their economic and ecological importance are limited. Therefore, this study generates chromosome-scale assemblies of five Hongmu species in Leguminosae: Pterocarpus santalinus, Pterocarpus macrocarpus, Dalbergia cochinchinensis, Dalbergia cultrata, and Senna siamea, using a combination of short-reads, long-read nanopore, and Hi-C data. We obtained 623.86 Mb, 634.58 Mb, 700.60 Mb, 645.98 Mb, and 437.29 Mb of pseudochromosome level assemblies with the scaffold N50 lengths of 63.1 Mb, 63.7 Mb, 70.4 Mb, 61.1 Mb and 32.2 Mb for P. santalinus, P. macrocarpus, D. cochinchinensis, D. cultrata and S. siamea, respectively. These genome data will serve as a valuable resource for studying crucial traits, like wood quality, disease resistance, and environmental adaptation in Hongmu.
Similar content being viewed by others


Background & Summary
Leguminosae (Fabaceae) is the third-largest plant family with 770 genera and 19,500 species with substantial economic value1. The wood of the trees is divided into the outer layers of sapwood (SW) and the inner core of heartwood (HW). HW is the inner, dark-colored wood of a tree that is a valuable commodity, particularly in the timber industry due to its high durability, exquisite color, special scent, and rot and insect resistance properties with a long history2. Hongmu is a special term for precious timber trees in China, comprising 29 woody species, especially the legume genus such as Pterocarpus, Dalbergia, Senna, and Millettia of the Fabaceae family3. Despite their high economic value and medicinal properties, the lack of genome data hampers the in-depth understanding of genetic architecture and heartwood formation mechanisms4,5. Therefore, in this study, we selected five highly priced and high-quality heartwood-producing trees namely Pterocarpus santalinus, Pterocarpus macrocarpus, Dalbergia cochinchinensis, Dalbergia cultrata and Senna siamea for generating the genomic resource.
Pterocarpus santalinus (2n = 20)6, commonly known as zitan, red sandalwood or red sanders is mainly distributed in India, South and Southwest China. The plant is valued for its heartwood with excellent red wood color, texture, decay resistance and insect resistance7,8. It is classified as “endangered” in the IUCN red list of threatened species, because of illegal overharvesting. Heartwood exhibits medicinal properties, including its ability to alleviate fever, reduce inflammation, combat microbes, and act as an antioxidant, all of which are harnessed in traditional medicine. The major bioactive phyto-compounds extracted from the heartwood are santalins, flavonoids, terpenoids, phenolic compounds, alkaloids, saponins, tannins, and glycosides9.
Pterocarpus macrocarpus (2n = 22)10, commonly known as Burma padauk, is also an important timber of Southeast Asia, with distribution in Myanmar and Thailand11. Its reddish HW is expensive and used for making furniture and handicrafts, because of superior wood properties, including high density and resistance to termite attack12.
Dalbergia cochinchinensis (2n = 20)13 (Thai Rosewood), distributed in Thailand, Cambodia, Vietnam, and Laos is listed as Critically Endangered in the IUCN red list of threatened species. Its reddish heartwood is valuable due high density, unique aroma and resistance to termites14.
Dalbergia cultrata (2n = 20) is a rosewood species also recognized as Burmese blackwood, and is distributed in a tropical and subtropical zone in Indo-China peninsula, and the south of Yunnan province in China. Heartwood is also valued for its quality, dark purplish-brown color, special scent and resistance to insects and disease. However, this species was threatened by overexploitation and listed in the IUCN red list of threatened species15.
Senna siamea (2n = 28)16, commonly known as kassod tree, cassod tree, and cassia tree in South or Southeast Asia, and is widely planted throughout the tropics. The HW is black-brown in texture, with high density and resistance to termites17. In Thailand, the young leaves and fruits are used as vegetables or traditional medicine18.
The heartwood properties of rot and insect resistance, durability and colors are largely defined by secondary metabolites19. Enhancing the quantity of secondary metabolites in the heartwood could potentially serve as a solution for reducing decay, increasing resistance to insects, and enhancing the durability of trees during breeding. Despite their economic and ecological importance, relatively little is known about the genetics and genomics of these heartwood Leguminosae species. Most previous studies have focused on molecular markers, such as microsatellites and amplified fragment length polymorphisms (AFLPs), which provide limited information about the genome structure and function20,21,22. The high-quality genome data provides a valuable resource for studying the genetic basis of important traits, such as wood quality, disease resistance, and environmental adaptation23,24,25,26,27. In this study, we provide chromosome-level genomes of five heartwood Hongmu species in Leguminosae: Pterocarpus santalinus, Pterocarpus macrocarpus, Dalbergia cochinchinensis, Dalbergia cultrata, and Senna siamea, using a combination of short-reads, long-read nanopore, and Hi-C data. This information can be used to improve breeding and conservation efforts for these species, as well as to develop new biotechnological applications. Additionally, the genome data can help shed light on the evolutionary history and relationships among the Leguminosae family, which is one of the largest and most diverse families of flowering plants.
Methods
Sample preparation and sequencing
The fresh leaves of Pterocarpus santalinus, Pterocarpus macrocarpus, Dalbergia cochinchinensis, Dalbergia cultrata, Senna siamea were collected form the Xishuangbanna Tropical Botanical Garden (XTBG), Yunnan, China, and were subjected for DNA extraction using CTAB (Cetyltrimethylammonium bromide) method28, then purified with QIAGEN Genomic kit (Cat#13343, QIAGEN). Furthermore, the DNA quality was checked by using NanoDrop (Thermo Fisher Scientific, USA) with OD260/280 ranging from 1.8-2.0 and OD260/230 between 2.0–2.2 was considered pure. Next, Qubit 4.0 (Invitrogen, USA) was used for DNA quantification. Subsequently, the long DNA fragments were selected by PippinHT system (Sage Science, USA) for each sample, and the ends were repaired by using NEBNext Ultra II End Repair/dA-tailing Kit (Cat# E7546). At last, the SQK-LSK109 kit (Oxford Nanopore Technologies, UK) was used for the adapter ligation reaction. Then the DNA libraries were performed on the Nanopore GridION X5 sequencer (Oxford Nanopore Technologies, UK). Finally, we generated 49, 44, 49, 61, and 53 Gb raw Oxford Nanopore long-reads of P. santalinus, P. macrocarpus, D. cochinchinensis, D. cultrata and S. siamea. The genome sequencing depth was more than 60x for each species. A total of 111, 168, 150, 155, and 162 Gb raw short insert-size reads of P. santalinus, P. macrocarpus, D. cochinchinensis, D. cultrata and S. siamea were generated by BGI-DIPSEQ sequencing platform. Subsequently, the extracted were digested using MboI according to the standard Hi-C library preparation protocol, then sequenced on the BGI-DIPSEQ platform, which generated 131, 132, 144, 178 and 171 Gb data for P. santalinus, P. macrocarpus, D. cochinchinensis, D. cultrata and S. siamea, respectively (Table S1).
For the RNAseq experiment, TIANGEN Kit was used for total RNA extraction from fresh leaves and stems. After quality control check, library construction and sequencing were performed on the Illumina platform which generated 11, 14, 22, 13, and 11 Gb raw data for P. santalinus, P. macrocarpus, D. cochinchinensis, D. cultrata and S. siamea, respectively, and on the other hand BGI-DIPSEQ platform generated a total of 74, 73, 69 and 77 Gb raw data for stem samples of P. santalinus, D. cochinchinensis, D. cultrata and S. siamea, respectively (Table S2).
Estimation of genome size
The short DNA reads were used to filter the adapter, duplicated and low-quality reads by trimmomatic (v3.0)29 using the parameters (adapter:2:30:10:8:true LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:50). The clean data were used for genome size estimation based on kmerfreq 16 bit (Version 2.4) and GCE software (Table S3)30. The result showed that the estimated genome size ranged from 673 to 693 Mb for P. santalinus, 657 to 682 Mb for P. macrocarpus, 672 to 705 Mb for D. cochinchinensis, 650 to 670 Mb for D. cultrata and 476 to 482 Mb for S. siamea.
De novo genome assembly and evaluation
The nanopore long reads of P. santalinus, P. macrocarpus, D. cochinchinensis and S. siamea were assembled by using NECAT31, while for D. cultrata nextdenovo32 software was used. Then all five assemblies were polished by short reads with NextPolish software33. Finally, the genomes were moved to the contig overlaps by using the purge dups (v.1.2.3)34 with the default parameters. As a result, we generated 623.76 Mb, 634.38 Mb, 700.50 Mb, 645.68 Mb, 437.21 Mb genome assemblies of P. santalinus, P. macrocarpus, D. cochinchinensis, D. cultrata and S. siamea, with the contig N50 lengths were 28.2 Mb, 12.2 Mb, 17.8 Mb, 41.1 Mb and 14.7 Mb, respectively (Table 1, Table S4).
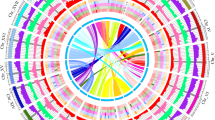
Further, we used the Hi-C data to anchor the contig assemblies to the chromosomes, the Juicer software35 was used to extract the uniquely mapped and non-PCR duplicated Hi-C contact reads, then 3D-DNA36 was used to integrate the assembled genome into a pseudochromosome level assembly. Finally, the Hi-C assembly result was visualized by Juicebox and manually improved according to the Hi-C contact map. As a result, we obtained 623.86 Mb, 634.58 Mb, 700.60 Mb, 645.98 Mb, 437.29 Mb of pseudochromosome level assemblies which were anchored to 10 chromosomes in P. santalinus, P. macrocarpus, D. cochinchinensis, D. cultrata, and 14 chromosomes in S. siamea, with the scaffold N50 lengths of 63.1 Mb, 63.7 Mb, 70.4 Mb, 61.1 Mb and 32.2 Mb, respectively. More than 96% of scaffolds were anchored into the pseudochromosomes of each species, which is consistent with the reported chromosome number of each species (2n = 20 for P. santalinus, P. macrocarpus, D. cochinchinensis and D. cultrata, 2n = 28 for S. siamea) (Fig. 1a–c, Table 1, Table S4, S5, Fig. 2)

Circos plot and phylogenetic tree of five Hongmu species in Leguminosae. The distribution of genomic features along the chromosomes (scale is in Mb) (a–c). I, Pseudochromosomes. II, the density of gene number. III, the density of GC content. IV, the density of transposable elements. V, the density of transposable elements LTR. VI, the density of transposable elements LTR/Copia. VII, the density of Gypsy of LTR transposable elements. VIII, the collinearity of the genome. Senna tora is used for comparison only, the genome data is not generated in this study. (d) The phylogenetic tree of 11 representative legume species. All nodes exhibit 100% bootstrap support based on maximum likelihood analysis. All the species sequenced in the present study are highlighted in red color.

Hi-C map showing genome-wide all-by-all interactions. The map shows a high resolution of individual chromosomes that are scaffolded and assembled independently. The heat map colors ranging from white to dark red indicate the frequency of Hi-C interaction links from low to high (0–8).
Repeat annotation
We combined the de novo and homolog-based methods to find the repeat element in the genomes of five species. For de novo prediction, we used LTR_FINDER37, RepeatModeler38 to detect the repeat elements and then built a non-redundant library to identify the repeat element by RepeatMasker39. For the homolog-based methods, we used TRF to find the tandem repeats, and RepeatMasker was used to search the repeat element against the RepBase (v.21.12). In total, 49.07%, 49.49%, 62.58%, 48.88%, and 47.14% of the genome sequences were identified as repetitive sequences in P. santalinus, P. macrocarpus, D. cochinchinensis, D. cultrata and S. siamea, respectively (Table 2, Table S6). Long terminal repeats (LTRs) showed the highest proportions, comprising 39.49%, 39.90%, 55.88%, 43.27%, and 39.97% in P. santalinus, P. macrocarpus, D. cochinchinensis, D. cultrata and S. siamea respectively. The two Dalbergia legume wood trees showed higher LTRs than other three legume trees. Among the LTRs, the Gypsy LTRs (28.69%, 28.81%, 38.93%, 30.59%, 26.25%) were the most abundant in the P. santalinus, P. macrocarpus, D. cochinchinensis, D. cultrata and S. siamea respectively. Meanwhile, the two Dalbergia legume wood trees showed the highest number of Gypsy compared to the other three trees (Table S6).
Protein-coding genes prediction and Non-coding RNA annotation
The protein-coding genes prediction was performed using BRAKER2 pipeline40, resulting in the identification of 34651, 34924, 33654, 34647, and 31038 protein-coding genes in P. santalinus, P. macrocarpus, D. cochinchinensis, D. cultrata and S. siamea, respectively (Table 2, Table S7), of which the complete BUSCOs were more than 97.7%. 97.9%, 98.1%, 97.7%, and 97.9% in these species (Table S8). All protein-coding genes were blast against NR, SwissProt, KOG, and KEGG databases with the 1e-05 E-value cutoff, resulting in 98.61%, 96.52%, 99.26%, 99.28%, and 92.84% functionally annotated genes (Table S9).
Ribosomal RNA (rRNA) genes were searched against the plant rRNA database by using BLAST. MicroRNAs (miRNA) and small nuclear RNA (snRNA) were searched against the Rfam 12.0 database. tRNAscan-SE was also used to scan for tRNAs41. As a result, a total of 2005, 2283, 1654, 1659, and 5437 ncRNAs were identified in P. santalinus, P. macrocarpus, D. cochinchinensis, D. cultrata and S. siamea genome, respectively (Table S10). In particular, the number of rRNAs in S. siamea was higher than the other four legume wood trees.
Analysis of the phylogeny tree
OrthoFinder (v2.3.14)42 software was used for comparative genome analysis between the predicted protein-coding sequences of 13 representative plant species (including P. santalinus, P. macrocarpus, D. cochinchinensis, D. cultrata, S. siamea, and other published genomes of Dalbergia odorifera, Glycine max, Pongamia pinnata, Sophora japonica, Senna tora, Faidherbia albida, Populus trichocarpa and Arabidopsis thaliana) (TableS 11).
The sequences of each 302 low-copy orthogroups were extracted and aligned by MAFFT (v 7.310)43 after filtering the gaps. The aligned coding protein sequences of each species were then concatenated to a supergene sequence. The phylogenetic tree was subsequently constructed by IQ-Tree (v 1.6.1)44 with the parameters ‘-bb 1000 -alrt 1000’ (Fig. 1d).
Data Records
All the genomic sequencing raw data are deposited in the Genome Sequence Archive in National Genomics Data Center (NGDC) Genome Sequence Archive (GSA) database with the accession number CRA01138945 under the BioProject accession number PRJCA01748646. The Chromosome-scale genome assemblies were submitted to the GenBank at NCBI under the accession number GCA_031439595.147, GCA_031439585.148, GCA_031216125.149, GCA_031216105.150, GCA_031216115.151 of P. santalinus, P. macrocarpus, D. cochinchinensis, D. cultrata and S. siamea, respectively. The raw sequencing data are also submitted to the CNGB Sequence Archive (CNSA) of China National GeneBank DataBase (CNGBdb) under accession No. CNP0003804. Genome annotation of gene structure is available via Figshare52.
Technical Validation
The completeness and contiguity of genomes were assessed by BUSCO (V3.0.2)53 software with the Embryophyta odb10, and the analysis suggested 97.9%, 98.1%, 97.9%, 97.7%, and 97.7% of complete embryophyte BUSCOs in the genome of P. santalinus, P. macrocarpus, D. cochinchinensis, D. cultrata and S. siamea, respectively (Table S12). The DNA short reads were mapped to the genomes by BWA (v.2.21) and showed a high mapping rate to the genome (99% for P. santalinus, P. macrocarpus, D. cultrata, S. siamea. 98.83% for D. cochinchinensis), the RNA short reads were also showed more than 90% mapping rate to the genome by using HISAT2 (V.2.1.0) (Table S13).
Furthermore, LAI (LTR Assembly Index) was used to evaluate the contiguity of the genome assembly by assessing the assembly of LTR sequences. First, LTRharvest54 was used to detect the LTR sequences with the parameter ‘-minlenltr 100 -maxlenltr 7000 -mintsd 4 -maxtsd 6 -motif TGCA -motifmis 1 -similar 85 -vic 10 -seed 20’, then combined with the previous LTR_FINDER result. Finally, the LTRretriever (v.2.8)55 was used to obtain the high-confidence LTR retrotransposons with default parameters. At last, the LAI score was calculated by using the LTRretriever with the default settings. The LAI values were 12.24, 11.86, 11.01, 15.16, and 10.2 for P. santalinus, P. macrocarpus, D. cochinchinensis, D. cultrata and S. siamea, respectively (Table S14). The high quality, contiguity, and completeness of the assembled genome were supported by various evidences56.
Code availability
The parameters of the software were default. No specific script was used in this work.
References
Abusaief, H. M. A. & Boasoul, S. H. A taxonomic study of twelve wild forage species of Fabaceae. Heliyon 7, e06077 (2021).
Ye, Z.-H. & Zhong, R. Molecular control of wood formation in trees. Journal of Experimental Botany 66, 4119–4131 (2015).
GB/T18107-2017 Standard of the People’s Republic of China (2017).
Sahu, S. K. et al. Chromosome-scale genome of Indian Rosewood (Dalbergia sissoo). Frontiers in Plant Science 14, 1218515 (2023).
Sahu, S. K. et al. Chromosome-scale genomes of commercial timber trees (Ochroma pyramidale, Mesua ferrea, and Tectona grandis). Scientific Data 10, 512 (2023).
Agasthikumar, S. et al. De novo transcriptome assembly and development of EST-SSR markers for Pterocarpus santalinus L. f. (Red sanders), a threatened and endemic tree of India. Genetic Resources and Crop Evolution 69, 2469–2484 (2022).
Walpola, B. C., Subasinghe, S., Yoon, M.-H. & Arunakumara, K. K. I. U. Pterocarpus santalinus Linn. f. (Rath handun): A review of its botany, uses, phytochemistry and pharmacology. Journal of the Korean Society for Applied Biological Chemistry 54, 495–500 (2011).
Rajan, L. J., Ramanan, S. S. & Anoop, E. Physical and Anatomical-wood properties variation among the provenances of Red Sanders (Pterocarpus santalinus). Indian Association of Hill Farming 32, 162–168 (2019).
Bulle, S., Reddyvari, H., Nallanchakravarthula, V. & Vaddi, D. R. Therapeutic Potential of Pterocarpus santalinus L.: An Update. Pharmacognosy reviews 10, 43–49 (2016).
Rojas-Sandoval, J. Pterocarpus macrocarpus (Burma padauk). Vol. CABI Compendium (CABI International, 2022).
Doungyotha, Y. & Owens, J. N. The Reproductive Biology and Reproductive Success of Pterocarpus macrocarpus Kurz. Biotropica 34, 58–67 (2002).
Morimoto, M., Fukumoto, H., Hiratani, M., Chavasiri, W. & Komai, K. Insect antifeedants, pterocarpans and pterocarpol, in heartwood of Pterocarpus macrocarpus Kruz. Bioscience, biotechnology, and biochemistry 70, 1864–1868 (2006).
Hung, T. H. et al. Reference transcriptomes and comparative analyses of six species in the threatened rosewood genus Dalbergia. Scientific Reports 10, 1–14 (2020).
Seng, M. & Cheong, E. J. Comparative study of various pretreatment on seed germination of Dalbergia cochinchinensis. Forest Science and Technology 16, 68–74 (2020).
Liu, Y. et al. MaxEnt modelling for predicting the potential distribution of a near threatened rosewood species (Dalbergia cultrata Graham ex Benth). Ecological Engineering 141, 105612 (2019).
Resende, K. F. M. D., Davide, L. C. & Torres, G. A. Chromosome number and meiosis in populations of Senna species (Caesalpinioideae – Fabaceae) from Southeast Brazil. Caryologia 66, 1–5 (2013).
Ma, F. & Huang, A.-M. Rapid identification and quantification three chicken-wing woods of Millettia leucantha, Millettia laurentii and Cassia siamea by FT-IR and 2DCOS-IR. Journal of Molecular Structure 1166, 164–168 (2018).
Huang da, W., Sherman, B. T. & Lempicki, R. A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37, 1–13 (2009).
Celedon, J. M. & Bohlmann, J. An extended model of heartwood secondary metabolism informed by functional genomics. Tree Physiology 38, 311–319 (2017).
Zwirchmayr, J. et al. Biochemometry identifies suppressors of pro-inflammatory gene expression in Pterocarpus santalinus heartwood. Phytochemistry 212, 113709 (2023).
Özkan, G. et al. Determining Genetic Diversity and Population Structure of Common Bean (Phaseolus vulgaris L.) Landraces from Türkiye Using SSR Markers. Genes (Basel) 13 (2022).
Sahu, S. K. Comparative analyses of 3654 plastid genomes unravel insights into evolutionary dynamics and phylogenetic discordance of green plants. Frontiers in Plant Science 13, 829 (2022).
Sahu, S. K. & Liu, H. Long-read sequencing (method of the year 2022): The way forward for plant omics research. Molecular Plant 16, 791–793 (2023).
Xian, L. et al. The draft genome and multi-omics analyses reveal new insights into geo-herbalism properties of Citrus grandis ‘Tomentosa’. Plant Science 325, 111489 (2022).
Wang, S. et al. The chromosome-scale genomes of Dipterocarpus turbinatus and Hopea hainanensis (Dipterocarpaceae) provide insights into fragrant oleoresin biosynthesis and hardwood formation. Plant Biotechnol J 20, 538–553 (2022).
Guo, X. et al. Chloranthus genome provides insights into the early diversification of angiosperms. Nature communications 12, 6930 (2021).
Sahu, S. K. et al. Draft genomes of two artocarpus plants, jackfruit (A. heterophyllus) and breadfruit (A. altilis). Genes 11, 27 (2019).
Sahu, S. K., Thangaraj, M. & Kathiresan, K. DNA extraction protocol for plants with high levels of secondary metabolites and polysaccharides without using liquid nitrogen and phenol. International Scholarly Research Notices 2012, 205049–205054 (2012).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Liu, B. et al. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. arXiv preprint arXiv:1308.2012, (2013).
Chen, Y. et al. Efficient assembly of nanopore reads via highly accurate and intact error correction. Nature Communications 12, 60 (2021).
Hu, J. et al. An efficient error correction and accurate assembly tool for noisy long reads. bioRxiv, 2023.2003.2009.531669 (2023).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255 (2020).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898 (2020).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell systems 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic acids research 35, W265–W268 (2007).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences 117, 9451–9457 (2020).
Tarailo‐Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Current protocols in bioinformatics 25, 4.10. 11–14.10. 14 (2009).
Brůna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR genomics and bioinformatics 3, lqaa108 (2021).
Lowe, T. M. & Chan, P. P. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic acids research 44, W54–W57 (2016).
Emms, D. M. & Kelly, S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome biology 16, 157 (2015).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular biology and evolution 30, 772–780 (2013).
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Molecular Biology and Evolution 32, 268–274 (2014).
NGDC Genome Sequence Archive https://bigd.big.ac.cn/gsa/browse/CRA011389 (2023).
NGDC BioProject https://ngdc.cncb.ac.cn/bioproject/browse/PRJCA017486 (2023).
NCBI Assembly https://identifiers.org/insdc.gca:GCA_031439595.1 (2023).
NCBI Assembly https://identifiers.org/insdc.gca:GCA_031439585.1 (2023).
NCBI Assembly https://identifiers.org/insdc.gca:GCA_031216125.1 (2023).
NCBI Assembly https://identifiers.org/insdc.gca:GCA_031216105.1 (2023).
NCBI Assembly https://identifiers.org/insdc.gca:GCA_031216115.1 (2023).
Liu, M. Five leguminosae species. figshare https://doi.org/10.6084/m9.figshare.22580914 (2023).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC bioinformatics 9, 1–14 (2008).
Ou, S. & Jiang, N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant physiology 176, 1410–1422 (2018).
Lawniczak, M. K. et al. Standards recommendations for the earth BioGenome project. Proceedings of the National Academy of Sciences 119, e2115639118 (2022).
Cheng, S. et al. 10KP: A phylodiverse genome sequencing plan. GigaScience 7, 1–9 (2018).
Acknowledgements
This work was supported by the Shenzhen Municipal Government of China (KCXFZ20201221173013035), the Major Science and Technology Projects of Yunnan Province (Digitalization, development and application of biotic resource, 202002AA100007), the Key Laboratory of Genomics, Ministry of Agriculture, BGI-Shenzhen, Shenzhen 518120, China, Guangdong Provincial Key Laboratory of core collection of crop genetic resources research and application, and Shenzhen Engineering Laboratory of Crop Molecular Design and Breeding, BGI-Shenzhen, Shenzhen 518120, China. This work is part of the 10KP project57 and is also supported by China National GeneBank (CNGB).
Author information
Authors and Affiliations
Contributions
H.L., S.X.L. and S.G.W. led and designed this project. H.L., J.L.Y., M.L. and S.K.S. conceived the study. J.M.L. and L.C. collected the leaf and tissue samples. S.K.S., M.L. contributed to the sample preparation and performed the genome and chromosome-scale assembly. M.L., R.L., G.W. and J.L.Y., performed annotation and comparative genomic analyses. J.L.Y., S.K.S. and M.L. wrote the original draft manuscript. J.L.Y., M.L., S.K.S., X.G. and H.L., revised and edited the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, J., Liu, M., Sahu, S.K. et al. Chromosome-scale genomes of five Hongmu species in Leguminosae. Sci Data 10, 710 (2023). https://doi.org/10.1038/s41597-023-02593-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02593-2
This article is cited by
-
Beyond NGS data sharing for plant ecological resilience and improvement of agronomic traits
Scientific Data (2024)
