
Abstract
Soybean aphid (Aphis glycines; SBA) and soybean cyst nematode (Heterodera glycines; SCN) are two major pests of soybean (Glycine max) in the United States of America. This study aims to characterize three-way interactions among soybean, SBA, and SCN using both demographic and genetic datasets. SCN-resistant and SCN-susceptible soybean cultivars with a combination of soybean aphids (biotype 1) and SCN (HG type 0) in a randomized complete block design (RCBD) with six blocks were used to evaluate the three-way interactions in a greenhouse setup. Treatments receiving SCN were infested at planting with 2000 nematode eggs, and the treatments with soybean aphids were infested at second trifoliate growth stage (V2) with 15 soybean aphids. The whole roots were sampled from plants at 5 and 30 days post SBA infestation for RNA sequencing using Illumina Hiseq. 3000. The data comprises of 47 libraries that are useful for further analyses of important genes, which are involved in interaction effects of SBA and SCN on soybean.
Design Type(s) | transcription profiling design • randomized complete block design • strain comparison design • stimulus or stress design |
Measurement Type(s) | transcription profiling assay |
Technology Type(s) | RNA sequencing |
Factor Type(s) | cultivar • temporal_interval • experimental condition • biological replicate |
Sample Characteristic(s) | Glycine max • root • cropland biome |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others

Background & Summary
Soybean [Glycine max (L.) Merr.], considered as the source of high-quality sugar, protein, and oil, is one of the most important crops worldwide1. Soybean aphid (SBA), Aphis glycines Matsumura (Hemiptera: Aphididae) and soybean cyst nematode (SCN), Heterodera glycines Ichinohe (Tylenchida: Heteroderidae) are the two most economically important pests of soybean in the Midwestern United States2,3. Soybean aphid, an aboveground herbivore (pest), feeds on phloem sap whereas SCN, a belowground pest, infests the soybean roots. These infestations can co-occur and amplify further reduction in soybean yield4,5. In the United States, annual economic losses due to the SBA and SCN have been estimated to be approximately $4 billion and $1.3 billion, respectively6,7,8. To counteract these devastating pests, farmers rely on various management strategies that include host plant resistance and chemical measures9,10,11. For SBA, dependency on the use of chemical management has resulted in pyrethroid resistance in SBA populations in Iowa, Minnesota, North Dakota and South Dakota as well as the impacts on non-target beneficial organisms12,13. In addition, the long-term use of SCN resistance has resulted in SCN populations that are capable of overcoming the resistance genes (i.e., HG types)14. Although host plant resistance has not been implemented on a large scale for SBA management, multiple virulent SBA biotypes have been discovered in the U.S. Virulent SBA biotypes and SCN races threaten the sustainability of host plant resistance for these two pests14,15,16,17. Thus, genetic data generated from greenhouse experiments on the effects of SBA and SCN on soybean cultivars are of tremendous importance for unraveling resistance genes and regulatory networks that can potentially be used for developing durable resistance in soybean to both pests.
Although above- and below- ground herbivores are spatially segregated, they both share the host plant through systemic tissues and are able to influence each other18. Previously, the influence of SCN on soybean aphid infestation or vice versa has been studied on soybean using demographic datasets4,5,19,20,21. McCarville, et al.4 conducted experiments on various soybean cultivars [SCN susceptible (DK 28–52, IA 3018, IA 3041) and SCN resistant (DK 27–52, AG 2821 V, IA 3028)] to understand the effect of SBA, SCN, and fungus Cadophora gregata (Allington & Chamberlain) Harrington & McNew on soybean. Their study showed 5.24 times increase in SCN reproduction in the presence of soybean aphid and the fungus. In contrast, the aphid population decreased by 26.4% in the presence of SCN and C. gregata and the aphid exposure reduced by 19.8% in SCN resistant cultivars. Later, McCarville, et al.5 demonstrated the relationship between the aboveground feeding of soybean aphid and belowground reproduction of SCN in the SCN resistant Dekalb 27–52 (PI 88788 derived) cultivar, and SCN susceptible Kenwood 94 cultivar. In 30 days, both SCN eggs and the number of females increased by 33% in SCN-resistant cultivar and reduced by 50% in the SCN-susceptible cultivar. In 60 days, the number of SCN eggs and female count remained unaffected in the resistant cultivar but decreased in the susceptible cultivar. The authors concluded that soybean aphid feeding improved the quality of soybean as a host for SCN but this result was varied significantly with the cultivar and length of the experiment. Apart from these demographic studies, molecular characterization of SBA-SCN-soybean interaction has not been reported previously.
RNA-Sequencing (RNA-Seq) has been a standard tool for studying qualitative and quantitative gene expression assays that provide information on transcript abundance with their variation22,23. The major objective of this study was to evaluate differential gene expression of soybean plants that are infested with SCN in the presence or absence of SBA. To achieve the objective, we conducted experiments on two genotypes of G. max [H. glycines susceptible Williams 82 (PI518671), and H. glycines resistant MN1806CN] that were infested with biotype 1 SBA and HG Type 0 SCN for RNA-sequencing. More than 1.1 billion reads (61.4 GB) of transcriptomic data were obtained from 47 samples derived from the experiment using whole roots of G. max. An overview of the experimental design, methods and transcriptome analysis pipeline is shown in Fig. 1a–c, respectively. A comprehensive understanding of these transcriptome data will enhance our understanding of interactions among soybean, SBA, and SCN at the molecular level. The rapid advancement of bioinformatics tools is facilitating the search of candidate genes and their function that might play a crucial role in various pathways for host resistance against both herbivores.

An overview of greenhouse experiments and transcriptomic data analysis pipeline. (a) A randomized complete block design (RCBD) using two water baths (Water bath I and Water bath II), (b) A flow chart representing experimental methods used for soybean cyst nematode and soybean aphid interaction using two cultivars of soybean, and (c) A flow chart showing RNA-seq data analysis pipeline.
Methods
Plant material, soybean aphid, and SCN
Two cultivars of soybean– Williams 82 (PI518671) and MN1806CN were used in this experiment. Williams 82 is susceptible to both HG Type 0 (race 3) of the SCN and SBA. MN1806CN is resistant to HG Type 0 (race 3) of the SCN but susceptible to SBA. Soybean aphid biotype 1 populations were originally obtained from the Ohio State University and were reared on susceptible cultivar LD12-15838R at South Dakota State University. This biotype is defined by an avirulent response to all known SBA resistance (Rag) genes and was first identified in Illinois24. The SCN population used was HG type 0, which is defined by having less than 10% reproduction documented by studies of SCN resistance and is avirulent to all SCN resistance genes in soybean.
Experimental design and sample collection
A greenhouse experiment was designed using a randomized complete block design (RCBD) with eight treatments (four treatments per cultivars) with eight experimental units (plants) in six blocks. The treatments were factors of soybean genotype, SBA infestation, and SCN infestation. For examples, each of the soybean genotypes received one of the following combinations: SCN:no SBA, no SCN:SBA, SCN:SBA, or no SBA:no SCN (control).
For this experiment, the soil-sand mixture was prepared by adding construction sand and clay soil including SCN infested clay soil in the ratio of 3:1. The 125 cc of the mixture was distributed in cone-tainers (diameter of 3.8 cm, a depth of 21 cm and a volume of 164 cc; Greenhouse Megastore, USA). For SCN included treatments, each cone-tainer received approximately 2,000 SCN eggs. The cone-tainers with three soybean seeds were arranged in a 2.0 U.S. gallon (7.57 liter) plastic buckets (Leaktite, USA) filled with construction sand (Quikrete, GA). These buckets were kept in a water bath for maintaining soil temperature between 26.7 °C and 28.9 °C to ensure the reproduction of SCN (i.e. ~30 days)5. The temperature of the water bathes were regularly monitored using thermometers. The plants were grown under 16:8 (L:D) in a greenhouse with a temperature of 28 °C and 45% relative humidity. The plants were thinned down to one plant per cone-tainer upon reaching the second vegetative growth stage (V2). The V2-staged plants with the SBA included treatments were infested with 15 mixed age (i.e., fourth instar nymphs and adults) biotype 1 SBA using a 000 fine tip paintbrush (Winsor & Newton, England). The SBA were applied on the abaxial surface of the first trifoliate of V2-staged plants. All plants in each bucket were covered with a large no-see-um mesh net (Quest Outfitters, Sarasota, FL) to prevent inter-bucket movement of aphids. After SBA infestation, soybean plants were regularly checked to confirm the successful establishment of soybean aphids. Soybean aphid populations were counted at 5, 15, and 30 days post infestation (dpi). For SBA only treatment, the populations on the two soybean varieties were not significantly different, indicating that both lines were susceptible to SBA. SCN eggs were sampled at 30 dpi. The whole roots were collected on 5 and 30 dpi by snap freezing in liquid Nitrogen and stored at −80 °C for further analysis. The 5 dpi and 30 dpi root samples treated with each treatment were collected from Water bath I and Water bath II, respectively, representing each plant from three blocks (three biological replicates). The SCN soil and SCN infested roots were used for SCN cysts collection (except root samples collected for transcriptomic study) and the soil was examined for SCN counts.
RNA extraction, library construction, and RNA sequencing
RNA was extracted from all samples representing three biological replicates of each treatment that constituted 24 samples collected at 5 and 30 dpi each. As the major foci of the project were to determine whether the gene expression differed between SCN resistant and SCN susceptible soybeans, and to evaluate the gene expression of soybeans that were dual infested with SCN and SBA, we selected two timepoints (5 and 30 dpi). We selected 30 dpi to observe gene expression of treatment effects on a single generation of SCN reproduction keeping 5 dpi as a reference in the presence or absence of SBA. Frozen root samples from each treatment were grounded in liquid nitrogen with a mortar and pestle to a fine powder followed by total RNA extraction using PureLink RNA mini kit (Invitrogen, USA). RNA samples were treated with TURBOTM DNase (Invitrogen, USA) to remove any DNA contamination following the manufacturer’s instructions. Assessment of the isolated RNA integrity was performed by 1% agarose gel electrophoresis, and RNA concentration was measured by Nanodrop 2000 (Thermo Fisher Scientific, USA). The cDNA libraries were constructed using NEBNext Ultra II RNA library 96 single index prep kit and sequenced using Illumina HiSeq. 3000 (single read end utilizing 100 bases read length) at Iowa State University Sequencing Facilities.
Pre-processing of sequencing data
Quality control of reads was assessed using FastQC program (version 0.11.3) (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/)25. The FastQC results were visualized using MultiQC v1.326, and low quality bases (QC value < 20; 5-bp window size) were removed by trimming in the program Btrim64 (version 0.2.0)27. High-quality single-end reads were mapped against the primary coding sequences of G. max. The coding sequences (Gmax: Gmax_275_Wm82.a2.v1.transcript_primaryTranscriptOnly.fa.gz) were obtained from the Phytozome database and aligned using Salmon ver.0.9.128 accessed from Bioconda29. Downstream analyses of the quantified transcript reads were performed using integrated Differential Expression and Pathway analysis (iDEP 0.81, R/Bioconductor packages)30. The quantified transcript reads were filtered with 0.5 counts per million (CPM) in at least one sample and transformed using regularized log (rlog), which is implemented in the DESeq. 231 package.
Data Records
All sequence reads were deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (accession SRR8427366-SRR8427408) under Bioproject PRJNA514200 (Project ID: SRP178193)33 (Table 1). The raw transcript abundance counts for all the samples was deposited at the Gene Expression Omnibus (GEO) database, GSE12510334. The transformed transcript abundance counts, hierarchical clustering, correlation matrices, and clusters are available in figshare at: https://doi.org/10.6084/m9.figshare.7755152.v332.
Technical Validation
Quality control
Forty-eight RNA libraries were prepared and sequenced with the sequencing depth ranging from 9,847,269 to 57,520,568 reads (see Table 1). Sequencing of one of the replicates of control resistant cultivar (MN1806CN collected at 30 dpi) came in error. Therefore, total reads of more than 1.1 billion from 47 libraries were subjected to FastQC analysis, which helped determine the data quality using various quality metrics such as mean quality scores (Fig. 2a), per sequence quality scores (Fig. 2b), per sequence GC content (Fig. 2c), and sequence length distribution (Fig. 2d). Phred quality scores per-base for all samples were higher than 30 and GC content ranged from 43 to 45%, following a normal distribution. After trimming, more than 99% of the reads were retained as clean and good quality reads. Upon mapping these reads, a high mapping rate of 73.8% to 94.3% was obtained. Among these, 67.1% to 87.6% reads were uniquely mapped.

Quality metrics of G. max sequencing data. (a) Mean quality scores per position, (b) Per sequence quality scores, (c) GC content distribution, and (d) Read length distribution.
Assessment of transcriptomic data
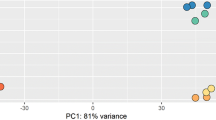
The 43,122 genes passed the filter upon filtering with 0.5 CPM in at least one sample. To reduce the mean dependent variance, the quantified transcript reads were transformed as shown in Fig. 3a–c and available in Figshare32 (the transformed transcript abundance count for all the samples). The transformed data were subjected to hierarchical clustering and principal component analysis (PCA) followed by visualization using t-SNE map35 in order to assess the global transcriptomic data. The hierarchical clustering of top 6000 variable genes based on two time points (5 dpi and 30 dpi) showed distinct clustering except for some samples [Fig. 4a; Figshare32 (the hierarchical clustering of top 6,000 variable genes)]. Figure 4b represents the standard deviation (SD) distribution of the top variable 6,000 genes. Figure 4c represents the Pearson’s correlation between the samples using the top 75% genes and available in Figshare32 (the correlation between the samples using the top 75% genes). The t-SNE map revealed four clusters (A, B, C, and D) for 6,000 variable genes [Fig. 4d; Figshare32 (the four clusters for 6,000 variable genes)]. Regarding the PCA, PC1 is correlated with time (P = 1.16e-06) with 28% variance, and PC2 is correlated with Treatment (P = 2.02e-08) with 15% variance (Fig. 4e).

Pre-processing of transcriptomic data. (a) Distribution of transformed data, (b) Density plot of transformed data, and (c) Scatter plot of the first two samples (SCNS5d_1 vs SCNS5d_2).

Assessment of transcriptomic data. (a) Heatmap of top 6,000 variable genes, (b) Gene SD distribution, (c) Correlation matrix, (d) Visualization of top 6,000 genes shown in the t-SNE map, and (e) A principal components analysis (PCA) plot.
Usage Notes
This data represents the first publicly available transcriptomic data for soybean roots from the three-way interaction among G. max, H. glycines, and A. glycines. The raw compressed fastq files (fastq.gz) were submitted to the National Center for Biotechnology Information (NCBI) and are available with accession numbers (SRR8427366-SRR8427408; http://identifiers.org/ncbi/insdc.sra:SRP178193)33. The data could be retrieved using fastq-dump tool SRA toolkit (https://www.ncbi.nlm.nih.gov/sra). There are various tools such as Trimmomatic36, cutadapt37, Fastq_clean38 that could be used for trimming purpose. Apart from the Salmon tool for the alignment and quantification of reads, other tools such as STAR aligner (https://github.com/alexdobin/STAR), Bowtie39, HISAT240, TopHat241, Cufflinks with HTSeq can be employed, which requires reference genome of G. max and annotation file in gff3 format. For differential gene expression analysis, EdgeR42 and limma43 could be used instead of DEseq. 231. Apart from the standalone tools like iDEP30, Galaxy (https://usegalaxy.org), CyVerse (http://www.cyverse.org), MeV (http://mev.tm4.org)44, and integrated RNA-seq interpretation system for gene expression data analysis tool (http://bmbl.sdstate.edu/IRIS/)45 could also be used for both analysis and visualization of RNA-seq data.
Code Availability
Codes used for RNA-seq data processing in the current study are available as supplementary material in Figshare at: https://doi.org/10.6084/m9.figshare.7755152.v332 (Codes used for RNA-seq data processing).
References
Yu, X., Yuan, F., Fu, X. & Zhu, D. Profiling and relationship of water-soluble sugar and protein compositions in soybean seeds. Food Chem. 196, 776–782 (2016).
Hartman, G. et al. Occurrence and distribution of Aphis glycines on soybeans in Illinois in 2000 and its potential control. Plant Health Progr. 2, 17, https://doi.org/10.1094/PHP-2001-0205-01-HN (2001).
Wrather, J. A. & Koenning, S. R. Estimates of disease effects on soybean yields in the United States 2003 to 2005. J. Nematol. 38, 173 (2006).
McCarville, M., O’Neal, M., Tylka, G., Kanobe, C. & MacIntosh, G. A nematode, fungus, and aphid interact via a shared host plant: implications for soybean management. Entomol. Exp. Appl. 143, 55–66 (2012).
McCarville, M. T., Soh, D. H., Tylka, G. L. & O’Neal, M. E. Aboveground feeding by soybean aphid, Aphis glycines, affects soybean cyst nematode, Heterodera glycines, reproduction belowground. PloS one 9(1) e86415 (2014).
Hill, C., Chirumamilla, A. & Hartman, G. Resistance and virulence in the soybean-Aphis glycines interaction. Euphytica 186, 635–646 (2012).
Koenning, S. R. & Wrather, J. A. Suppression of soybean yield potential in the continental United States by plant diseases from 2006 to 2009. Plant Health Progr, https://doi.org/10.1094/PHP-2010-1122-01-RS (2010).
Niblack, T., Lambert, K. & Tylka, G. A model plant pathogen from the kingdom animalia: Heterodera glycines, the soybean cyst nematode. Ann. Rev. Phytopathol. 44, 283–303 (2006).
Olson, K. D., Badibanga, T. M. & DiFonzo, C. Farmers’ awareness and use of IPM for soybean aphid control: report of survey results for the 2004, 2005, 2006, and 2007 crop years. Staff Papers 45803, University of Minnesota, Department of Applied Economics 1701-2016-138883, 1–29 (2008).
Niblack, T. Soybean cyst nematode management reconsidered. Plant Dis. 89, 1020–1026 (2005).
Ragsdale, D. W., Landis, D. A., Brodeur, J., Heimpel, G. E. & Desneux, N. Ecology and management of the soybean aphid in North America. Annu. Rev. Entomol 56, 375–399 (2011).
Koch, R. L. et al. Biology and economics of recommendations for insecticide-based management of soybean aphid. Plant Health Progr. 17, 265–269 (2016).
Varenhorst, A. J. & O’Neal, M. E. The response of natural enemies to selective insecticides applied to soybean. Environ. Entomol. 41, 1565–1574 (2012).
Tylka, G. L. Understanding soybean cyst nematode HG types and races. Plant Health Progr. 17, 149–151 (2016).
Mitchum, M. G., Wrather, J. A., Heinz, R. D., Shannon, J. G. & Danekas, G. Variability in distribution and virulence phenotypes of Heterodera glycines in Missouri during 2005. Plant Dis. 91, 1473–1476 (2007).
Hesler, L. S. et al. Performance and prospects of Rag genes for management of soybean aphid. Entomol. Exp. Appl. 147, 201–216 (2013).
Mitchum, M. G. Soybean resistance to the soybean cyst nematode Heterodera glycines: an update. Phytopathology 106, 1444–1450 (2016).
Megías, A. G. & Müller, C. Root herbivores and detritivores shape above‐ground multitrophic assemblage through plant‐mediated effects. J. Anim. Ecol. 79, 923–931 (2010).
Hong, S., Donaldson, J. & Gratton, C. Soybean cyst nematode effects on soybean aphid preference and performance in the laboratory. Environ. Entomol. 39, 1561–1569 (2010).
Hong, S., MacGuidwin, A. & Gratton, C. Soybean aphid and soybean cyst nematode interactions in the field and effects on soybean yield. J. Econ. Entomol. 104, 1568–1574 (2011).
Heeren, J. et al. The interaction of soybean aphids and soybean cyst nematodes on selected resistant and susceptible soybean lines. J. Appl. Entomol. 136, 646–655 (2012).
Griffith, M., Walker, J. R., Spies, N. C., Ainscough, B. J. & Griffith, O. L. Informatics for RNA sequencing: a web resource for analysis on the cloud. PLoS Comp. Bio. 11, e1004393 (2015).
Shan, X. et al. Transcriptome profile analysis of maize seedlings in response to high-salinity, drought and cold stresses by deep sequencing. Plant Mol. Biol. Rep. 31, 1485–1491 (2013).
Kim, K. S., Hill, C. B., Hartman, G. L., Mian, M. & Diers, B. W. Discovery of soybean aphid biotypes. Crop Sci. 48, 923–928 (2008).
Andrews, S. FastQC: a quality control tool for high throughput sequence data, http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2010).
Ewels, P., Magnusson, M., Lundin, S. & Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016).
Kong, Y. J. G. Btrim: a fast, lightweight adapter and quality trimming program for next-generation sequencing technologies. Genomics 98, 152–153 (2011).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417 (2017).
Grüning, B. et al. Bioconda: sustainable and comprehensive software distribution for the life sciences. Nat. Methods 15, 475 (2018).
Ge, S. X., Son, E. W. & Yao, R. iDEP: an integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinformatics 19, 534 (2018).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq. 2. Genome Biol. Evol. 15, 550 (2014).
Neupane, S., Mathew, F. M., Varenhorst, A. J. & Nepal, M. P. Transcriptome profiling of interaction effects of soybean cyst nematodes and soybean aphids on soybean. figshare, https://doi.org/10.6084/m9.figshare.7755152.v3 (2019).
NCBI Sequence Read Archive, http://identifiers.org/ncbi/insdc.sra:SRP178193 (2019).
Neupane, S., Varenhorst, A. J. & Nepal, M. P. Transcriptome profiling of interaction effects of soybean cyst nematodes and soybean aphids on soybean. Gene Expression Omnibus, https://identifiers.org/geo:GSE125103 (2019).
Maaten, L. V. D. & Hinton, G. Visualizing data using t-SNE. J. Mach. Learn Res. 9, 2579–2605 (2008).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet journal 17, 10–12 (2011).
Zhang, M. et al. Fastq_clean: an optimized pipeline to clean the illumina sequencing data with quality control. In Bioinformatics and Biomedicine (BIBM), IEEE International Conference (IEEE), 44–48 (2014).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. Evol. 10, R25 (2009).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357 (2015).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. Evol. 14, R36 (2013).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47–e47 (2015).
Howe, E. A., Sinha, R., Schlauch, D. & Quackenbush, J. RNA-Seq analysis in MeV. Bioinformatics 27, 3209–3210 (2011).
Monier, B. et al. IRIS-EDA: An integrated RNA-Seq interpretation system for gene expression data analysis. PLOS Comput. Biol. 15, e1006792 (2019).
Acknowledgements
Authors would like to acknowledge Dr. Emmanuel Byamukama (South Dakota State University) for providing H. glycines HG Type 0 infested soil as a source of SCN for the experiment. Also, Philip Rozeboom and Alyssa Vachino are acknowledged for their assistance in greenhouse experiments. The greenhouse experiments and RNA sequencing were funded through South Dakota Soybean Research and Promotion Council (SDSRPC-SA1800238). The analysis, data interpretation and writing of the manuscript were funded through USDA-NIFA hatch Projects (SD00H469-13 and SD00H659-18 to M.P.N.).
Author information
Authors and Affiliations
Contributions
S.N. conducted the experiment, performed data analyses and drafted the manuscript. M.P.N. and A.J.V. conceived the project and helped designed the experiments. M.P.N., A.J.V., and F.M.M. edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
ISA-Tab metadata file
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Neupane, S., Mathew, F.M., Varenhorst, A.J. et al. Transcriptome profiling of interaction effects of soybean cyst nematodes and soybean aphids on soybean. Sci Data 6, 133 (2019). https://doi.org/10.1038/s41597-019-0140-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-019-0140-4
