
Abstract
Microglia are central players in Alzheimer’s disease pathology but analyzing microglial states in human brain samples is challenging due to genetic diversity, postmortem delay and admixture of pathologies. To circumvent these issues, here we generated 138,577 single-cell expression profiles of human stem cell-derived microglia xenotransplanted in the brain of the AppNL-G-F model of amyloid pathology and wild-type controls. Xenografted human microglia adopt a disease-associated profile similar to that seen in mouse microglia, but display a more pronounced human leukocyte antigen or HLA state, likely related to antigen presentation in response to amyloid plaques. The human microglial response also involves a pro-inflammatory cytokine/chemokine cytokine response microglia or CRM response to oligomeric Aβ oligomers. Genetic deletion of TREM2 or APOE as well as APOE polymorphisms and TREM2R47H expression in the transplanted microglia modulate these responses differentially. The expression of other Alzheimer’s disease risk genes is differentially regulated across the distinct cell states elicited in response to amyloid pathology. Thus, we have identified multiple transcriptomic cell states adopted by human microglia in a multipronged response to Alzheimer’s disease-related pathology, which should be taken into account in translational studies.
Similar content being viewed by others


Main
Microglia are a central part of the cellular response in Alzheimer’s disease (AD) pathogenesis1,2, in particular in the early response to amyloid-β (Aβ) pathology3,4,5,6. Murine microglia transit from a homeostatic state toward ‘reactive’ disease-associated microglia (DAM), also called activated response microglia (ARM) or neurodegenerative microglia (MGnD)4,5. This response is partially Triggering receptor expressed in myeloid cells 2 (TREM2) and Apolipoprotein E (APOE)-dependent and elicited by Aβ plaques in mice3,4,7. Ageing, tauopathy, amyotrophic lateral sclerosis4,5,8,9, apoptotic neurons5, Danish amyloid10 and even peripheral lipid dyshomeostasis11 can elicit a similar response, suggesting that the DAM state is a generic response of mouse microglia to damage. However, it is somewhat controversial as to what extent the DAM response is conserved in the human brain12,13. Mouse and human microglia are evolutionarily divergent, for instance, regarding the expression of relevant AD genetic risk7,14,15.
In addition, the microglial cell states reported by postmortem samples are inconsistent. Mathys et al.16 found 22 genes upregulated in microglia from patients with AD (only five overlapping with the DAM signature). Grubman et al.17 reported eight genes, whereas Zhou et al.18 identified 11 DAM genes enriched in patients with AD compared with controls. Del-Aguila et al.19 analyzed single-nucleus transcriptomes from three patients with AD and were unable to recapitulate an activation signature. A large study by Gerrits et al.20 analyzed 482,472 single nuclei from nondemented control and AD brains and indicated several distinct transcriptomic states of microglia encompassing DAM-like (ITGAX, SPP1) and pro-inflammatory (IL1B, NFKB1) states in AD. A more recent study by Sun et al.21 analyzed 194,000 single nuclei from 443 AD and healthy subjects, reporting 12 distinct microglial transcriptional states with clear pro-inflammatory profiles (IL1B, CCL3), but no clear enrichment for DAM signatures. The lack of congruency in these human postmortem brain studies can be partially explained by technical issues, such as postmortem time12, or intrinsic lesser resolution of single-nuclei sequencing approaches13, heterogeneity in pathological samples or technical shortcomings including sample size. Additionally, there are inherent limitations to postmortem studies as they reflect only terminal stages of a complex, decade-long disease process, which often encompasses additional concomitant neuropathologies (Tau, TDP-43, vascular dementia or alpha-synuclein).
Here, we circumvent these multiple drawbacks by using a xenotransplantation model where we engrafted stem cell-derived microglia in the brain of the AppNL-G-F model of amyloid pathology and of wild-type (WT) controls14. We provide a full transcriptomic characterization of the different cell states adopted by human microglia in response to Aβ pathology, including the elusive component of soluble Aβ oligomers (Aβo) that have been linked to neuroinflammation, neurodegeneration and cognitive impairment22,23,24,25. We show that human microglia react very differently from mouse microglia to Aβ pathology, with several branches all differentially enriched in AD risk genes, and which can be selectively disturbed by introducing the TREM2 risk variant or APOE polymorphisms.
Results
Response of human microglia to Aβ pathology
We transplanted microglial precursors in the mouse brain according to the MIGRATE (microglia in vitro generation refined for advanced transplantation experiments) protocol26. We used three different host genetic backgrounds, that is Rag2−/− Il2rγ−/− hCSF1KI AppNL-G-F (with progressive Aβ plaque accumulation from 3 months after birth, thereafter called AppNL-G-F), Rag2−/− Il2rγ−/− hCSF1KI Apphu/hu mice (humanized App controls27 for the AppNL-G-F mice that we called AppWT in the current manuscript) and Rag2−/− Il2rγ−/− hCSF1KI AppNL-G-F ApoE−/− (AppNL-G-F ApoE−/−) mice (Fig. 1a). The AppWT mice are a better control than the initially proposed AppNL mouse, as discussed by Serneels et al.27. We also studied stem cell-derived microglia from three different human genetic backgrounds, that is, UKBIO11-A, BIONi010-C and H9 (Methods and Table 1). Microglia were transplanted in 4-day-old (P4) neonates and isolated at 6–7 months after transplantation by fluorescence-activated cell sorting (FACS) (CD11b+hCD45+; Fig. 1a and Supplementary Fig. 1b). We obtained high-quality single-cell transcriptomes from 138,577 microglia across 106 mice, excluding CNS-associated macrophages (CAMs), proliferative cells, other myeloid cells, and low-quality cells and doublets (Supplementary Figs. 2 and 3).

a, Experimental design used in this study. b, UMAP plot visualizing 138,577 single xenografted human microglial cells sorted from mouse brain (CD11b+hCD45+) ~6 months after transplantation. Cells are colored according to clusters identified: HM, RM, DAM, IRM, HLA, CRM-1 and CRM-2, and tCRM. The assignment of different clusters to distinct cell types or states is based on previous experimental data from our and other laboratories3,4,7,14 (see c,d and Extended Data Figs. 3 and 4). c, UMAP plots as in b, colored by the combined level of expression of groups of genes that characterize distinct microglial transcriptional states. d, Top ten most differentially expressed genes in each cluster (normalized expression scaled by gene is shown). FC, fold change; GO, gene ontology; MP, microglial precursor; SC, stem cell.
We identified five distinct microglial clusters, which we annotated as Homeostatic (HM), Cytokine response-1 and -2 (CRM-1 and -2), Interferon response (IRM), Disease-associated response (DAM) and Antigen-presenting response (HLA), as well as two intermediate clusters which we named Ribosomal microglia (RM), enriched in ribosomal genes, and Transitioning CRM (tCRM) microglia, which show high levels of homeostatic genes but also express CRM markers (Fig. 1b–d, Supplementary Fig. 4 and Table 1). The DAM and IRM clusters have similar counterparts in mice3,4 (Supplementary Fig. 4b). Using existing datasets3,4, we observed that only 20–25% of the previously described mouse DAM signature overlapped with the transcriptome of human xenografted microglia (Extended Data Fig. 3d and Supplementary Table 2). In addition, and contrary to mouse cells, human microglia do not seem to display the DAM1/DAM2 duality4. Although CRM and HLA cell states have been described before in mouse models of trauma and as a response to neuronal death28, they were not identified before in mouse microglia derived from mouse models of Aβ pathology, including AppNL-G-F (ref. 3). CRM is defined by the upregulation of genes encoding cytokines and chemokines (CCL4, CCL3, CCL42L, CCL3L1, GADD45B, CCL2, CH25H, IL1B, ZFP36, SGK1), while HLA encompasses HLA-DRA, HLA-DPA1, HLA-DRB5, CD74, HLA-DPB1, HLA-DMA, MS4A6A and HLA-DMB expression (Fig. 1d and Supplementary Fig. 4). Interestingly, whereas the DAM profile shows a clear downregulation of homeostatic microglial genes, these are much less downregulated in CRM, HLA and IRM clusters (Fig. 1d).
As a quality control, we compared the transcriptomic profiles of cells transplanted into AppWT mice (n = 10, 3 independent cell lines) with those previously obtained from microglia transplanted into the brain of Rag2−/− Il2rγ−/− hCSF1KI, and from primary human microglia freshly isolated from surgical resections14. As expected, we found a vast overlap between transplanted and primary cells, with all clusters represented to similar extents in the different experimental groups, confirming that the engrafted cells acquire a mature transcriptomic profile resembling that of human adults. Of note, a small cluster that represented 4% of the dataset was enriched only in the primary human cells (Extended Data Fig. 1 and Supplementary Table 3).
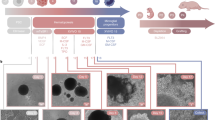
Next, we compared the transcriptomic profiles of human microglia transplanted in AppNL-G-F (n = 23) and AppWT (n = 10) (three independent cell lines per group) mice to evaluate to what extent different microglial phenotypes were induced by Aβ pathology. DAM, HLA and CRM-1 clusters were significantly enriched in AppNL-G-F compared with AppWT mice (Fig. 2a,b and Extended Data Fig. 2a,b). This is only partially consistent with a previous study by Hasselman et al.7,29 (Extended Data Fig. 3d), where DAM but not antigen-presenting (HLA/MHC-II) or chemokine transcriptional profiles were correlated to Aβ pathology in a human chimera model. Nevertheless, the authors used only n = 1 mice per group and a different amyloid mouse model. We performed immunostaining using human-specific markers to confirm that both human DAM (CD9) and HLA (HLA-DQ/DR) microglia clustered around Aβ plaques (Fig. 2c), as well as the intermediate RM state (FTH1) (Extended Data Fig. 2d). Expression of various genes of the HLA cluster has been associated with dense cored Aβ plaques in human AD brain, and was detected also in demyelinating regions in multiple sclerosis cases30.

a, Density plots displaying the average distribution of human microglia transplanted in AppNL-G-F (n = 23) and AppWT (n = 10) mice (density is normalized to sample size). b, Distribution and proportion of cells across all identified clusters (box plots are limited by lower and upper quartiles and midline indicates median; whiskers show minimum to maximum values. Each dot represents a single mouse. Unpaired t-test with Welch’s correction, two-tailed, alpha = 0.05, significance was set as P < 0.05). c, Representative images and quantification of human microglia engrafted in the brain of AppNL-G-F mice at 6 months of age, labeled with human-specific antibodies for P2RY12 (HM microglia), CD9 (DAM), HLA-DR/DQ (HLA microglia), as well as X-34 for Aβ plaques. The shift in immunofluorescent signal in the proximity of Aβ plaques was performed using a modified Sholl analysis where the fluorescent intensity of microglial markers P2RY12, CD9 and HLA was measured through concentric rings (annuli) of increasing diameter surrounding the X-34 plaque center. Intensities of each channel were scaled for comparison using z-score normalization. Intensity over distance (µm) was plotted using Loess nonparametric regression in R with estimated standard error for each predicted value. For comparison of intensities near and distant from the plaque center, the means of the inner and outer three annuli were independently calculated (near, 0–10 mm, distant, 70–80 mm from the plaque; n = 3 per group; bar plots represent mean ± s.e.m.; unpaired t-test, one-tailed, alpha = 0.05, significance was set as P < 0.05). Scale bars, 50 μm. MFI, median fluorescent intensity.
Trajectory analysis indicated that microglia follow three main activation routes from homeostatic states toward four distinct transcriptional cell states: DAM, HLA, CRM and IRM (Fig. 3a,b and Extended Data Figs. 4 and 5). Whereas the DAM to HLA and IRM trajectories partially overlap, CRM deflects early during the phenotypic transition, resulting in an apparently independent response program (Fig. 3a,b and Extended Data Figs. 4 and 5). HLA appears as a continuation of the DAM response, resulting in a DAM to HLA trajectory (Fig. 3a,b and Extended Data Fig. 5). To confirm the trajectory inference, we performed a time course analysis using one of the cell lines (BIONi-010C) to analyze the transcriptomic changes of microglia isolated from 3- (onset of plaque deposition) and 6-month-old AppNL-G-F mice (Fig. 3c,d and Extended Data Fig. 4). We confirm the same three independent trajectories (Extended Data Fig. 4a–c), with an age-dependent increase in DAM and HLA clusters, correlating with amyloid deposition (Extended Data Fig. 4d–f). The changes over time are not simple shifts in cell numbers across the transcriptional profiles, because overall levels of expression of homeostatic markers within the HM cluster specifically decreased (WT > 3 months > 6 months), and disease-associated and antigen-presenting markers within DAM and HLA clusters specifically increased (WT < 3 months < 6 month) over the disease course (Fig. 3e). In addition, we performed costaining of DAM (CD9) and HLA (HLA-DR/DQ) and observed that whereas all HLA-positive cells showed CD9 expression, not all CD9-positive cells showed HLA signal, thus supporting the prediction of a phenotypic continuum between DAM and HLA (Fig. 3f).

a, Phenotypic trajectory followed by the human microglia after exposure to Aβ in vivo, obtained by an unbiased pseudotime ordering with Monocle 3. b, Distribution of cells from different host mouse genetic backgrounds (y axis) across the two main transcriptional trajectories, DAM and HLA (top panel) and CRM (bottom panel), colored by clusters shown in a. Note the shift in transcriptional states in AppNL-G-F versus AppWT mice (box plots are limited by lower and upper quartiles and midline indicates median; whiskers extend from the box to the smallest or largest value no further than 1.5 × interquartile range. Each dot represents a single cell, n = 10 mice (10,663 cells) for AppWT, n = 23 mice (32,436 cells) for AppNL-G-F. Unpaired t-test with Welch’s correction, two-tailed, alpha = 0.05, significance was set as P < 0.05). c,d, Expression profile of selected genes across the HM–DAM–HLA pseudotime axis from the entire dataset (c), and divided into the different experimental groups: AppWT, AppNL-G-F at 3 months and AppNL-G-F at 6 months (d). e, Top ten most differentially expressed genes in each cluster divided by experimental group: AppWT, AppNL-G-F at 3 months and AppNL-F-G at 6 months (normalized expression scaled by gene is shown). f, Representative images of human microglia engrafted in the brain of AppNL-G-F mice at 6 months of age and labeled with human-specific CD9 (DAM) and HLA-DR/DQ (HLA microglia) antibodies. Scale bars, 25 μm (upper panel) and 50 μm (lower panel). APP–NLGF_3m, AppNL-G-F 3 months; APP–NLGF_6m, AppNL-G-F 6 months; APP–WT_6m, AppWT 6 months.
Interestingly, both the presence and magnitude of HLA and CRM phenotypic programs appear to be human-specific features, as these clusters have not been reported previously in mouse models even in advanced stages of disease3,4,5,9. To confirm this further, we compared the transcriptional profile of engrafted human microglia with mouse cells isolated from 6-month-old Rag2−/− Il2rγ−/− hCSF1KI AppNL-G-F (n = 1,024 cells, 2 mice) and with a dataset of immunocompetent AppNL-G-F and WT mice3 that we published previously (n = 2,779 cells, 4 mice). We integrated these two datasets, performed clustering analysis and investigated whether the HLA and CRM transcriptomic signatures were present. A small number of cells were consistent with HLA and CRM profiles, but, unlike human microglia, they did not seem to increase in response to Aβ pathology (Extended Data Fig. 3a,b). This low percentage of MHC-II-expressing cells is consistent with previous reports31.
We also explored whether the relationship between DAM and HLA could be the equivalent to that of DAM1 versus DAM2 observed in mice4. Direct comparison between these responses confirmed that the HLA response is unique to human microglia, and different from the murine DAM2 (Extended Data Fig. 3c). Both HLA and DAM2 showed increased expression of DAM genes (such as CD9, CSTD and GPNMB) compared with homeostatic cells, but these slightly decrease in the DAM–HLA human transition while they keep increasing in the mouse DAM1–DAM2 trajectory. Moreover, HLA is characterized by a specific induction of antigen-presenting molecules (including HLA-DQB1, HLA-DRB5 and CD74) and a smaller enrichment in ribosomal genes (such as RPS3, RPS9 and RPS19) which are instead downregulated in DAM2. This indicates that human microglia display an elaborate response to Aβ pathology. The HLA response suggests that they engage in antigen presentation of phagocyted material (for example, Aβ). Although the potential interaction of HLA microglia and T cells cannot be assessed in the chimeric model system, our findings emphasize the need of further research efforts in that direction. Overall human microglia display a complex, multipronged response to Aβ pathology, including some programs that share features of the previously described DAM, IRM and TRM responses3,4, but also two human-specific CRM and HLA transcriptional states.
TREM2 and APOE control microglial cell-state transition
We investigated the molecular mechanisms governing the transition toward different transcriptional cells states in human microglia in vivo. TREM2 is one of the main cell-autonomous mediators of the microglial response to Aβ plaques in mice4,5,32, and thus we wondered whether it was able to modify human microglial cell states in a similar way. We analyzed 3,282 H9 (n = 2) and 1,301 H9-TREM2−/− (n = 3) microglia from 6-month-old AppNL-G-F mice and observed a profound suppression of both DAM and HLA responses, but no significant alterations in the CRM profile (Fig. 4a–c and Extended Data Fig. 6a–c). These data confirm that TREM2 is necessary for the DAM and HLA responses in human microglia, confirming and extending a previous study showing that TREM2-deficient human microglia have an impaired DAM phenotype33.

a,d, Density plots displaying the average distribution of human H9-WT (n = 2) and H9-TREM2−/− (n = 3) (a), and human APOE3/3 (n = 6) and APOE−/− (n = 7) (d) microglia transplanted in AppNL-G-F mice. Density is normalized by sample size. b,e, Distribution and proportion of cells across all identified clusters for H9-WT (n = 2) and H9-TREM2−/− (n = 3) (b), and APOE3/3 (n = 6) and APOE−/− (n = 7) (e) transplanted microglia. Dots represent single mice. c,f, Phenotypic trajectory followed by H9-WT (n = 2 mice and 3,282 cells) and H9-TREM2−/− (n = 3 mice and 1,301 cells) (c), and APOE3/3 (n = 6 mice and 7,894 cells) and APOE−/− (n = 7 mice and 14,012 cells) (f) human microglia obtained by an unbiased pseudotime ordering with Monocle 3. Proportion of cells (y axis) at different stages of the pseudotime trajectory (x axis), colored as shown in Fig. 1a. Dots represent single cells. g, Correlation analysis of the logFC in microglia transplanted in AppNL-G-F ApoE−/− versus AppNL-G-F mice (y axis from Fig. 4) and H9-TREM2−/− versus H9-WT from AppNL-G-F mice (x axis) (Pearson’s correlation, R = 0.17, differentially expressed genes adjusted using Bonferroni correction and colored as in Fig. 1b; ‘activated’ indicates that differential expression was performed comparing cell states reactive to pathology, excluding homeostatic or transitioning clusters). h, Correlation analysis of the logFC in TREM2−/− (x axis) and APOE−/− (y axis) microglia transplanted in AppNL-G-F mice (Pearson’s correlation, R = −0.11, differentially expressed genes adjusted using Bonferroni correction and colored as in Fig. 1b; ‘activated’ indicates that differential expression was performed comparing reactive cell states, excluding homeostatic or transitioning clusters). Box plots in b,c,e,f are limited by lower and upper quartiles and midline indicates median; whiskers show minimum to maximum value (b,e) or extend from the box to the smallest or largest value no further than 1.5× interquartile range (c,f). Unpaired t-test with Welch’s correction, two-tailed, alpha = 0.05, significance was set as P < 0.05.
ApoE is critical for the transition to DAM in mouse models5. Therefore, we transplanted and analyzed the transcriptomes of 7,894 APOE3/3 (n = 6) and 11,799 APOE−/− (n = 7) human microglia isolated from 6-month-old AppNL-G-F mice. We observed that the ablation of APOE had mild effects on the microglial cell states, with no impact on DAM or CRM profiles, but caused a significant reduction in HLA microglia (Fig. 4d–f and Extended Data Fig. 6d–f). This suggests that although microglial APOE may not be necessary to initiate the response to Aβ plaques, it is required for the transition to the HLA state.
Our data show that TREM2 is a powerful inductor of the phenotypic transition to both DAM and HLA states in human microglia and place APOE as a subsequent factor needed for the shift from DAM to HLA, highlighting species-specific differences in how microglia react against the Aβ present in plaques.
Different activation programs against distinct forms of Aβ
We wondered whether the distinct transcriptional trajectories adopted by human microglia might be caused by different forms of Aβ pathology. We used a genetic approach to reduce the Aβ plaques in the mouse brain by knocking out ApoE (AppNL-G-F ApoE−/−). Note that in these experiments, the endogenous mouse ApoE gene is inactivated, as opposed to the experiments described above where only the APOE gene in the microglia was deleted. As previously reported, this leads to a significant reduction of Aβ plaques34, as well as a diminished plaque size (Supplementary Fig. 5b–e). We analyzed 22,387 microglia isolated from 6-month-old AppNL-G-F (n = 11) and AppNL-G-F ApoE−/− mice (n = 6, 2 independent induced pluripotent stem cells (iPSC) lines per group) (Supplementary Figs. 2 and 3). Clustering analysis revealed a significant reduction in the recruitment of microglia into DAM and HLA phenotypes, with no alterations in the CRM response (Fig. 5a,b and Extended Data Fig. 7). Trajectory analysis confirmed a strong reduction in the proportion of cells transitioning into the DAM–HLA transcriptional axis (Fig. 5c and Extended Data Fig. 7). These observations indicate that there is a non-cell-autonomous effect of ApoE secreted, for instance, from astroglia on microglia. This effect could be indirect via the modulation of Aβ aggregation or Aβ fibril structure or by modulating other cells or cellular processes.

a,d, Density plots displaying the average distribution of human microglia transplanted in AppNL-G-F (n = 11) and AppNL-G-F ApoE−/− (n = 6) mice (a), and human microglia transplanted in AppWT mice and challenged with scrambled peptide (Scr, n = 7) or Aβo (n = 13) (d). Density is normalized by sample size. b,e, Distribution and proportion of cells across all identified clusters for microglia transplanted in AppNL-G-F (n = 11) and AppNL-G-F ApoE−/− (n = 6) mice (b), and Scr/Aβo injected mice (e). Dots represent single mice. c,f, Phenotypic trajectory followed by human microglia transplanted in AppNL-G-F (n = 11 and 14,649 cells) and AppNL-G-F ApoE−/− (n = 6 and 7,738 cells) mice (c), and challenged with scrambled peptide (Scr, n = 7 and 10,967 cells) or Aβo (n = 13 and 18,691 cells) (f), obtained by an unbiased pseudotime ordering with Monocle 3. Proportion of cells from different mouse hosts (y axis) at different stages of the pseudotime trajectory (x axis), colored as shown in Fig. 1a. Dots represent single cells. g, Correlation analysis of the logFC in microglia either challenged with Aβο versus Src (y axis) or transplanted in AppNL-G-F ApoE−/− versus AppNL-G-F mice (x axis) (Pearson’s correlation, R = 0.05, differentially expressed genes were adjusted using Bonferroni correction and colored according to clusters in Fig. 1a). h, Correlation analysis of the logFC in microglia either challenged with Aβ soluble aggregates (Aβο) versus scrambled peptide (Src, y axis) or transplanted in AppNL-G-F mice (x axis) (Pearson’s correlation, R = 0.06, differentially expressed genes adjusted using Bonferroni correction). Box plots in b,c,e,f are limited by lower and upper quartiles and midline indicates median; whiskers show minimum to maximum values (b,e) or extend from the box to the smallest or largest value no further than 1.5× interquartile range (c,f). Unpaired t-test with Welch’s correction, two-tailed, alpha = 0.05, significance was set as P < 0.05. Scr, scrambled peptide.
To further assess whether the remaining CRM response is mediated by soluble Aβo, we injected recombinant soluble Aβo (5 μl at 10 μM, n = 13) or scrambled peptide (Scr, n = 7, 2 independent cell lines per group) in the ventricle of xenotransplanted ~3-month-old AppWT mice14. We isolated and sequenced 29,658 microglia 6 h after injection (Supplementary Fig. 2). Whereas microglia exposed to Scr peptides remain largely homeostatic, microglia challenged with soluble Aβo adopt a CRM transcriptional state (Fig. 5d,e and Extended Data Fig. 7). Contrary to Aβ plaques, and despite that these soluble Aβo partially shifted cells into a DAM state, they did not induce an HLA response. Trajectory analysis confirmed that soluble Aβo-treated cells undergo an almost complete phenotypic transition across CRM-1 and CRM-2 clusters (Fig. 5f). CRM-2 is at the extreme of this trajectory and the dominant cell state after acute injection of high levels of Aβo (Fig. 5f and Extended Data Fig. 7), consistent with our previous observations14. Several variables can affect the response to these aggregates, including concentration of Aβ, levels of aggregation or traces of bacterial endotoxin from the manufacturing process. We analyzed independent batches of soluble Aβ aggregates and could exclude that these sources of variation affected the induction of CRM. Thus, overall the microglial response consists of independent cell states that coexist in the AppN-L-GF model for AD and are elicited by Aβ plaques (DAM, HLA) and soluble Aβo species (CRM), respectively (Fig. 5g,h).
The response of engrafted microglia mimics the human brain
We wondered whether the two main modalities of human microglial response to Aβ pathology are preserved in AD human brain. We extracted microglial single-nuclei RNA sequencing data from the four most recent independent single-nuclei sequencing studies investigating the transcriptome of human microglia in the AD brain, providing 176,136 nuclei from Gerrits et al.20, 28,767 from Sayed et al.35, 3,978 from Zhou et al.18 and 194,000 from Sun et al.21, respectively. A fifth study from Olah et al.36 provided 16,242 single-cell transcriptomes from freshly isolated microglia from surgical resection. We reproduced their original clustering analysis (Supplementary Fig. 6 and Extended Data Fig. 8) and re-analyzed these data using the transcriptomic profiles from transplanted microglia (Fig. 6). Despite discrepancies in the numbers and proportions of clusters reported across these studies, we were able to identify populations of nuclei weakly (R = 0.2–0.4) to moderately (R = 0.4–0.6) or strongly (R > 0.6) correlating to all the microglial cell states, especially HM, DAM, HLA and CRM (Fig. 6 and Extended Data Fig. 8), as well as a clear IRM signature in the fourth datasets from single cells (Fig. 6). Of note, none of the postmortem datasets alone covered the full phenotypic diversity in response to Aβ as observed in the xenotransplanted microglia (Fig. 3d). For example, in Gerrits et al.20 DAM corresponds to AD1, and CRM to Inflammation, but the HLA signature is absent (Extended Data Fig. 3d and Fig. 6a,d). In Zhou et al.18, all signatures are present, but they converge in a limited number of clusters (Micro1, Micro4 and Micro3). In Sayed et al.35, although the main Aβ-induced signatures are present, DAM and HLA, and IRM and CRM show partial overlap (Fig. 6b,d). For Sun et al.21, we were able to correlate our HM (MG0 and MG1), DAM (MG3, MG4 and MG10), HLA (MG4) and CRM (MG6 and MG7) states. Certain transcriptomic states from primary samples that were correlated with tau pathology (for example, AD2)20 overlap with our HM signature. In Olah et al.36, we also observed an overlap between the transcriptional states of transplanted microglia and those from human biopsies36, consistent with our HM (MG2), DAM (MG1), HLA (MG8), CRM (MG3) and IRM (MG4) clusters, respectively (Extended Data Fig. 3d and Fig. 6d,e). Microglial states resembling HLA and CRM were shown before in brains from AD subjects by Lau et al.37. Additional cell states described in some of these postmortem studies are not captured in our xenotransplantation model, which might reflect the response to additional pathologies in the late phase of AD or the heterogeneity of pathology in old age (Fig. 6d and Extended Data Fig. 8). Overall, it appears that the cell states in transplanted microglia are present in the human AD brain and that the data generated here in the well-controlled xenograft system are useful to understand the response of human microglia to amyloid pathology in patients.

a–e, Human snRNA-seq datasets from Gerrits et al.20 (n = 176,136) (a), Sayed et al.35 (n = 28,767) (b), Zhou et al.18 (n = 3,978) (c), Olah et al.36 (n = 16,242) (d) and Sun et al.21 (n = 194,000) (e) re-analyzed to reproduce their original embeddings and annotated with our xenotransplanted microglial profiles as in Fig. 1b. f, Pairwise Pearson correlation between logFC of all differentially expressed genes (logFC cut-off set at 0.25, P < 0.05) of each microglial subtype and logFC of all differentially expressed genes (P < 0.05) of clusters from each human snRNA-seq study, with significance set at a P-adjusted value < 0.05 (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001). Only positive correlations are depicted here (r > 0). Additional correlations are shown in Extended Data Figs. 6 and 7. scRNA-seq, single cell RNA-sequencing.
AD risk genes modulate microglial cell states in vivo
Our model is very well suited to explore how AD genetic risk modulates human microglial responses. As proof-of-concept, we investigated some of the major genetic risk factors for AD, hypothesizing that they would modulate microglia according to their role in AD risk, that is, APOE2/2 should increase protective and/or decrease damaging responses, whereas APOE4/4 and TREM2 mutations should increase damaging or reduce protective responses1, to the extent that their pathological effect is restricted to microglia. We therefore tested first the effect of the clinical mutation TREM2R47H in the H9 background (Table 1). We profiled 3,282 H9 (n = 2) and 5,845 H9-TREM2R47H (n = 3) microglia transplanted in AppNL-G-F mice (Supplementary Figs. 2 and 3). The TREM2R47H mutation induced a significant reduction in the fraction of cells recruited into HLA, but also an unexpected reduction of the CRM-1-cluster (Fig. 7a,b and Extended Data Fig. 9a–d). Trajectory analysis confirmed that this clinical mutant remains largely locked in a homeostatic state (Fig. 7c and Extended Data Fig. 9c,d). We compared the gene expression alterations induced by complete loss-of-function of TREM2 and the clinical TREM2R47H mutant (Fig. 7d). Both genotypes show a strong correlation (R = 0.66) and a common downregulation of DAM and HLA genes, which is supported by morphological evidence in TREM2R47H knock-in mice showing a reduction in the number of plaque-associated microglia38,39. However, whereas TREM2−/− retains expression of CRM genes (including IL1B, CCL3L3 or CCL3L1), TREM2R47H shows only spared expression of IRM genes (such as IFIT1, IFIT3, IFI6 or IFITM3). Our data indicate that TREM2R47H results in an inability of microglia to engage in both HLA and CRM transcriptional programs.

a,e, Density plots displaying the average distribution of human H9-WT (n = 2) and H9-TREM2R47H (n = 3) (a), and APOE2/2 (n = 6), APOE3/3 (n = 6) and APO4/4 (n = 6) human microglia (e) transplanted in AppNL-G-F mice. b,f, Distribution and percentage of cells across all identified clusters for H9-WT (n = 2) and H9-TREM2R47H (n = 3) (b), and APOE2/2 (n = 6), APOE3/3 (n = 6) and APO4/4 (n = 6) (f). Dots represent single mice. c,g, Phenotypic trajectory followed by H9-WT (n = 2) and H9-TREM2R47H (n = 3) (c), and APOE2/2 (n = 6), APOE3/3 (n = 6) and APO4/4 (n = 6) (g) human microglia obtained by an unbiased pseudotime ordering with Monocle 3. Proportion of cells (y axis) over the binned pseudotime trajectory (x axis), colored by genotypes shown in a. Dots represent single mice (*P < 0.05). d,h, Correlation of the logFC in TREM2R47H versus H9-WT (y axis) and H9-TREM2−/− versus H9-WT (x axis) (d), and TREM2R47H versus H9-WT (y axis) and APOE4/4 versus APOE3/3 (x axis) (h) microglia transplanted in AppNL-G-F mice (Pearson’s correlation, R = 0.17, differentially expressed genes were adjusted using Bonferroni correction and colored as in Fig. 1b). i,j, Comparison of transcriptomic profiles of APOE4/4 (versus APOE3/3) versus APOE−/− (versus APOE3/3) (i), and APOE4/4 (versus APOE2/2) versus APOE−/− (versus APOE2/2) (j) microglia transplanted in AppNL-G-F mice (Pearson’s correlation, differentially expressed genes were adjusted using Bonferroni correction and colored according to clusters in Fig. 1b). Box plots in b,c,f,g are limited by lower and upper quartiles and midline indicates median; whiskers show minimum to maximum values (b,f) or extend from the box to the smallest or largest value no further than 1.5 × interquartile range (c,g). Unpaired t-test with Welch’s correction, two-tailed, alpha = 0.05, significance was set as P < 0.05 (b,c); or one-way ANOVA with Tukey’s multiple comparisons as post hoc test, alpha = 0.05, significance was set as P-adjusted value < 0.05 (f,g). In d,h,i, ‘activated’ indicates that differential expression was performed comparing reactive cell states, excluding homeostatic or transitioning clusters.
Upregulation of ApoE in microglia is one of the main transcriptional changes induced by Aβ plaques in mouse systems3,4,5. This suggests that the APOE allelic variants could be important regulators of human microglia. We used a series of isogenic APOE2/2, 3/3 and 4/4 iPSC lines (UKBIO11-A, European Bank for induced Pluripotent Stem Cells (EBiSC)). We also obtained a second series of the APOE alleles in an APOE knockout iPSC line (BIONi010-C, EBiSC) with APOE2/0, 3/0 and 4/0 genotypes (Supplementary Figs. 2 and 3). Both series have very similar APOE expression levels (Supplementary Fig. 6) and we therefore grouped them together for our analyses. We transplanted microglial precursors from all these lines into AppNL-G-F mice (n = 3 per cell line). Unlike TREM2R47H cells, clustering analysis did not reveal large differences between cells harboring the different APOE allelic variants. APOE4/4-expressing microglia show, however, a significant reduction in the proportion of cells acquiring the HLA phenotype (P < 0.05) and a tendency toward an increase in CRM response (P = 0.17; Fig. 7e,f and Extended Data Fig. 9e,f). The response of APOE4 cells to Aβ pathology strongly correlates with that of APOE−/− cells (Fig. 7i), suggesting that APOE4 results in a loss-of-function phenotype in microglia. This downregulation of HLA genes is not observed in APOE2 microglia (Fig. 7j). This is consistent with recent observations in mouse models, where microglial APOE4 has a negative impact in the acquisition of the MGnD phenotype40 and interferes with complement and lysosomal pathways41. Trajectory analysis confirmed this and revealed that APOE4 fails to transition toward the HLA state (Fig. 7g and Extended Data Fig. 9g,h), suggesting an inability to engage in a full response to Aβ plaques. Despite APOE4 being the major genetic risk factor for AD, our data suggest that its cell-autonomous role in microglia is rather limited and consistent with the effect we observed in APOE−/− microglia. As mentioned before, ApoE has a major effect on Aβ aggregation that might indirectly influence the function of microglia34. Nevertheless, the changes induced by TREM2R47H and APOE4 show a common impairment in activating the human-specific HLA (rather than DAM) transcriptional program and together suggest that the HLA response to Aβ plaques might be a beneficial aspect of the response of human microglia to AD pathology (Fig. 7h).
AD risk genes are expressed in different cell states
Finally, we extracted a list of 85 genome-wide association studies (GWAS) significant gene candidates (nearest gene to significant genetic loci, P < 5 × 10−8) from the most recent meta-analysis of AD complex genetics42. We found that almost 90% of these genes were detected in our dataset (Supplementary Table 4 and Extended Data Fig. 7a) while more than half were differentially expressed in the conditions we tested, confirming that a large part of the genetic risk for AD is harbored by microglia and that expression of these genes is modified in the context of Aβ pathology6 (Fig. 8a and Extended Data Fig. 10). We observed that different subsets of AD risk genes were enriched in all cell states in response to Aβ pathology. Whereas genes such as ABI3, INPP5D, SORL1, GRN, BIN1 and MAF were enriched in homeostatic microglia, both the response to Aβ plaques and Aβo soluble aggregates were associated with distinctive groups of genes, including TREM2, APOE, COX7C and HLA-DQA1 in DAM/HLA; and SPl1, SPPL2A, TNIP1, LILRB2, SEC61G, NCK2 and CTSH in the response to Aβο (Fig. 8a and Extended Data Fig. 10). We also observed altered expression of subsets of these AD risk genes after introducing clinically relevant mutation APOE4 or TREM2R47H. Consistent with their inability to transition toward responsive cell states, both TREM2−/− and TREM2R47H microglia showed higher expression of those risk genes present in homeostatic cells, such as SORL1, GRN, CSTB, BIN1 and MAF; and reduced expression of the DAM and HLA genes APOE and HLA-DQA1 (Fig. 8a). APOE2/2 and APOE4/4 microglia showed milder enrichment of risk genes when compared with APOE3/3, but in a similar direction and with decreased association to DAM and HLA genes (Fig. 8a and Extended Data Fig. 10). Our data show that the contribution of AD genetics to the biology of microglia is complex, likely having an impact on multiple cell states. It is interesting to see how known genetic risk factors affect the expression of other genetic risk factors in microglia, providing a strong basis for the concept of polygenic risk and providing a framework to understand its functional implications (Fig. 8b).

a, Analysis of genome-wide association studies (GWAS) genes enrichment in xenotransplanted microglia. The black bars represent the number of cells (in thousands) with detectable expression (≥1 read per cell) for each candidate gene from Bellenguez et al.42. The heatmap summarizes the deregulated expression (logFC, color scale) of these genes across cell states (each cluster compared with all others), as well as after exposure to Aβ plaque pathology, upon injection of soluble Aβ aggregates or altering the genetic background of the mice or the transplanted cells. The genes are ranked in rows based on hierarchical clustering. We identify three sets of genes that display a common profile across cell states (based on their enrichment in the specific microglial phenotypic transcriptional states HM, DAM and HLA, and CRM), Aβ pathology and genetic risk, and we group these profiles as: microglial homeostasis, plaque-induced genes and soluble aggregates-induced genes. The remaining genes did not show a clear enrichment in cell states or other conditions. All differential expressions were significant after adjusting P values using Bonferroni correction (FDR < 0.05). Only genes that are significantly changing in at least one of the tested conditions are reported, see Supplementary Table 4 and Extended Data Fig. 10 for further details. b, Illustration of the complex microglial AD genetics by cell-state profiles and driven by different Aβ pathologies as found in a and Extended Data Fig. 10.
Discussion
We provide a full transcriptomic characterization of the different cell states adopted by human microglia in response to Aβ pathology by using a xenotransplantation model where we engrafted stem cell-derived human microglia in the brain of the AppNL-G-F model of amyloid pathology14. Human microglia respond to AD’s multi-faceted Aβ pathology with a complex, multipronged response covering, among others, a late HLA response and a strong CRM response, which are not clearly recapitulated in mouse microglia. Even the human DAM response, which has also been observed in mouse microglia as a reaction to Aβ plaques, is only 20–25% similar to the mouse DAM response. This should be carefully considered when using preclinical models to investigate the role of blocking or boosting this putative protective cell state for potential therapeutic development. Interestingly, TREM2 and APOE have different, complementary effects in mediating human microglial activation. Similar to mouse4,5,43, human TREM2 appears a master inductor of DAM and HLA responses against Aβ plaques. APOE plays a more subtle role, affecting mostly the phenotypic switch from DAM to HLA.
We demonstrate here that genetic risk of AD significantly alters the response of human microglia to Aβ pathology. APOE4 or the TREM2R47H mutation hampers the HLA profile, suggesting a protective role of this response against AD. On the other hand, it is unclear how the CRM response against soluble Aβo species contributes to AD pathology. CRM is characterized by the upregulation of classical inflammatory cytokines and chemokines such as IL1B, CCL2, CCL3, CCL4 and components of the NFKB pathway. It is also enriched with several AD risk genes. Soluble Aβ species appear early during the disease process long before Aβ plaques and have been linked to early neuronal dysfunction44. Our data suggest that microglia might engage with this Aβo already in the very early disease stages of AD. The question remains as to whether the upregulated cytokines and chemokines characteristic for this response affect neurons or other brain cells, inducing the cellular responses in AD that ultimately result in neurodegeneration45.
One strength of the current dataset appears to be that it helps to better classify the multipronged response of microglia as recorded in single nuclei isolated from AD postmortem samples. The transplantation system therefore provides a complementary tool to analyze the human-specific microglial contribution to AD. In addition, its great flexibility makes it possible to analyze the effects of genetic or pharmacological manipulation of human microglia in the context of disease in an unprecedented way. A major shortcoming of the model is the lack of adaptive immunity, which is needed to allow the xenotransplant, which at the same time may preclude the evaluation of the contribution of the adaptive immune system to the microglial responses in AD.
In conclusion, our findings unravel the complex responses of human microglia to early and late Aβ pathology in AD. The results indicate that therapeutic targeting of microglia needs to be implemented with care as it might differentially affect their cell states and modify the disease course in unpredictable ways. The xenograft model provides a good way to investigate this question and to further explore the genetics underlying the multipronged responses of human microglia to amyloid pathology in AD.
Methods
Mice
The App single knock-in mouse model (AppNL-G-F; Takaomi Saido)47 does not overexpress APP as in classical APP mouse models, but contains the humanized Aβ sequence, as well as Swedish (NL), Arctic (G) and Iberian (F) mutations. AppNL-G-F mice accumulate Aβ plaques and suffer from learning, memory and attention impairments from 6 months onwards47,48. The humanized Apphu/hu mice (named AppWT in the main text) were recently generated in our laboratory to serve as controls27. Both strains were crossed with homozygous Rag2tm1.1Flv Csf1tm1(CSF1)Flv Il2rgtm1.1Flv Apptm3.1Tcs mice (Jackson Laboratory, strain 017708) to generate the Rag2−/− Il2rγ−/− hCSF1KI AppNL-G-F and the Rag2−/− Il2rγ−/− hCSF1KI Apphu/hu used in this study. In total, we transplanted >150,000 cells across 11 different cell lines, and three mouse background genotypes. For all experiments, we used Total-Seq A hashing antibodies (BioLegend) so as to be able to demultiplex individual mouse replicates (Supplementary Fig. 2). Mice had access to food and water ad libitum and were housed with a 14/10-h light/dark cycle at 21 °C and 32% humidity, in groups of 2–5 animals. All experiments were conducted according to protocols approved by the local Ethical Committee of Laboratory Animals of KU Leuven (Ethische Commissie Dierproeven project no. P177/2017) following country and European Union guidelines.
Generation of Rag2 −/− Il2rγ −/− hCSF1 KI App NL-G-F ApoE −/− mice
ApoE knockout mice were generated in zygotes from homozygous Rag2tm1.1Flv Csf1tm1(CSF1)Flv Il2rgtm1.1Flv Apptm3.1Tcs mice using CRISPR–Cas9 technology by targeting exon 4 of the mouse ApoE gene. The RNA guide 5′-CCTCGTTGCGGTACTGCCCGAGT-3′ was selected using the CRISPOR web tool. Ribonucleoproteins containing 0.3 μM purified Cas9HiFi protein (Integrated DNA Technologies), 0.3 μM CRISPR RNA (crRNA) and 0.3 μM trans-activating crRNA (Integrated DNA Technologies) were injected into the pronucleus of 72 embryos by microinjection in the Mouse Expertise Unit of KU Leuven. Two candidate pups were identified by PCR analysis with several primer combinations. One founder was selected for breeding and an allele with a chromosomal deletion of 335 base pairs (bp) (corresponding to 148 bp of intronic sequence and the first 187 bp of exon 4 sequence) (Extended Data Fig. 4) was selected to establish the colony. The founder mouse was backcrossed over two generations before a homozygous colony was established, which was designated AppNL-G-F ApoE−/−. The strain was maintained on the original C57Bl6J:BalbC background. Standard genotyping for the ApoE allele was performed by PCR with primers 5′-GCTCCCAAGTCACACAAGAA-3′ and 5′-CTCACGGATGGCACTCAC-3′, resulting in a 755-bp amplicon for the WT allele and a 420-bp amplicon for the ApoE knockout allele.
Differentiation of microglial progenitors
UKBIO11-A, BIONi010-C and H9-WA09 and their isogenic modifications (Table 1) were differentiated into microglial precursors and transplanted following our recently published protocol, MIGRATE26. In brief, stem cells were plated and maintained in human Matrigel-coated six-well plates and in E8 flex media until reaching ~70–80% confluence. Once confluent, stem cell colonies were dissociated into single cells and plated into U-bottom 96-well plates at a density of ~10,000 per well in mTeSR1 medium with BMP4 (50 ng ml−1), VEGF (50 ng ml−1) and SCF (20 ng ml−1) for 4 d. On day 4, embryoid bodies were transferred into six-well plates (~20 embryoid bodies per well) in X-VIVO (+supplements) medium supplemented with SCF (50 ng ml−1), M-CSF (50 ng ml−1), IL-3 (50 ng ml−1), FLT3 (50 ng ml−1) and TPO (5 ng ml−1) for 7 d with a full change of medium on day 8. On day 11, differentiation medium was replaced with X-VIVO (+supplements) with FLT3 (50 ng ml−1), M-CSF (50 ng ml−1) and GM-CSF (25 ng ml−1). On day 18, human microglial precursors were collected and engrafted into P4 mouse brains (0.5 million cells per pup) as previously described. Before transplantation, mouse microglia were depleted by inhibiting CSF1 receptor (CSF1R) with BLZ945 (dose of 200 mg kg−1) at P2 and P3 as previously described14,26. To confirm the biological activity of known essential mouse cytokines on human microglia, we cultured microglial progenitors in TIC medium49 for 9 d, supplemented with either hCSF1, hIL-34, hTGFb and hCX3XR1, or hCFS1, mIL-34, mTGFb and mCX3CR1, and measured the expression of several microglial makers by quantitative PCR (qPCR). We did not find any differences in the levels of P2RY12, CX3CR1, C1Q, HEXB, TGFbR1 and TREM2 (Supplementary Fig. 1a).
Genetic modification of stem cell lines
Generation of TREM2−/− and TREM2+/R47H from H9-WT (WA09) human embryonic stem cells was done as described by Claes et al.46. Briefly, the TREM2+/R47H CRISPR–Cas9 nickases and the two guide RNAs (gRNAs) (gRNA A and B) that target exon 2 of TREM2 nearby the location of R47H (G>A) and a genomic TTAA were purchased from Addgene. A donor plasmid was made, comprising homology arm 1 (HA1) of TREM2 (with the R47H mutation), a selection cassette (CAGG promoter, HYG/TK, green fluorescent protein) and HA2 of TREM2 exon 2. To create TREM2−/− human pluripotent stem cells (hPSCs), a CRISPR–Cas9, gRNA B and the same donor plasmid were used. To create TREM2+/R47H hPSCs, 2 × 106 single cells of the heterozygously targeted clone were nucleofected with 4 μg of piggyBac transposase plasmid and negative selection with fialuridine, also known as 1-(2-deoxy-2-fluoro-1-d-arabinofuranosyl)-5-iodouracil (FIAU) (1:8,000–1:2,500; 0.5 mM in water), was applied to select for cells wherein the selection cassette was removed. Of note, the H9-WT line from which the TREM2+/R47H and TREM2−/− lines were created carries an APOE ε3/ε4 genotype.
To get the gRNA into the cells, nucleofection was performed. Briefly, 2 × 106 single-cell H9s were preincubated with Revitacell (Life Technologies) and nucleofected using the Amaxa Nucleofector II on setting F16 with 2.5 μg of CRISPR–Cas9, 2.5 μg of gRNA (A and) B and 5 μg of donor template to create TREM2−/− hPSCs. Selection was initiated after 2–3 d with 25–150 μg ml−1 Hygromycin B (Sigma-Aldrich) and maintained for 10–15 d. Recombinant colonies were manually picked and expanded for further characterization.
gRNA A | F: CACCGACCCAGGGTATCGTCTGTGA | R: AAACTCACAGACGATACCCTGGGTC |
gRNA B | F: CACCGCACTCTCACCATTACGCTG | R: AAACCAGCGTAATGGTGAGAGTGC |
The H9 (WA09) embryonic stem cell line was also modified to express the CAS9 in a doxycycline-dependent manner (iCas9), using TALENS at the AAVS1 locus to facilitate the generation of targeted gene deletion. H9-iCas9 was used only in libraries 14 and 16 and doxycycline was never administered.
APOE expression using qPCR
The levels of APOE expression in the grafted stem cell lines were checked by collecting cell pellets. Using the RNeasy Micro Kit, RNA was extracted according to manufacturer’s instructions and the RNA was reverse transcribed with the High-Capacity cDNA Reverse Transcription Kit. A qPCR was performed with SensiFast SYBR reagent and custom-made primers for GAPDH (FW: tcaagaaggtggtgaagcagg; RV: accaggaaatgagcttgacaaa) and APOE (we average the level of expression of multiple primers spanning the whole gene, see table below).
hAPOE_1_F | F: TAGAAAGAGCTGGGACCCT | R: CACAGAACCTTCATCTTCCT |
hAPOE_2_F | F: GTTGCTGGTCACATTCCTG | R: GCAGGTAATCCCAAAAGCGA |
hAPOE_3_F | F: CTGGGTCGCTTTTGGGATTA | R: GTCAGTTGTTCCTCCAGTTC |
hAPOE_4_F | F: AATCACTGAACGCCGAAG | R: TTATTAAACTAGGGTCCACC |
hAPOE_qPCR_F | F: GTTGCTGGTCACATTCCTGG | R: GCAGGTAATCCCAAAAGCGAC |
Soluble Aβ preparation and intracerebroventricular injections
Soluble Aβ aggregates (10 μM) or scrambled peptides (10 μM) were prepared as previously14,50. Briefly, recombinant Aβ (1–42) or scrambled peptides were thawed during 30 min at room temperature and dissolved in hexafluoroisopropanol at 2 mg ml−1. Hexafluoroisopropanol was fully evaporated with a gentle stream of N2 gas and resulting peptides were dissolved in dimethylsulfoxide at 2 mg ml−1. Dimethylsulfoxide medium was removed using HiTrap Desalting column 5kD and peptides were eluted in Tris-EDTA buffer. Of note, Tris-EDTA buffer was composed of 50 mM Tris buffer and 1 mM EDTA at pH 7.5. Tris-EDTA-eluted Aβ or scrambled peptides were quantified using Bradford assay before aggregation. Peptides were left to aggregate for 2 h at room temperature. After 2 h, Aβ (1–42) or scrambled aggregates were diluted to a final concentration of 10 μM in Tris-EDTA buffer, snap frozen and stored at −80 °C. Following a similar approach as previously described14, at 12 weeks of age, AppWT mice engrafted with the full isogenic series of UKBIO11-A or BIONi010-C were anesthetized with isoflurane and injected intracerebroventricularly with 5 μl of soluble aggregates of Aβ (10 μM) or scrambled peptides (10 μM). Stereotactic coordinates from Bregma: anteroposterior: −0.22 mm; mediolateral: −1 mm; dorsoventral: −2.74 mm. After surgery, mice were placed on a thermal pad until recovery. At 6 h after injection, AppWT mice were euthanized, and human microglia were isolated using FACS for transcriptomics analysis.
Human microglia isolation from mouse brain for single-cell transcriptomics
At 6–7 months of age AppNL-G-F, AppNL-G-F ApoE−/− and AppWT mice xenotransplanted with H9, UKBIO11-A, BIONi010-C (Table 1) and their isogenic modifications were killed with an overdose of sodium pentobarbital and immediately perfused with ice-cold 1 × DPBS (Gibco, Cat. no. 14190-144) supplemented with 5 U of heparin (LEO). After perfusion, one hemisphere of each mouse brain without cerebellum and olfactory bulbs was placed in FACS buffer (1 × DPBS, 2% FCS and 2 mM EDTA) + 5 μM actinomycin D (ActD; Sigma, Cat. no. A1410-5MG) for transcriptomics. Brains were mechanically and enzymatically dissociated using Miltenyi Neural Tissue Dissociation Kit P (Miltenyi, Cat. no. 130-092-628) supplemented with 5 μM ActD. Next, samples were passed through a 70-μm strainer (BD2 Falcon), washed in 10 ml of ice-cold FACS buffer + 5 μM ActD and spun at 300g for 15 min at 4 °C. Note that 5 μM ActD was kept during collection and enzymatic dissociation of the tissue to prevent artificial activation of human microglia during the procedure as previously reported12. ActD was removed from the myelin removal step to prevent toxicity derived from long-term exposure. Following dissociation, myelin was removed by resuspending pelleted cells in 30% isotonic Percoll (GE Healthcare, Cat. no. 17-5445-02) and centrifuging at 300g for 15 min at 4 °C. Accumulating layers of myelin and cellular debris were discarded and Fc receptors were blocked in FcR blocking solution (1:10, Miltenyi, Cat. no. 130-092-575) in cold FACS buffer for 10 min at 4 °C. Next, cells were washed in 5 ml of FACS buffer and pelleted cells were incubated with the following antibodies: PE-Pan-CD11b (1:50, Miltenyi, Cat. no. 130-113-806), BV421-mCD45 (1:500, BD Biosciences, Cat. no. 563890), APC-hCD45 (1:50, BD Biosciences, Cat. no. 555485), Total-Seq A cell hashing antibodies (1:500, BioLegend) and viability dye (1:2,000, eFluor 780, Thermo Fisher Scientific, Cat. no. 65-0865-14), in cold FACS buffer during 30 min at 4 °C. After incubation, cells were washed, and the pellet was resuspended in 500 μl of FACS buffer and passed through a 35-μm strainer before sorting. For sorting, the cell suspension was loaded into the input chamber of a MACSQuant Tyto Cartridge and human cells were sorted based on CD11b and hCD45 expression at 4 °C (Supplementary Fig. 1). FACS data were analyzed using FCS Express 7 software.
Histology
When killing and collecting brains of mice for single-cell sequencing, one hemisphere of AppNL-G-F, AppNL-G-F ApoE−/− and AppWT mice xenotransplanted with H9, UKBIO11-A and BIONi010-C was also preserved and postfixed in 4% PFA overnight at 4 °C. After 24 h, PFA was removed and they were washed and kept in 1 × DPBS at 4 °C until further processing. For sectioning, olfactory bulbs and cerebellum were discarded and brains were cut coronally (40-µM thickness) with a vibrating microtome (Leica). Each sample was collected under free-floating conditions in a series of six sections and stored in cryoprotectant solution (40% PBS, 30% ethylene glycol, 30% glycerol) at −20 °C. For staining, sections are washed in 1 × DPBS and permeabilized for 15 min at room temperature in PBS with 0.2% Triton. After permeabilization, sections were stained with X-34 staining solution (10 µM X-34 (Sigma-Aldrich), 20 mM NaOH (Sigma-Aldrich) and 40% ethanol) for 20 min at room temperature. Sections were washed several times with 40% ethanol for 2 min and with PBS + 0.2% Triton for 5 min. For the staining of microglia with anti-hP2RY12 (HPA014518, Sigma-Aldrich, 1:2,000), CD9 (312102, BioLegend, 1:100), FTH1 (PA5-1905, Invitrogen, 1:500) and HLA antibodies (ab7856, Abcam, 1:200), sections were blocked with 5% normal donkey serum in PBS + 0.2% Triton for 1 h at room temperature. For the costaining of CD9/FTH1 with P2RY12, primary antibody incubation was done overnight at 4 °C. For the HLA staining, signal was enhanced using a Tyramide SuperBoost kit (B40915, Thermo fisher) according to the manufacturer’s instructions. Briefly, after overnight incubation with HLA primary antibody, sections were incubated with a poly-HRP-conjugated secondary antibody. Tyramid solution was then added to the slices for 5 min and they were washed in PBS + 0.2% Triton after the reaction was stopped. The HLA sections were later costained with P2RY12 as previously described. Secondary antibodies were incubated for 1 h at room temperature. Finally, sections were mounted with Mowiol mounting medium (Sigma-Aldrich) or DAKO mounting medium (Agilent). Images at ×4 and ×20 magnification were taken on a Nikon A1R Eclipse confocal microscope. To measure the shift in microglial cell states at the site of Aβ plaques, we used a modified Sholl analysis where the fluorescent intensity of microglial markers P2RY12, CD9 and HLA was measured through concentric rings (annuli) of increasing diameter surrounding the X-34 plaque center. The analysis was performed using ImageJ software after determining a threshold for background correction. Intensities of each channel were scaled for comparison using z-score normalization. Intensity over distance (µm) was plotted using Loess nonparametric regression in R with estimated standard error for each predicted value. For comparison of intensities near and distant from the plaque center, the means of the inner and outer three annuli were independently calculated. Bar plots were generated in Prism v.10.
Single-cell library preparation and sequencing
For single-cell RNA sequencing, 15,000–20,000 human microglia (CD11b+, hCD45+) from each mouse were sorted on the MACSQuant Tyto (Supplementary Fig. 1) and diluted to a final concentration of 1,000 cells per µl. Since all the samples were individually hashed using Total-Seq A cell hashing antibodies, 2,000 human microglia per animal were pooled and loaded onto the Chromium Next GEM Chip G (PN no. 2000177). The DNA library preparations were generated following the manufacturer’s instructions (CG000204 Chromium Next GEM Single Cell 3′ Reagent Kits v3.1). In parallel, the hashtag oligo libraries were prepared according to the manufacturer’s instructions (BioLegend, Total-Seq A Antibodies and Cell Hashing with 10x Single Cell 3′ Reagent Kit v3 3.1 Protocol) using 16 cycles for the index PCR. A total of 20 libraries containing 95 biological replicates were sequenced, targeting a 90% messenger RNA and 10% hashtag oligo library (50,000 reads per cell), on a HiSeq4000 or NovaSeq6000 (Illumina) platform with the recommended read lengths by the 10X Genomics workflow.
Statistics and reproducibility
Statistical analysis of the distribution of different experimental groups across clusters was performed using each mouse as a single replicate. Normality and equal variance were tested, and the data were normalized if needed. We used both t-test and one-way analysis of variance (ANOVA) when comparing two or more than two groups, respectively. Statistical significance was set at P < 0.05, and multiple comparisons Bonferroni was applied when necessary. No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those reported in previous publications3,14. All the mice were randomized across the different experimental groups. Data collection and analysis were performed by blind researchers. Immunohistochemical data were repeated at least five times with similar results.
Analysis of single-cell RNA sequencing datasets
Alignment and software
The raw BCL files were demultiplexed and aligned by Cellranger (v.3.1.0) against the human genome database (hg19, Ensembl 87). Raw count matrices were imported in R (v.4.1.3) for data analysis. Datasets were analyzed using the Seurat R package pipeline (v.4.0.1). For specific statistical tests and visualizations, we also used GraphPad Prism v.9.0, Python, R and Bioconductor.
Quality control of cells and samples
For each library included in this study, we excluded low-quality cells (poorly sequenced, damaged or dead cells) by filtering out cells with <1,000 reads or <100 genes detected. We also excluded cells with >15% of reads aligning to mitochondrial genes. Doublets were first excluded by removing cells with a number of reads or genes more than 3 s.d. from the library mean (Supplementary Fig. 2a). Doublets removal was further refined with cell hashing information by using Seurat’s function MULTIseqDemux() to assign cells to singlets, doublets or negatives (Supplementary Fig. 2b). Only singlets were retained, as negative cells cannot be demultiplexed and assigned with certainty to the sample of origin. For one library out of 20 sequenced for this study (library 9), the counts related to one hash (sample MG452) were high across all samples in the library and MULTIseqDemux() failed to demultiplex many cells. We used the function HTODemux() instead to demultiplex library 9 which performed better. Sample MG452 was entirely removed in further quality control steps (see below). Genes detected in fewer than three cells were excluded from the count matrices. At this step, when quality control of single cells was completed, the dataset consisted of 154,624 cells across 101 independent mice and 20 sequencing libraries (Supplementary Fig. 2d,c and Tables 5–7). For detailed sequencing statistics per library see Supplementary Tables 1–8.
Normalization and integration
After quality control, each library was individually normalized and scaled using SCTransform(). For all libraries, we selected the 3,000 most variable features for downstream integration. We determined a list of common integration anchors across libraries with FindIntegrationAnchors() that we used as an input for integration. To integrate all the libraries, we used the IntegrateData() function of the Seurat package to correct for any potential library batch effect. Integrated matrix was used for downstream analysis (Supplementary Fig. 2c,d).
In the integrated dataset, we performed principal components analysis and found that the highest variability in the dataset was explained by the separation of CAMs from the microglial cell states, while integrated sequencing libraries did not show any batch effects in the dataset (Supplementary Fig. 2c,d). We selected 27 dimensions for dimensionality reduction by Uniform Manifold Approximation and Projection (UMAP), which we performed with the RunUMAP() function. To produce the initial UMAP as in Supplementary Fig. 3a we used the following parameters in RunUMAP(): dims = 1:27, n.neighbors = 30L, n.epochs = 200, min.dist = 0.01. To identify clusters, we first used the function FindNeighbors() (parameters for Supplementary Fig. 3a: dims = 1:20, k.param = 100, nn.method = “annoy”, annoy.metric = “cosine”) and then performed unbiased clustering by using FindClusters() (parameters for Supplementary Fig. 3a: resolution = 0.6, n.iter = 1000, n.start = 10, algorithm = 1, group.singletons = T). This led to the identification of 14 clusters, ten of which represented unique microglial/myeloid cell-state identities, three of which were merged into one homeostatic cluster for their overlapping transcriptomic signature and one of which resulted in a small low-quality cell cluster. The specific parameters used for UMAP and clustering were defined after assessment of a wide range of possible parameters, which were evaluated in light of cell-state annotations and differential expression results. We start from underclustering and we progressively increase the resolution by identifying further heterogeneity in the data, but we prevent overclustering by assessing that high resolutions lead to the definition of extra clusters that do not significantly differ in gene expression from the existing ones. Cell states were annotated by means of differential expression (FindAllMarkers() function for overall differential expression, FindMarkers() for side by side comparison) and using the AddModuleScore() function with a large number of published datasets, gene ontology categories, pathways and signatures as input3,4,7,9,14,20 (Supplementary Fig. 3b). Out of 14 clusters, nine clustered together and showed high expression of human microglial genes (P2RY12, CX3CR1, P2RY13 and so on). Three of these clusters were merged into an HM cell state for their common signature, while unique cell states were assigned to the other microglial clusters (Supplementary Fig. 3b,c, see main text and Fig. 1b–d for details). The remaining five clusters were enriched in nonmicroglial markers. Out of those, one small population clustered away from the main microglial clusters and expressed high levels of CAM markers (9,645 cells, marked by CD163, MRC1, RNASE1; Supplementary Fig. 3c,d) as previously described by Mancuso et al.14. In total, 3,396 cells clustered apart and expressed proliferation markers (high in TOP2A, MKI67, STMN1; Supplementary Fig. 3c,d). Although the dataset was previously quality controlled, a population of Low Quality/Doublets (3,517 cells) was still present. A small cluster of secretory cells was defined by sharing the ‘secretory’ signature previously identified also in Hasselman et al.7 (1,156 cells, high in AGR2, MNDA; Supplementary Fig. 3c,d). Finally, we defined a small cluster of other myeloid cells characterized by low expression of microglial homeostatic genes and expression of macrophage/monocyte markers (CD14, NEAT1, MAFB) and pro-inflammatory markers (IL1B, CCL2, CCL3) (3,078 cells; Supplementary Fig. 3c). The top differentially expressed genes from each defined cell state confirmed the unique signatures and no overclustering (top 20 marker genes per cluster are visualized in Supplementary Fig. 3c).
Microglia subsetting, re-clustering and annotation
For the final analysis of high-quality isolated human microglia, we subsetted out other myeloid and low-quality clusters (CAM, Other Myeloid, Secretory, Proliferating and Doublets/Low Quality). After trimming, 127,755 human microglia were retained for downstream analysis. Using the previously integrated dataset, we performed principal components analysis and selected 30 dimensions for dimensionality reduction by UMAP, as described above. No library-dependent batch effect was observed (Supplementary Fig. 2d). To produce the final UMAP as in Fig. 1b, we used the following parameters in RunUMAP(): dims = 1:30, n.neighbors = 30L, n.epochs = 500, min.dist = 0.05. To identify clusters, we first used the function FindNeighbors() (parameters for Fig. 1b: dims = 1:20, k.param = 100, nn.method = “annoy”, annoy.metric = “cosine”) and then performed unbiased clustering by using FindClusters() (parameters for Fig. 1b: resolution = 0.7, n.iter = 1000, n.start = 10, algorithm = 1, group.singletons = T). This led to the identification of 11 clusters, four of which were merged into an HM cell state for their overlapping transcriptomic signature (Fig. 1b,c and Supplementary Fig. 3c), and the other seven represented cell states defined by unique or transitory profiles. With the higher resolution provided by the microglia subclustering, we could identify two distinct CRM populations (CRM-1 and CRM-2) that were initially grouped together (Supplementary Fig. 3a,c) but actually represent two consecutive stages on the same phenotypic trajectory, and which can be differentially modified by Aβ pathologies and genetic backgrounds (Figs. 1b–d, 3a,b, 5d–f and 7). The specific parameters used for UMAP and clustering were defined after assessment of a wide range of possible parameters, which were evaluated in light of cell-state annotations, differential expression results and sample distributions. Cell-state annotation was performed as described for the full dataset, by means of iterative clustering, differential expression and signature scores (Fig. 1c and Supplementary Fig. 4b). We finally defined eight microglial cell states that included HM, CRM-1 and CRM-2, IRM, DAM, HLA, RM and tCRM (Fig. 1b–d). The expression profiles of the top differentially expressed genes from each defined cell state (top ten markers on heatmap in Fig. 1d, top three markers on UMAP in Supplementary Fig. 4c and all markers of differential expression statistics in Supplementary Table 8) and the signature scores calculated with AddModuleScore() (Supplementary Fig. 4a) confirmed the unique transcriptomic profiles of these clusters and that no overclustering was performed. We excluded from this dataset six mice that showed signs of infection, extremely low cell numbers and/or mice with the vast majority of cells mapping to one unique cell state. The final high-quality microglia dataset consisted of 127,755 cells from 95 independent mice and 20 sequencing libraries (Supplementary Fig. 2d,e). To extend our pseudotime analysis (Fig. 3 and Extended Data Figs. 4 and 5), we added three sequencing libraries with 10,822 single microglial cells across 11 independent mice. The total final dataset therefore includes 138,577 single cells from 106 mice and 23 sequencing libraries. For detailed sequencing statistics per library see Supplementary Table 5. For the number of sequenced cells per replicate/condition and all metadata of this study see Supplementary Tables 6 and 7.
Differential expression
Differentially expressed genes were found by applying the FindAllMarkers() function for overall differential expression between each cluster and the rest of the dataset and FindMarkers() for side by side comparisons of two groups, both from the Seurat R package. All the reported comparisons in the manuscript were performed with the following parameters: assay = “SCT”, test.use = “wilcox”, min.pct = 0.01, logfc.threshold = 0.1. We used the Wilcoxon rank-sum test to calculate P values. We performed differential expression on the ‘SCT’ assay calculated from SCTransform(), since Pearson residuals resulting from regularized negative binomial regression effectively mitigate depth-dependent differences in differential expression, as described by Hafemeister and Satija51. We tested only genes that were detected in a minimum fraction of 1% in either of the two populations. We limited testing to genes that showed, on average, at least 0.1-fold difference (log-scale) between the two groups of cells. Only genes with their adjusted P < 0.05 (post hoc, Bonferroni correction) were considered as significant. The complete list of differentially expressed genes from all comparisons is displayed in Supplementary Tables 1, 3 and 8.
Pseudotime
Pseudotime analysis was performed in the final human microglia dataset to infer the phenotypic transitions happening between the different microglial cell states. Unsupervised single-cell trajectory analysis was performed with Monocle 3, an algorithm that allows us to learn the sequence of gene expression changes each cell must go through as part of a dynamic biological process. We used SeuratWrappers to convert our microglial Seurat object into a Monocle object with as.cell_data_set(). We kept the UMAP embeddings previously calculated with RunUMAP() to estimate the phenotypic transitions between our annotated cell states. We ran cluster_cells() and learn_graph() (parameters used: close_loop = T, learn_graph_control = list(36an.k = 100, prune_graph = TRUE, orthogonal_proj_tip = F, minimal_branch_len = 50)) to learn the trajectory. To infer how resting microglia transition into reactive cell states, we set the roots of the trajectory with order_cells() by selecting the ten most homeostatic cells in our dataset (based on our previously defined HM signature score), to avoid limiting the selection of the root to a manually picked single cell and to account for heterogeneity of the HM cluster. Similar trials that set the origin of the trajectory to different HM cells led to comparable results, always identifying the main axes of phenotypic transitions described in Fig. 3a. To generate the trajectory displayed in Extended Data Figs. 4 and 5 including the 3-month data, we used the following parameters for learn_graph(): close_loop = F, learn_graph_control = list(rann.k = 100, prune_graph = TRUE, proj_tip = F, branch_len = 20), and we set the roots of the trajectory with order_cells() by selecting the five most homeostatic cells in the dataset.
Gene name conversion from human to mouse orthologs
Mouse to human orthologs tables were downloaded from Ensembl/Biomart (release 106). For the analysis reported in Extended Data Fig. 3c,d and Supplementary Table 2, mouse genes were converted to human genes with the most conservative criteria: if multiple orthologues of a mouse gene existed in human, the one with highest log2FC (fold change) in human DAM was selected. If multiple mouse genes converted to the same human gene, the one with highest log2FC in mouse DAM was selected. The overlap shown is therefore the highest that it is possible to achieve and can only be an overestimation. Note that even with the most inclusive criteria, the overlap between human and mouse DAM was very limited, and any other stricter approach (such as considering only bidirectional one-to-one orthologs) would find only lower correlations while losing information from the mouse genes.
Data exclusion
We excluded six mice from this dataset that showed signs of infection or extremely low cell numbers, and/or mice with most cells mapping to one unique cell state.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data generated in this study are available at the Gene Expression Omnibus (GEO) database with accession number GSE216999 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE216999 (ref. 52). An online platform for user-friendly access and visualization of the data shown in this study is available as a beta version both in the De Strooper lab website (data.bdslab.org/Mancuso2022) and in the Mancuso lab website (mancusolab.bioinf.be/ShinyRenzoAlfaBeta/). Other datasets included in the manuscript can be found at GEO (Mancuso et al., GSE137444; Sala Frigerio et al., GSE127893; Hasselman et al., GSE133433; Gerrits et al., GSE148822; Sayed et al., GSE183068; Zhou et al., GSE140511; Keren-Shaul et al., GSE98969; Friedman et al., GSE89482). The raw data files of Olah et al. are available through Synapse (https://www.synapse.org/#!Synapse:syn21438358). The list of GWAS risk genes for Fig. 8a was obtained according to the criteria of nearest protein coding genes to SNPs as identified in ref. 42 (Supplementary Table 4). The list of GWAS risk genes for Extended Data Fig. 10b,c was obtained as the union of candidate genes from Bellenguez et al. and a candidate gene list selected in our previous publication14 (see Supplementary Table 4 for complete list).
Code availability
The code used for the transcriptomic and imaging data analysis is available upon request.
References
Sierksma, A., Escott-Price, V. & De Strooper, B. Translating genetic risk of Alzheimer’s disease into mechanistic insight and drug targets. Science 370, 61–66 (2020).
Song, W. M. & Colonna, M. The identity and function of microglia in neurodegeneration. Nat. Immunol. 19, 1048–1058 (2018).
Sala Frigerio, C. et al. The major risk factors for Alzheimer’s disease: age, sex, and genes modulate the microglia response to Aβ plaques. Cell Rep. 27, 1293–1306.e6 (2019).
Keren-Shaul, H. et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290.e17 (2017).
Krasemann, S. et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, 566–581.e9 (2017).
Sierksma, A. et al. Novel Alzheimer risk genes determine the microglia response to amyloid‐β but not to TAU pathology. EMBO Mol. Med. https://doi.org/10.15252/emmm.201910606 (2019).
Hasselmann, J. et al. Development of a chimeric model to study and manipulate human microglia in vivo. Neuron https://doi.org/10.1016/j.neuron.2019.07.002 (2019).
Mancuso, R. et al. CSF1R inhibitor JNJ-40346527 attenuates microglial proliferation and neurodegeneration in P301S mice. Brain 142, 3243–3264 (2019).
Friedman, B. A. et al. Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of Alzheimer’s disease not evident in mouse models. Cell Rep. 22, 832–847 (2018).
Coomaraswamy, J. et al. Modeling familial Danish dementia in mice supports the concept of the amyloid hypothesis of Alzheimer’s disease. Proc. Natl Acad. Sci. USA 107, 7969–7974 (2010).
Jaitin, D. A. et al. Lipid-associated macrophages control metabolic homeostasis in a Trem2-dependent manner. Cell 178, 686–698.e14 (2019).
Marsh, S. E. et al. Dissection of artifactual and confounding glial signatures by single-cell sequencing of mouse and human brain. Nat. Neurosci. 25, 306–316 (2022).
Thrupp, N. et al. Single-nucleus RNA-seq is not suitable for detection of microglial activation genes in humans. Cell Rep. 32, 108189 (2020).
Mancuso, R. et al. Stem-cell-derived human microglia transplanted in mouse brain to study human disease. Nat. Neurosci. 22, 2111–2116 (2019).
Geirsdottir, L. et al. Cross-species single-cell analysis reveals divergence of the primate microglia program. Cell 179, 1609–1622.e16 (2019).
Mathys, H. et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337 (2019).
Grubman, A. et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci. 22, 2087–2097 (2019).
Zhou, Y. et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med. 26, 131–142 (2020).
Del-Aguila, J. L. et al. A single-nuclei RNA sequencing study of Mendelian and sporadic AD in the human brain. Alzheimers Res. Ther. https://doi.org/10.1186/s13195-019-0524-x (2019).
Gerrits, E. et al. Distinct amyloid‑β and tau‑associated microglia profiles in Alzheimer’ s disease. Acta Neuropathol. https://doi.org/10.1007/s00401-021-02263-w (2021).
Sun, N. et al. Human microglial state dynamics in Alzheimer’s disease progression. Cell 186, 4386–4403.e29 (2023).
De, S. et al. Different soluble aggregates of Aβ42 can give rise to cellular toxicity through different mechanisms. Nat. Commun. 10, 1541 (2019).
De, S. et al. Soluble aggregates present in cerebrospinal fluid change in size and mechanism of toxicity during Alzheimer’s disease progression. Acta Neuropathol. Commun. 7, 120 (2019).
Sideris, D. I. et al. Soluble amyloid beta-containing aggregates are present throughout the brain at early stages of Alzheimer’s disease. Brain Commun. 3, fcab147 (2021).
Benilova, I., Karran, E. & De Strooper, B. The toxic Aβ oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat. Neurosci. https://doi.org/10.1038/nn.3028 (2012).
Fattorelli, N. et al. Stem-cell-derived human microglia transplanted into mouse brain to study human disease. Nat. Protoc. https://doi.org/10.1038/s41596-020-00447-4 (2021).
Serneels, L. et al. Modeling the β-secretase cleavage site and humanizing amyloid-beta precursor protein in rat and mouse to study Alzheimer’s disease. Mol. Neurodegener. 15, 1–11 (2020).
Mathys, H. et al. Temporal tracking of microglia activation in neurodegeneration at single-cell resolution. Cell Rep. 21, 366–380 (2017).
McQuade, A. et al. Development and validation of a simplified method to generate human microglia from pluripotent stem cells. Mol. Neurodegener. 13, 1–13 (2018).
Hendrickx, D. A. E., van Eden, C. G., Schuurman, K. G., Hamann, J. & Huitinga, I. Staining of HLA-DR, Iba1 and CD68 in human microglia reveals partially overlapping expression depending on cellular morphology and pathology. J. Neuroimmunol. 309, 12–22 (2017).
Ellwanger, D. C. et al. Prior activation state shapes the microglia response to antihuman TREM2 in a mouse model of Alzheimer’s disease. Proc. Natl Acad. Sci. USA 118, 1–12 (2021).
Götzl, J. K. et al. Opposite microglial activation stages upon loss of PGRN or TREM2 result in reduced cerebral glucose metabolism. EMBO Mol. Med. 11, e9711 (2019).
McQuade, A. et al. Gene expression and functional deficits underlie TREM2-knockout microglia responses in human models of Alzheimer’s disease. Nat. Commun. https://doi.org/10.1038/s41467-020-19227-5 (2020).
Rao, E. et al. Lack of apolipoprotein E dramatically reduces amyloid b-peptide deposition. Nat. Genet. 15, 57–61 (1997).
Sayed, F. A. et al. AD-linked R47H-TREM2 mutation induces disease-enhancing microglial states via AKT hyperactivation. Sci. Transl. Med. 13, eabe3947 (2022).
Olah, M. et al. Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer’s disease. Nat. Commun. 11, 6129 (2020).
Lau, S. F., Cao, H., Fu, A. K. Y. & Ip, N. Y. Single-nucleus transcriptome analysis reveals dysregulation of angiogenic endothelial cells and neuroprotective glia in Alzheimer’s disease. Proc. Natl Acad. Sci. USA 117, 25800–25809 (2020).
Cheng-Hathaway, P. J. et al. The Trem2 R47H variant confers loss-of-function-like phenotypes in Alzheimer’s disease. Mol. Neurodegener. 13, 1–12 (2018).
Wood, J. I. et al. Plaque contact and unimpaired Trem2 is required for the microglial response to amyloid pathology. Cell Rep. https://doi.org/10.1016/j.celrep.2022.111686 (2022)
Yin, Z. et al. APOE4 impairs the microglial response in Alzheimer’s disease by inducing TGFβ-mediated checkpoints. Nat. Immunol. https://doi.org/10.1038/s41590-023-01627-6 (2023).
Liu, C.-C. et al. Cell-autonomous effects of APOE4 in restricting microglial response in brain homeostasis and Alzheimer’s disease. Nat. Immunol. https://doi.org/10.1038/s41590-023-01640-9 (2023).
Bellenguez, C. et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 54, 412–436 (2022).
Leyns, C. E. G. et al. TREM2 function impedes tau seeding in neuritic plaques. Nat. Neurosci. https://doi.org/10.1038/s41593-019-0433-0 (2019).
Busche, M. A. et al. Tau impairs neural circuits, dominating amyloid-β effects, in Alzheimer models in vivo. Nat. Neurosci. 22, 57–64 (2019).
De Strooper, B. & Karran, E. The cellular phase of Alzheimer’s disease. Cell 164, 603–615 (2016).
Claes, C. et al. Human stem cell-derived monocytes and microglia-like cells reveal impaired amyloid plaque clearance upon heterozygous or homozygous loss of TREM2. Alzheimers Dement. https://doi.org/10.1016/j.jalz.2018.09.006 (2019).
Saito, T. et al. Single App knock-in mouse models of Alzheimer’s disease. Nat. Neurosci. 17, 661–663 (2014).
Shah, D. et al. Spatial reversal learning defect coincides with hypersynchronous telencephalic BOLD functional connectivity in APPNL-F/NL-F knock-in mice. Sci. Rep. 8, 1–11 (2018).
Bohlen, C. J. et al. Diverse requirements for microglial survival, specification, and function revealed by defined-medium cultures. Neuron 94, 759–773.e8 (2017).
Kuperstein, I. et al. Neurotoxicity of Alzheimer’s disease Aβ peptides is induced by small changes in the Aβ42 to Aβ40 ratio. EMBO J. 29, 3408–3420 (2010).
Hafemeister, C. & Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 1–15 (2019).
Mancuso, R. et al. Xenografted human microglia display diverse transcriptomic states in response to Alzheimer’s disease-related Aβ pathology. GEO https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE216999 (2024).
Acknowledgements
This project received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Programme (grant agreement no. ERC-834682 CELLPHASE_AD). This work was also supported by UKRI-MRC (grant no. MR/Y014847/1), the UK Dementia Research Institute, the Flanders Institute for Biotechnology (VIB vzw), a Methusalem grant from KU Leuven and the Flemish Government, the Fonds voor Wetenschappelijk Onderzoek, KU Leuven, the Queen Elisabeth Medical Foundation for Neurosciences, the Opening the Future campaign of the Leuven Universitair Fonds, the Belgian Alzheimer Research Foundation (SAO-FRA) and the Alzheimer’s Association USA. R.M. has funding from the ERC under the European Union’s Horizon 2020 Research and Innovation Programme (project no. 101041867—XenoMicrogliaAD) and Fonds voor Wetenschappelijk Onderzoek (grants no. G0C9219N, G056022N and G0K9422N), and is a recipient of a postdoctoral fellowship from the Alzheimer’s Association USA (fellowship nos. 2018-AARF-591110 and 2018-AARF-591110-RAPID). He also receives funding from BrightFocus Foundation (grant no. A2021034S), SAO-FRA (grant no. 2021/0021) and the University of Antwerp (grant no. BOF-TOP 2022-2025). B.D.S. holds the Bax-Vanluffelen Chair for Alzheimer’s Disease. N.F. is the recipient of a PhD fellowship from Fonds voor Wetenschappelijk Onderzoek (fellowship no. 1139520N). A.M.-M. is supported by a fellowship from the Alzheimer’s Association USA (fellowship no. AARF-20-684397) and an FWO-Marie Skłodowska-Curie Actions—Seal of Excellence Postdoctoral Fellowship (fellowship no. 12ZX621N). C.V. receives funding from Fonds voor Wetenschappelijk Onderzoek (grants no. G0B5819 and S001221N). J.P. is the recipient of a PhD fellowship from Fonds voor Wetenschappelijk Onderzoek (fellowship no. 1122423N). B.B. is the recipient of a PhD fellowship from the University of Antwerp (DOCPRO fellowship no. FFB210307). We thank V. Hendrickx and A. Claes for animal husbandry. Schemes and drawings in Figs. 1a and 8b were created with BioRender.com.
Author information
Authors and Affiliations
Contributions
R.M., N.F., A.M.-M. and B.D.S. conceived and designed the study and wrote the manuscript. R.M., N.F. and A.M.-M. performed experiments and analyzed data together with B.D.S. E.D. provided bioinformatic support and performed the analyses on publicly available datasets. L.W. performed experiments. J.V.D.D. performed the experiments involving TREM2 mutant microglia. I.G. performed mouse brain surgery and injections of Aβ soluble aggregates in the brain. P. Preman assisted in the experiments involving ApoE−/− mice. L.S. generated the Apphu/hu and ApoE−/− mouse lines. S.P. assisted in the generation of cDNA libraries for sequencing. S.B. generated and provided the iCas9-H9 stem cell line. C.V. provided supervision to J.V.D.D. M.F. provided supervision to E.D. J.P. and P. Polanco performed and quantified the immunostaining of human microglia in mouse brain. B.B. generated the samples and processed the data from mouse single cells. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
B.D.S. is or has been a consultant for Eli Lilly, Biogen, Janssen Pharmaceutica, Eisai, AbbVie and other companies. B.D.S. is also a scientific founder of Augustine Therapeutics and a scientific founder and stockholder of Muna Therapeutics. R.M. has scientific collaborations with Alector, Nodthera and Alchemab, and Roche, and has been a consultant for Sanofi. I.G. and L.W. are currently employed by Muna Therapeutics but were part of the De Strooper lab when this work was performed. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Neuroscience thanks Li-Huei Tsai and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Comparison of human microglia transplanted in AppWT and Rag2−/− hCSFKI mice, and primary microglia isolated from human surgical resections published before14.
a, UMAP plot of the 39,538 cells passing quality control, coloured by annotated cell states after removal of CAMs, other myeloid, low quality and proliferating clusters. b, UMAP plots as in a, coloured by sample. c, UMAP plots as in a, split between the three experimental groups. d, Box plot showing proportion of cells across all identified clusters (box plots are limited by lower and upper quartiles and midline indicates median; whiskers show minimum to maximum value. Each dot represents a single replicate, n = 13 for AppWT, n = 1 for Rag2−/−/hCSF1KI, n = 7 for primary MG. One-way ANOVA with Tukey’s multiple comparisons as post-hoc test, alpha = 0.05, significance was set as P-adjusted value < 0.05). e, Heatmap displaying the top20 most upregulated genes in each cluster as in a. f, Heatmap depicting the expression levels of homeostatic microglial markers across all experimental groups (normalized expression scaled by gene is shown).
Extended Data Fig. 2 Extended analysis of the cell state transition in AppNL-G-F and AppWT mice.
a, Volcano plot showing a paired comparison between AppNL-G-F and AppWT mice (Wilcoxon rank-sum test, P-values adjusted with Bonferroni correction based on the total number of genes in the dataset). The number of significant genes per condition is reported in brackets in the legend (Log2 Fold-Change threshold > 0.1). Adjusted p-value threshold < 0.05 (ns = not significant). b, Volcano plot showing a paired comparison between AppNL-G-F and AppWT mice (Wilcoxon rank-sum test, P-values adjusted with Bonferroni correction based on the total number of genes in the dataset), coloured by clusters as in Fig. 1 (HM = red squares, CRMs (CRM-1 & CRM-2) = ochre circles, DAM = olive triangles, HLA = cyan diamonds, other markers = grey circles). The number of significant genes per condition is reported in brackets in the legend (Log2 Fold-Change threshold > 0.1). Adjusted p-value threshold < 0.05 (ns = not significant). c, Phenotypic trajectory followed by the human microglia obtained by an unbiased pseudotime ordering with Monocle 3. Proportion of cells (y-axis) present in each cell state as in Fig. 1a (rows) at different stages of the binned pseudotime trajectory (x-axis) in AppNL-G-F and AppWT mice (box plots are limited by lower and upper quartiles and midline indicates median; whiskers extend from the box to the smallest or largest value no further than 1.5*inter-quartile range. Each dot represents a single mouse, n = 10 for AppWT, n = 23 AppNL-G-F. Unpaired t-test with Welch’s correction, two-tailed, alpha = 0.05, significance was set as P-value < 0.05; *p < 0.05, **p < 0.01, ****p < 0.0001). d, Representative images of human microglia engrafted in the brain of AppNL-G-F mice at 6 months of age and labelled with human specific antibodies FTH1 (RM). Human P2RY12 was stained in green, and amyloid-β plaques were stained with X34 in white. Scale bars are 25 μm (top row) and 50 μm (bottom row).
Extended Data Fig. 3 Extended comparison of the response of human and mouse microglia to amyloid pathology.
a, UMAP plot of the 3,803 mouse microglia isolated from AppNL-G-F and Rag2−/− Il2rγ−/− hCSF1KI AppNL-G-F passing quality control, coloured by annotated cell states and after removal of CAMs, other myeloid, low quality and proliferating clusters. b, Distribution and proportion of cells across all identified clusters (box plots are limited by lower and upper quartiles and mid line indicates median; whiskers show minimum to maximum value. Each dot represents a single mouse, n = 10 for xMG AppWT 6 m, n = 23 for xMG AppNL-G-F 6 m, n = 2 for AppNL-G-F). One-way ANOVA with Tukey’s multiple comparisons as post-hoc test, alpha = 0.05, significance was set as P-adjusted value < 0.05). For cell states that only had 2 groups (tCRM and CRM-1), we applied an unpaired t-test with Welch’s correction, alpha = 0.05, two-tailed, significance was set as P-value < 0.05. Note that whereas mouse cells polarize to DAM, HLA and CRM, proportions do not increase. c, Correlation analysis of the logFC in DAM2 vs. DAM1 clusters defined by Keren-Shaul et al.4 (y-axis) and HLA vs. DAM clusters defined in this study (x-axis). Pearson’s correlation, R = −0.44, differentially expressed genes were adjusted using Bonferroni correction and colored according to clusters in Fig. 1a. d, Overlap in the number of genes between our DAM signature and that from mouse systems (KarenShaul DAM and SalaFrigerio ARM), other human microglia xenotransplantation studies (Hasselman DAM), and postmortem single nuclei samples (Gerrits AD1, and Olah DAM). The arrow depicts the direction used to calculate the percentage overlap.
Extended Data Fig. 4 Extended pseudotime analysis, including transplanted microglia isolated from 3 months old AppNL-G-F mice.
a, UMAP plot of the 38,360 mouse microglia isolated from 3 and 6 months AppNL-G-F and AppWT mice passing quality control, coloured by annotated cell states and after removal of CAMs, other myeloid, low quality and proliferating clusters. b, Phenotypic trajectory followed by the human microglia after exposure to amyloid-β in vivo, obtained by an unbiased pseudotime ordering with Monocle 3. c, Density plots displaying the average distribution of human microglia transplanted in 3 months old AppNL-G-F (n = 11) 6 months old AppNL-G-F (n = 9) and AppWT (n = 8) mice (density is normalized to sample size). d, Box plot showing proportion of cells across all identified clusters (eEach dot represents a single mouse, n = 8 for AppWT, n = 11 for AppNL-G-F 3 m, n = 9 for AppNL-G-F 6 m). e, Distribution of cells from different host mouse genetic backgrounds and ages (y-axis) across the DAM and HLA transcriptional trajectories colored by clusters shown in a (Each dot represents a single cell, n = 8 mice (8,974 cells) for AppWT, n = 11 mice (10,822 cells) for AppNL-G-F 3 m, n = 9 mice (12,197 cells) for AppNL-G-F 6 m). f, Phenotypic trajectory followed by the human microglia obtained by an unbiased pseudotime ordering with Monocle 3. Proportion of cells (y-axis) present in each cell state as in a (rows) at different stages of the binned pseudotime trajectory (x-axis) in 3 and 6 months old AppNL-G-F and AppWT mice (Each dot represents a single mouse, n = 8 for AppWT, n = 11 for AppNL-G-F 3 m, n = 9 for AppNL-G-F 6 m; *p < 0.05, ****p < 0.001). Box plots in d, e and f are limited by lower and upper quartiles and midline indicates median; whiskers show minimum to maximum value (d) or extend from the box to the smallest or largest value no further than 1.5*inter-quartile range (e and f). One-way ANOVA with Tukey’s multiple comparisons as post-hoc test, alpha = 0.05, significance was set as P-adjusted value < 0.05.
Extended Data Fig. 5 Extended pseudotime analysis, including transplanted microglia isolated from 3 months old AppNL-G-F mice (continuation).
a, b, Heatmaps displaying the cells ordered by the (a) HM-DAM-HLA and (b) HM-CRM pseudotime axis, coloured by single-cell levels of expression of the top markers per cluster. c, d, Expression profile of selected genes across the (c) HM-DAM-HLA and (d) CRM pseudotime axis averaging all experimental groups in the dataset. e, Comparison of the expression profile of selected CRM markers across the trajectory. f, Expression profile of selected genes across the CRM pseudotime axis as in d divided into the different experimental groups: AppWT, AppNL-G-F 3 M and AppNL-F-G 6 M.
Extended Data Fig. 6 Extended analysis on the differential impact of TREM2 and APOE deficiency in the transplanted microglia.
a, Phenotypic trajectory followed by TREM2 deficient and control microglia, obtained by an unbiased pseudotime ordering with Monocle 3. Proportion of cells (y-axis) present in each cell state as in Fig. 1b (rows) at different stages of the binned pseudotime trajectory (x-axis), colored by host genotypes (each dot represents a single mouse, n = 2 mice H9-WT, n = 3 mice H9-TREM2−/−). b, c, Volcano plots showing a paired comparison between TREM2−/− and WT microglia transplanted in AppNL-G-F, coloured by (b) cells genotype and (c) cluster as in Fig. 1b. The number of significant genes per condition is reported in brackets in the legend (Log2 Fold-Change threshold > 0.1). Adjusted p-value threshold < 0.05 (Wilcoxon rank-sum test, P-values adjusted with Bonferroni correction based on the total number of genes in the dataset, ns = not significant). d, Phenotypic trajectory followed by APOE deficient microglia compared to isogenic APOE3/3 controls, obtained by an unbiased pseudotime ordering with Monocle 3. Proportion of cells (y-axis) present in each cell state as in Fig. 1b (rows) at different stages of the binned pseudotime trajectory (x-axis), colored by host genotypes (each dot represents a single mouse, n = 6 APOE3/3 and n = 7 for APOE−/−). e, f, Volcano plots showing a paired comparison between APOE−/− and APOE3/3 microglia transplanted in AppNL-G-F, coloured by (e) cells genotype and (f) cluster as in Fig. 1b. The number of significant genes per condition is reported in brackets in the legend (Log2 Fold-Change threshold > 0.1). Adjusted p-value threshold < 0.05 (Wilcoxon rank-sum test, P-values adjusted with Bonferroni correction based on the total number of genes in the dataset, ns = not significant). Box plots in a and d, are limited by lower and upper quartiles and midline indicates median; whiskers extend from the box to the smallest or largest value no further than 1.5*inter-quartile range. Unpaired t-test with Welch’s correction, two-tailed, alpha = 0.05, significance was set as P-value < 0.05, *p < 0.05, **p < 0.01.
Extended Data Fig. 7 Extended analysis on the differential effects of amyloid-β plaques and soluble aggregates on microglia cell states.
a, Phenotypic trajectory followed by the human microglia transplanted in AppNL-G-F ApoE−/− and AppNL-G-F, obtained by an unbiased pseudotime ordering with Monocle 3. Proportion of cells (y-axis) present in each cell state as in Fig. 1b (rows) at different stages of the binned pseudotime trajectory (x-axis), colored by host genotypes (dots represent single mice, n = 11 AppNL-G-F, n = 6 AppNL-G-F ApoE−/−). b, c, Volcano plots showing a paired comparison between microglia transplanted in AppNL-G-F ApoE−/− and AppNL-G-F mice, coloured by (b) host genotype and (c) cluster as in Fig. 1a. The number of significant genes per condition is reported in brackets in the legend (Log2 Fold-Change threshold > 0.1). Adjusted p-value threshold < 0.05 (Wilcoxon rank-sum test, P-values adjusted with Bonferroni correction based on the total number of genes in the dataset, ns = not significant). d, Phenotypic trajectory followed by the human microglia after injection of amyloid-b oligomers (Aβο) or scrambled peptide (Scr), obtained by an unbiased pseudotime ordering with Monocle 3. Proportion of cells (y-axis) present in each cell state as in Fig. 1a (rows) at different stages of the binned pseudotime trajectory (x-axis), colored by cells genotypes (dots represent single mice, n = 7 for Scr, n = 13 Aβο). e, f, Volcano plots showing a paired comparison between microglia transplanted in AppWT mice and treated with Aβο or Scr, coloured by (e) treatment and (f) cluster as in Fig. 1a. The number of significant genes per condition is reported in brackets in the legend (Log2 Fold-Change threshold > 0.1). Adjusted p-value threshold < 0.05 (Wilcoxon rank-sum test, P-values adjusted with Bonferroni correction based on the total number of genes in the dataset, ns = not significant). Box plots in a and d, are limited by lower and upper quartiles and midline indicates median; whiskers extend from the box to the smallest or largest value no further than 1.5*inter-quartile range. Unpaired t-test with Welch’s correction, two-tailed, alpha = 0.05, significance was set as P-value < 0.05, *p < 0.05, **p < 0.01, ***p < 0.001,****p < 0.0001.
Extended Data Fig. 8 Extended analysis of the single nuclei from human post-mortem brains (continuation).
a, UMAP plots as in Fig. 6, coloured by the combined level of expression of groups of genes that characterise distinct microglial transcriptional states from xenotransplanted microglia. b, Full set of pairwise Pearson correlations between logFC of all DE genes (logFC cut off set at 0.25, P-value < 0.05) of each microglia sub-type and logFC of all DE genes (p < 0.05) of clusters from each human snRNA-seq study, with significance set at a P-adjusted value < 0.05 (*p < =0.05, **p < =0.01, ***p < =0.001). c, Representative scatter plots showing the correlation analysis of the logFC between xenotransplanted and single nuclei post-mortem microglia. The labels are coloured by cell state as in Fig. 1.
Extended Data Fig. 9 Extended analysis of the impact of clinical mutations on microglial cell states.
a, b, Volcano plots showing a paired comparison between TREM2R47H and TREM2-WT microglia transplanted in AppNL-G-F mice and coloured by (a) cells genotype or (b) cluster as in Fig. 1a. The number of significant genes per condition is reported in brackets in the legend (Log2 Fold-Change threshold > 0.1). Adjusted p-value threshold < 0.05 (Wilcoxon rank-sum test, P-values adjusted with Bonferroni correction based on the total number of genes in the dataset, ns = not significant). c, d, Distribution of TREM2R47H and TREM2-WT cells across (c) DAM-HLA and (d) CRM transcriptional trajectories. Note the shift in transcriptional states in both the DAM-HLA and CRM axes (each dot represents a single cell, n = 2 (3,282 cells) H9-WT, n = 3 (5,845 cells) H9-TREM2R47H). e, f, Volcano plots showing a paired comparison between APOE4/4 and APOE3/3 microglia transplanted in AppNL-G-F mice and coloured by (e) cells genotype or (f) cluster as in Fig. 1a. The number of significant genes per condition is reported in brackets in the legend (Log2 Fold-Change threshold > 0.1). Adjusted p-value threshold < 0.05 (Wilcoxon rank-sum test, P-values adjusted with Bonferroni correction based on the total number of genes in the dataset, ns = not significant). g, h, Distribution of APOE4 and APOE3 cells across (g) DAM and HLA and (h) CRM transcriptional trajectories (each dot represents a single mouse, n = 6 (7,894 cells) for APOE3/3, n = 6 (10,151 cells) for APO4/4). i, Distribution and proportion of cells across all identified clusters as in Fig. 5e, coloured by the microglial APOE genotype (each dot represents a single mouse, n = 7 for Scr, n = 13 Aβο). Box plots in c, d, g, h and i, are limited by lower and upper quartiles and midline indicates median; whiskers show minimum to maximum value (i) or extend from the box to the smallest or largest value no further than 1.5*inter-quartile range (c, d, g and h). Unpaired t-test with Welch’s correction, two-tailed, alpha = 0.05, significance was set as P-value < 0.05.
Extended Data Fig. 10 Extended analysis of GWAS genes enrichment in xenotransplanted microglia.
a, Summary heatmap with the expression levels of the Top10 marker genes for each cluster (as in Fig. 1c), compared across all experimental groups used in this study. b, Expanded gene list with the canonical AD genes according to the latest GWAS study of Fig. 8a, with the addition of other genes of interest, that is the genes selected in our previous publication14 (see Supplementary Table 4 for the complete list). Raw counts (log scale) are reported on the x axis, while standard deviation of counts is shown on the y-axis. Dots are coloured by SCT-normalised expression (see Methods) and size reflects percentage of cells in our dataset where the gene is detected (see all values in Supplementary Table 4). All genes from (a) that are significantly changing in at least one of the tested conditions in this manuscript are reported in an extended heat map build as in Fig. 8a. The black bars represent the number of cells (in thousands) with detectable expression (>=1 read per cell) for each candidate gene. The heat map summarizes the deregulated expression (LogFC, colour scale) of these genes across cell states (each cluster compared to all others), as well as after exposure to amyloid-β plaque pathology, upon injection of soluble aggregates of Aβ, or altering the genetic background of the mice or the transplanted cells. The genes are ranked in rows based on hierarchical clustering. We identify 3 sets of genes that display a common profile across cell states (based on their enrichment in the specific microglial phenotypic transcriptional states HM, DAM and HLA, and CRM), amyloid-β pathology and genetic risk, and we group these profiles as: microglia Homeostasis, plaque-induced genes, and soluble aggregates-induced genes. The remaining genes did not show a clear enrichment in cell states or other conditions. All differential expressions were significant after adjusting P-values using Bonferroni correction (FDR < 0.05).
Supplementary information
Supplementary Information
Supplementary Figs. 1–7.
Supplementary Table 1
Differential expression between microglial cell states. Differential expression results comparing each cell state in Fig. 1b with all the others identify specific markers of microglial cell states reacting to amyloid pathology. We used the Wilcoxon rank-sum test to calculate P values. We tested only genes that are detected in a minimum fraction of 1% in either of the two populations. We limited testing to genes that show, on average, at least 0.1-fold difference (log-scale) between the two groups of cells. Only genes with their adjusted P value < 0.05 (post hoc, Bonferroni correction) were considered as significant.
Supplementary Table 2
Overlap of human and mouse DAM markers. List of DAM marker genes defined in this study and in other transplantation, human postmortem and mouse studies, as compared in Extended Data Fig. 3d.
Supplementary Table 3
Differential expression between all tested conditions. Differential expression results comparing side by side all the conditions tested in this study, including genotype of the host, type of injection and TREM2/APOE mutations. We used the Wilcoxon rank-sum test to calculate P values. We tested only genes that are detected in a minimum fraction of 1% in either of the two populations. We limited testing to genes that show, on average, at least 0.1-fold difference (log-scale) between the two groups of cells. Only genes with their adjusted P value < 0.05 (post hoc, Bonferroni correction) were considered as significant.
Supplementary Table 4
Alzheimer’s disease GWAS gene expression in human microglial cell states. The table reports how the list of GWAS genes was compiled from the most recent studies, the expression levels of the risk genes in our dataset as in Extended Data Fig. 10b and their differential expression across cell states and tested conditions as in Fig. 8a and Extended Data Fig. 10c.
Supplementary Table 5
Statistics and QC of the sequencing libraries. The table reports summary statistics such as average number of genes, reads and percentage of reads mapping to mitochondrial genes for each of the 23 sequencing libraries included in this study. The percentage of cells removed by applying our QC criteria is also indicated.
Supplementary Table 6
Metadata by transplanted mouse replicate. Detailed metadata of the 106 transplanted mice used in this study, including genotype of the host, genotype of the transplanted human cells, age, sex and type of injection. The number of sequenced cells for each tested condition is also indicated.
Supplementary Table 7
Metadata by single cells. Detailed metadata of the single cells sequenced in this study, including all metadata as in Supplementary Table 2, the assigned cell state and the UMAP coordinates as in Fig. 1b.
Supplementary Table 8
Differential expression between cell states before removal of nonmicroglial cells. Differential expression results comparing each cell state in Supplementary Fig. 3a with all the others identify markers of nonmicroglial cells excluded from this study. We used the Wilcoxon rank-sum test to calculate P values. We tested only genes that are detected in a minimum fraction of 1% in either of the two populations. We limited testing to genes that show, on average, at least 0.1-fold difference (log-scale) between the two groups of cells. Only genes with their adjusted P value < 0.05 (post hoc, Bonferroni correction) were considered as significant.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mancuso, R., Fattorelli, N., Martinez-Muriana, A. et al. Xenografted human microglia display diverse transcriptomic states in response to Alzheimer’s disease-related amyloid-β pathology. Nat Neurosci (2024). https://doi.org/10.1038/s41593-024-01600-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41593-024-01600-y
This article is cited by
-
Regulation of human microglial gene expression and function via RNAase-H active antisense oligonucleotides in vivo in Alzheimer’s disease
Molecular Neurodegeneration (2024)
