
Abstract
Primary hyperoxaluria (PH) is an inherited disorder that results from the overproduction of endogenous oxalate, leading to recurrent kidney stones, nephrocalcinosis and eventually kidney failure; the subsequent storage of oxalate can cause life-threatening systemic disease. Diagnosis of PH is often delayed or missed owing to its rarity, variable clinical expression and other diagnostic challenges. Management of patients with PH and kidney failure is also extremely challenging. However, in the past few years, several new developments, including new outcome data from patients with infantile oxalosis, from transplanted patients with type 1 PH (PH1) and from patients with the rarer PH types 2 and 3, have emerged. In addition, two promising therapies based on RNA interference have been introduced. These developments warrant an update of existing guidelines on PH, based on new evidence and on a broad consensus. In response to this need, a consensus development core group, comprising (paediatric) nephrologists, (paediatric) urologists, biochemists and geneticists from OxalEurope and the European Rare Kidney Disease Reference Network (ERKNet), formulated and graded statements relating to the management of PH on the basis of existing evidence. Consensus was reached following review of the recommendations by representatives of OxalEurope, ESPN, ERKNet and ERA, resulting in 48 practical statements relating to the diagnosis and management of PH, including consideration of conventional therapy (conservative therapy, dialysis and transplantation), new therapies and recommendations for patient follow-up.
Similar content being viewed by others

Introduction
Primary hyperoxaluria (PH) is a group of autosomal recessive disorders of glyoxylate metabolism that cause the overproduction of endogenous oxalate — a redundant metabolic end product that is excreted primarily via the kidneys. In high concentrations, it tends to form crystals with calcium in the renal tubules, leading to the formation of kidney stones, nephrocalcinosis or both. In PH, the combination of intra-tubular and interstitial deposits of calcium oxalate, chronic tubulo-interstitial inflammation and kidney obstruction by stones leads to kidney failure in more than 70% of patients. As soon as glomerular filtration rate (GFR) falls below 30–40 ml/min/1.73 m², hepatic oxalate production exceeds renal removal, leading to systemic oxalate storage in various tissues, including bone, heart, vessels, nerves and eye, and causing life-threatening multi-organ disease.
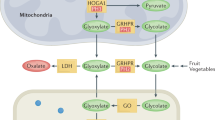
Three biochemically defined types of PH exist (Fig. 1) of which type 1 (PH1) is by far the most prevalent and has the worst prognosis. Timely diagnosis and disease management can be challenging for all three subtypes. Clinical practice guidelines for PH1 were published in 2012 (ref. 1); however, most recommendations in those guidelines were opinion based given the paucity of clinical data available at the time. In the past decade, large registry analyses have delivered new insights into the validity and pitfalls of diagnostic procedures, the outcomes of PH2 and PH3 and the impact of available therapies2,3. Importantly, the introduction of two new therapies based on RNA interference (RNAi) — both of which substantially lower endogenous oxalate production in patients with PH1 — have influenced the management of this disease.


a, Simplified overview of glyoxylate metabolism in patients with primary hyperoxaluria type 1 (PH1), characterized by a deficiency in alanine–glyoxylate aminotransferase (AGT), which leads to increased levels of glycolate, glyoxylate and oxalate. b, Simplified overview of glyoxylate metabolism in patients with PH type 2 (PH2), characterized by a deficiency in glyoxylate and hydroxypyruvate reductase (GRHPR), which leads to raised levels of glyoxylate, l-glycerate and oxalate. c, Simplified overview of glyoxylate metabolism in patients with PH type 3 (PH3). The exact mechanism by which 4-hydroxy-2-oxoglutarate (HOG) deficiency causes high oxalate levels is yet to be determined. Two hypothetical mechanisms are indicated: HOG could act as an inhibitor of GRHPR or it could be transported into the cytosol where it is converted into glyoxylate by an unknown cytosolic aldolase. d, Overview of the working mechanisms of lumasiran and nedosiran. Lumasiran inhibits the production of glycolate oxidase (GO); nedosiran inhibits the production of l-lactate dehydrogenase (LDH). AspAT, aspartate aminotransferase; DHG, 2,4-dihydroxyglutarate; HYPDH, hydroxyproline dehydrogenase; HOGA, 4-hydroxy-2-oxoglutarate aldolase; 1P5CDH, Δ1-pyrroline-5-carboxylate dehydrogenase.
In response to these developments, members of OxalEurope — a network of European scientists and physicians who specialize in PH — and the metabolic workgroup of the European Rare Kidney Disease Reference Network (ERKNet), formed a workgroup. Our goal was to update the 2012 guidelines and formulate new clinical practice recommendations for the diagnostic approach to patients with a suspicion of PH and the management of all types of PH with various stages of kidney dysfunction. We aim to make clinical practice recommendations for worldwide application and have therefore added statements for countries with restricted financial and medical means. We also propose key future research questions in the field, which may further help practitioners in clinical decision making. These guidelines are endorsed by the European Society of Paediatric Nephrology (ESPN), the European Renal Association (ERA) and ERKNet.
Methods
The core group and external voting panel
The recommendations presented here were assembled by a consensus development core group and voted on by an external voting panel. The core group consisted of paediatric nephrologists, geneticists, biochemical researchers, nephrologists, a paediatric urologist, an adult urologist and three PhD students working on PH, from eight European countries (Belgium, France, Germany, Lithuania, Italy, Netherlands, Serbia and the UK). All members of the core group were members of OxalEurope and ERKNet, except for two of the three PhD students (E.M. and L.D.). The voting panel consisted of 20 paediatric nephrologists, 11 nephrologists and 5 scientists or geneticists from 14 countries (Belgium, Dubai, France, Germany, Hungary, Israel, Italy, Netherlands, Norway, Spain, Switzerland, Turkey, the UK and the USA). All members of the voting panel had expertise in PH and were members of one or more of the metabolic or inherited disease workgroups of ESPN, ERA, ERKNet or OxalEurope. The core group was responsible for defining the scope of the project, formulating the key questions, performing the literature review, rating the quality of evidence, composing and grading recommendations, considering the rationale for the recommendations and drafting the initial and final versions of the manuscript. Six thematic workgroups were formed by members of the core study group to cover specific topics: the genetic basis of PH1–3 and its clinical implications; the diagnostic work-up and monitoring of PH in patients aged >1 year; the management of PH in patients aged >1 year; the specific management of infantile oxalosis; and the urological approach to patients with PH. In addition, two physicians from Egypt and Jordan (R.A., N.A.S.) added recommendations for the management of PH in low-resource settings.
Statement development
To ensure that the statements derived from this work could be translated into actionable advice, the core group developed clinical questions based on the elements of the PICO framework — the patient (or population) to whom the recommendation applies; the intervention under consideration; the comparator of the intervention under consideration (that is, compared with ‘no action’, placebo or an alternative intervention); and the outcomes affected by the intervention. The resulting questions were addressed through literature searches to identify papers published in the PubMed database between 1970 and 2022. We included randomized clinical trials (RCTs), prospective uncontrolled and observational studies irrespective of the number of patients, and registry studies, retrospective studies and case series, restricted to human studies in English (Supplementary Table 1). Each PICO question formed the basis for a statement, and all workgroups were asked to propose recommendations and provide a rationale for their statements.
Grading system
Forty-eight statements related to the management of PH were graded by individual members of the core group according to the system used by the American Academy of Paediatrics4 (Supplementary Figure 1). These gradings were circulated to other members of the core group, along with supporting evidence from the literature. Over the course of six virtual meetings, consensus was reached within the group. The statements and their gradings were then reviewed by the external voting panel. The members of the voting panel were asked to provide a level of agreement for all 48 statements on a 5-point scale (strongly disagree, disagree, neither agree nor disagree, agree, strongly agree — according to the Delphi method5) and to suggest rewording if appropriate.
All levels of agreement other than ‘I agree’ or ‘I strongly agree’ were defined as ‘no agreement’. If fewer than 70% of the voting panel agreed with a statement, the process was restarted by the core group. At least 70% agreement was achieved for 46 of 48 statements. For one of the statements with insufficient agreement (statement 8, relating to the assessment of calcium oxalate crystal volume (so-called ‘crystalluria’)), 13 of the 17 ‘no agreement’ votes were ‘I do not agree’ and ‘I do not disagree’ with most comments indicating insufficient availability and/or insufficient experience with the recommended approach. Hence, the core group decided to leave statement 8 unchanged, with a footnote that crystalluria cannot replace genetic testing or biochemical urinary assessment to establish a diagnosis of PH. This proposal was sent to the voting panel and accepted by all. The other statement for which agreement was insufficient (statement 21, relating to the early initiation of kidney replacement therapy) was revised, and subsequently agreed upon by more than 70% of the voting panel.
Management recommendations
These clinical practice recommendations have been developed to provide guidance to health-care professionals for the diagnosis and management of children and adults who are suspected to have, or are diagnosed with, PH, on the basis of the available evidence from studies and the opinions of experts in the field. Our clinical practice recommendations are intended as a guide, not as a dictate. We outline 48 statements that summarize our recommendations for the management of PH (Table 1) and outline the rationale for each of these in the separate sections. We also provide recommendations for the general monitoring of patients with PH (Box 1) along with reference values of biomarkers (Supplementary Tables 2 and 3). We further provide indications for RNAi therapy and recommendations for monitoring patients while on RNAi therapy (Table 2 and Box 2) and an algorithm for the management of patients with or suspected to have PH (Fig. 2).

The diagnostic work-up for patients with estimated glomerular filtration rate (eGFR) >30 ml/min/1.73 m2 and suspected primary hyperoxaluria (PH) should include at least two urine oxalate assessments (preferably from a 24 h urine collection). If genetic assessment reveals a mutation consistent with vitamin B6 (VB6; also known as pyridoxine) non-responsive PH1, RNA interference (RNAi) therapy is indicated. Patients who are partially responsive to VB6 therapy may also be eligible for RNAi therapy if hyperoxaluria persists. In patients with a suspicion of PH and eGFR <30 ml/min/1.73 m2, diagnostics should include plasma oxalate assessments. If genetic assessment reveals a mutation consistent with VB6 non-responsive PH1, RNAi therapy is indicated. Patients with (partial) VB6 responsive mutations might be eligible for RNAi therapy depending on plasma oxalate levels.
Diagnostic approach
Rationale for genetic assessment in PH
An exhaustive review of the genetic basis of PH has been published elsewhere6. In brief, we regard genetic testing as the gold standard for the diagnosis of all three types of PH. We therefore recommend that all patients who are suspected to have PH should undergo genetic assessment, as genetic confirmation of PH and typing are pivotal to the management of these patients, and assessment of biochemical parameters can be unreliable. PH1 has on average a far worse outcome than PH2 or PH3 and should be treated and monitored more vigorously2,3. New RNAi therapies have so far proved effective only in patients with PH1. Moreover, some PH1 genotypes are strongly associated with therapeutic response to pyridoxine; hence, genetic assessment can provide extremely important information for the clinical care of these patients, especially in patients with severe kidney failure in whom the biochemical response to pyridoxine can be difficult to measure.
Ideally, genetic testing for PH1–3 should be performed as early as possible, but within 30 days of a patient presenting with suspected PH and severe kidney failure (eGFR <30 ml/min/1.73 m2). Genetic confirmation of suspected PH with eGFR >30 ml/min/1.73 m2 should be carried out promptly, but note that turnaround times for such tests can vary between countries7. Genetic counselling for couples in which both partners are carriers of mutations that predispose to PH1 is important to enable early diagnosis and management of affected offspring.
PH1
PH1 results from a deficiency in the liver-specific, peroxisomal, pyridoxal phosphate-dependent enzyme, alanine–glyoxylate aminotransferase (AGT), which is encoded by AGXT (Fig. 1a). Although more than 200 mutations have been described in AGXT, p.Gly170Arg is the most frequent in Western populations and accounts for approximately 28–30% of mutant alleles8,9,10; c.33dupC is more common in other regions8,9,11,12.
PH2
PH2 results from a deficiency in the enzyme glyoxylate and hydroxypyruvate reductase (GRHPR), which is expressed in many tissues and is encoded by GRHPR (Fig. 1b). The most common mutation among Caucasian patients with PH2 is a single nucleotide deletion, c.103delG, which accounts for 31–35% of all cases in this population. By contrast, patients of Asian ancestry more often have a 4 bp deletion, c.404+3_404+6del (previously described as c.[403_404+2delAAGT])2,9.
PH3
PH3 results from the loss of function of the mitochondrial enzyme, 4-hydroxy-2-oxoglutarate aldolase (HOGA), which is predominantly found in the liver and kidney13 (Fig. 1c). A report based on data from a large European database of patients with PH3 identified 37 mutations in HOGA1. The splice mutation c.700+5G>T was the most common with an allelic frequency (AF) of 46%, followed by c.569C>T (AF 8%) and c.944-46delAGG (AF 5%)3. The c.944-46delAGG mutation was common among patients of Ashkenazi Jewish descent9. Of note, the mutation profile of patients with European ancestry differs from that of patients of Chinese descent in whom a splice site mutation, c.834_834+1GG>TT, accounted for 50% of mutant alleles in one series with no evidence of c.700+5G>T found in this cohort14.
Genotype–phenotype association
Evidence exists for associations between genotype, disease phenotype and therapeutic responsiveness in PH1. Patients with PH1 resulting from homozygous p.Gly170Arg or p.Phe152Ile mutations combined with a common polymorphism p.Pro11Leu — the so-called minor allele — are most likely to respond to pyridoxine therapy, resulting in a significant decrease and sometimes normalization of urinary oxalate levels. These patients also have a significantly higher median age of kidney failure onset than patients with PH1 with other, pyridoxine-insensitive mutations9,10,15, although kidney failure may still occur in infancy16. Other non-truncating PH1 genotypes may also be associated with pyridoxine responsiveness, but evidence in support of such associations is less clear. No biochemical or clinical genotype–phenotype correlation has been found for PH2 or PH3 (refs. 2,9,17).
Rationale for biochemical assessment
Biochemical assessment has an important role in the diagnostic work-up of patients with symptoms suggestive of PH and can focus genetic testing. It can also be used as an indication of therapeutic response. However, measurement of oxalate and relevant metabolites is not without difficulty and one must interpret the results carefully, taking all potential flaws into account.
Urine oxalate
Suspicion of PH in a patient with normal kidney function should be investigated initially by measurement of urine oxalate in a 24 h urine sample, collected into acid or acidified within 24 h after collection to achieve a pH of <2 to aid sample preservation and oxalate solubility18. Non-acidified 24 h collections must be acidified and well mixed in the laboratory to ensure that oxalate crystals are resolubilized before aliquoting as failure to resolubilize oxalate crystals can result in falsely low readouts (G. Rumsby, unpublished work). Samples with pH >8 are unsuitable for analysis of urine oxalate, as oxalogenesis can occur in vitro under such conditions19. Correction of oxalate level for body surface area to 1.73 m2 enables interpretation of paediatric results using the adult reference range, with accepted normal values of <0.46 mmol/24 h. As 24 h collections are inconvenient for patients and difficult for children, a random urine sample can also be used for preliminary analysis but it must be normalized to urinary creatinine level20. Acidification of such samples can be carried out in the laboratory provided the sample is kept at 4 °C after collection18. The ratio of urine oxalate to creatinine falls rapidly in the first year of life and plateaus at around 5 years of age. Thus, age-adjusted reference ranges are required. In adults, sex differences in creatinine excretion imply the existence of sex differences in urine oxalate-to-creatinine ratios21 (Supplementary Table 2). Preterm infants without PH have significantly higher urinary oxalate-to-creatinine ratios than infants without PH who are born at term22. Hyperoxaluria can also occur in patients who receive total parenteral nutrition, in patients with an excessive dietary intake of gelatin-rich foods (for example, sweets23) or oxalate-rich foods (for example, spinach, beetroot or dark chocolate) and in patients with increased intestinal absorption of oxalate owing to fat malabsorption — a situation that is defined as secondary hyperoxaluria24. A very-low-calcium diet can also increase intestinal oxalate absorption and therefore urinary oxalate level25. Of note, 24 h oxalate excretion does not correlate perfectly with oxalate-to-creatinine ratio, possibly as a consequence of imperfect urine collections and the effect of body size, which influences creatinine excretion and may therefore affect the oxalate-to-creatinine ratio21. However, available evidence suggests that either measurement can be used to monitor response to treatment26. The intra-individual biological coefficient of variation (CVbio) — that is, the natural day-to-day variation of oxalate excretion by one person — is normally high for both 24 h urine oxalate and the oxalate-to-creatinine ratio in healthy individuals (43% and 79%, respectively)27. In patients with PH, the mean CVbio is around 14% for 24 h urinary oxalate excretion with a corresponding mean reference change value of 32% (ref. 28). Thus, a decrease in urine oxalate of at least one-third is required to be certain of a definite response to treatment. We therefore recommend that urine oxalate measurements are repeated on at least two, but preferably three occasions to confirm that levels are elevated, particularly if findings are equivocal. The exclusion of high-oxalate foods for 24 h before sampling may resolve equivocal results. Urine oxalate above 1 mmol/1.73 m2 per day is strongly suggestive of PH. Exclusion of enteric causes of hyperoxaluria (for example, chronic pancreatitis, cystic fibrosis, inflammatory bowel syndrome or bariatric surgery) in which the degree of hyperoxaluria may overlap with PH is required before further metabolic or genetic investigations29. Agreement between laboratories in measurements of urine oxalate is typically good, reflecting the availability of calibration materials and external quality assurance schemes. Most laboratories measure oxalate after its conversion into hydrogen peroxide with oxalate oxidase, but liquid chromatography–tandem mass spectrometry (LC-MS) and gas chromatography–mass spectrometry (GC-MS) are also used and may have slightly different outcomes of no clinical significance30.
Urinary PH metabolites
Analyses of urinary PH metabolites can provide additional support for a preliminary diagnosis of PH and can aid triage of patients for genetic analysis. Unfortunately, measurement of these urinary metabolites — glycolate, l-glycerate, 4-hydroxy-2-oxoglutarate (HOG) and 2,4-dihydroxyglutarate (DHG) — are offered only by specialist laboratories but they can be analysed using the same sample as used for urine oxalate measurement Urine glycolate is elevated in approximately 75% of cases of PH1 (Supplementary Table 2); however, this metabolite is not specific for PH; levels can change in response to diet. In addition, urinary glycolate levels are grossly elevated in patients with glycolate oxidase deficiency31,32, which is a relatively benign disorder, although it was associated with hyperoxaluria in at least one case33. Urine l-glycerate is elevated in patients with PH2; however, false negatives can result from urinary organic acid screens, and use of a specific assay, which has been demonstrated to have 100% sensitivity, is therefore advised2. HOG and DHG are both markers of PH3; however, HOG is unstable34 and can lead to false negative results35 whereas DHG has 100% sensitivity36. Comparison of results between laboratories is currently impossible for these analytes as no international calibrators exist and one must therefore rely on local reference ranges.
Plasma oxalate
Plasma oxalate levels should only be used for the diagnosis of PH in patients with kidney failure. In these patients, oxalate excretion has declined to such an extent that urine results are misleading. Plasma oxalate results from different laboratories can vary substantially, reflecting the difficulty in measuring this analyte, a paucity of calibrators and lack of external quality assurance material37. Reference values can also vary significantly between laboratories. A comparison of the three most commonly used methods for measurement of plasma oxalate — GC-MS, ion chromatography–mass spectrometry (IC-MS) and oxalate oxidase — showed that IC-MS values were 33% lower than the GC-MS values but similar to those achieved with oxalate oxidase37. An explanation for these discrepancies may be that the oxalate oxidase and IC-MS methods both require deproteinization of the sample by ultrafiltration, causing reduced overall recovery.
Plasma oxalate levels increase with decreasing eGFR regardless of aetiology, but are typically higher in patients with PH38. Patients on dialysis who do not have PH can have average plasma oxalate levels of 50–60 µmol/l, although inter-individual differences and differences resulting from the method of assessment exist38,39,40,41,42. Plasma oxalate levels can decrease by half during a haemodialysis (HD) session41. Thus, especially in the era of RNAi therapies, evaluation of plasma oxalate levels in patients with PH1 who are undergoing dialysis should be compared with values from dialysis populations without hyperoxaluria rather than with those from healthy individuals.
Kidney stone analysis
Analyses of kidney stones in patients with PH1 have demonstrated that these are typically calcium oxalate monohydrate (whewellite) stones and have a peculiar morphology (white or pale yellow with a disorganized internal structure instead of brown with a radiating inner structure) reflecting their speed of formation43. Stones from patients with PH2 or PH3 frequently contain mixtures of calcium oxalate and calcium phosphate and therefore cannot be distinguished from those of idiopathic stone formers. Recognition of stone burden and their speed of recurrence are therefore more useful indicators of a metabolic cause in patients with PH2 or PH3 than analysis of stone composition.
Crystalluria
Crystalluria — that is the assessment of urinary calcium oxalate crystal volume and morphological analysis of urinary crystals — can be helpful in the diagnostic evaluation and assessment of therapeutic efficacy in stone formers in general44. The finding of >200 pure whewellite crystals per cubic millimetre in the urinary sediment is highly suggestive of PH1, especially in young children. The specificity of this finding is lower in the adult population. Nonetheless, this rapid, non-invasive and inexpensive test enables the rapid exclusion of other crystal species not normally found in the urine, such as cystine45,46. Oxalate crystal volume measurement can also be useful for post-transplantation monitoring in patients with PH1, as positive crystalluria indicates the risk of calcium oxalate deposits on the graft. The goal after transplantation is to achieve negative crystalluria or an oxalate crystal volume of <100 µm3/mm3 by means of hydration and other symptomatic measures47.
Therapy
Rationale for conservative therapy
Urine dilution is key to preventing the formation of calcium oxalate kidney stones in patients with PH48,49. A study of children with urolithiasis but without PH found that diuresis above 1 ml/kg/h nearly eliminated the risk of calcium oxalate supersaturation50; however, it is important to note that this advice accounts for non-PH stone formers and that this level of diuresis is probably insufficient to eliminate stone formation in patients with PH. To guarantee adequate urine dilution, the EAU advises a fluid intake of 3.5–4 l daily for adults and 1.5 l/m2 body surface area (BSA) for children with PH to achieve a urine volume of least 2.5 l per 24 h. As a fluid intake of 1.5 l/m2 BSA might not necessarily produce sufficient urine volume, we recommend a fluid intake of at least 2–3 l/m2 BSA for children with PH51. A gastrostomy tube may be indicated to meet this high fluid intake in infants. We recommend adapting fluid management to optimize urinary oxalate excretion as determined by a morning spot urine analysis. If possible, assessment of crystalluria can also be useful to monitor the efficacy of fluid management44.
Only one small cohort study has demonstrated a benefit of oral potassium citrate administration in a dosage of 0.1–0.15 g/kg in patients with PH52. Other studies have found that the use of urine alkalizers, including citrate, was not associated with improved renal outcomes in children with PH53. However, on the basis of the reasoning that citrate binds to calcium and may decrease calcium oxalate crystal formation, we do recommend including citrate in the therapeutic work-up of patients with PH.
Studies of the effect of dietary oxalate restriction are also contradictory. One case series from 2018 reported a 30–40% decrease in urinary oxalate excretion after restriction of dietary oxalate in two patients with PH, whereas another study failed to show beneficial effects of a low-oxalate diet23,54. Considering the impact of dietary oxalate restriction on quality of life, we do not recommend a low-oxalate diet, but we suggest limiting the intake of products that contain very high amounts of oxalate, such as spinach, rhubarb, chocolate and nuts.
Pyridoxine (vitamin B6) is effective in lowering urinary oxalate excretion in a subgroup of patients with PH1 (refs. 55,56,57,58,59). Pyridoxine responsiveness, defined as a >30% decrease in urinary oxalate excretion after at least 3 months of treatment with an optimal dose of pyridoxine28,60,61, is most often achieved in patients with p.Gly170Arg and p.Phe125Ile mutations; however, patients with other non-truncating genotypes, such as p.Gly41Arg mutation, may also (partially) respond56,58,62,63. We therefore recommend starting pyridoxine supplementation in all patients suspected to have PH and in all patients with genetically proven PH1. Earlier recommendations of dosages up to 20 mg/kg lack evidence. A 2005 study found no support for additional benefits of doses above 5 mg/kg55. As long-term, high-dose pyridoxine is potentially neurotoxic, we suggest administering a maximum of 5 mg/kg and only use higher doses in selected patients with close monitoring. We also recommend that all patients with PH1 are tested for pyridoxine responsiveness; urine oxalate measurements should be repeated on at least two occasions after at least 2 weeks of pyridoxine administration for evaluation of pyridoxine responsiveness, defined as a mean decrease of >30% between the two samples59. If responsive, the dose of pyridoxine should be gradually tapered to the lowest dose that maintains an optimal reduction in urinary oxalate level. Dose monitoring should each time be based on at least two assessments of urinary oxalate per dose. Frequent follow-up assessment of urine oxalate level is not indicated in patients who do not respond to pyridoxine and are not on RNAi therapy. However, in patients who do respond to pyridoxine, urine oxalate should be checked frequently until the adequate dose is determined. At later time points, urinary oxalate levels can be checked twice per year.
Indication and rationale for dialysis
Dialysis treatment may be indicated in patients with PH who have progressed to stage 4–5 CKD before the development of uraemia, depending on the potential risk of systemic oxalosis. Clinical indications for early onset of dialysis are high plasma oxalate levels despite oxalate-lowering therapy (RNAi therapy or pyridoxine) and signs of systemic oxalosis. Hard, direct evidence to support this statement is lacking, but circumstantial evidence suggests that oxalate storage is a key threat in patients with PH and stage 5 CKD, and that high plasma oxalate levels are indicative of tissue storage. In such patients, treatment should aim to minimize oxalate storage, and to achieve this goal, intensive dialysis may be necessary as a bridge to liver transplantation. Estimates of endogenous oxalate production in patients with PH1 range from 4 mmol daily to 7 mmol daily64, whereas mean oxalate removal rates with regular dialysis regimens are only 1.0–1.4 mmol daily65. Consequently, regular dialysis regimens are unable to counteract the high rate of oxalate production in these patients66 and systemic oxalate accumulation will continue. Liver transplantation should therefore be performed as soon as possible66. Mean, weekly rates of oxalate elimination are similar for conventional HD comprising thrice-weekly sessions and daily peritoneal dialysis (PD) (3.9 mmol/1.73 m² BSA and 3.5 mmol/1.73 m² BSA, respectively). However, the rate of oxalate removal per minute is significantly higher for HD than PD (mean 116 ml/min/1.73 m² BSA and 7 ml/min/1.73 m2, respectively)65. Moreover, a 2006 study found that administration of six, 4.5 h sessions of HD per week with a high-flux filter achieved the removal of 24 mmol/1.73 m2 per week, which approaches estimates of weekly oxalate production (28–37.7 mmol per week)64,66. We therefore recommend intensive HD over PD, preferably using a high-flux dialyser with maximal blood flow67. We suggest daily HD sessions if tolerated, combined with nocturnal PD if needed and tolerated, bearing in mind that such a dual approach to dialysis increases both the risk of infectious complications and the burden on the patient and carers. PD treatment can be optimized by adjusting the dwell time and dwell volume according to a peritoneal equilibration test. Increased numbers of cycles and exchanges per day while optimizing dwell time will result in greater removal of oxalate65,66.
For patients on HD, we suggest that pre-dialysis plasma oxalate levels are maintained at around 50–70 μmol/l, which is the concentration of plasma oxalate in patients on dialysis without PH, noting previous comments about assay differences in different populations. However, reference values for patients on dialysis without PH may vary between laboratories, and residual diuresis should be taken into account68 (E. Metry, unpublished work) (Supplementary Table 3). Oxalate accumulation is usually progressive in patients with PH on dialysis69, although one case report described the reversal of oxalosis with intensive dialysis70. For an intensive dialysis regimen, we recommend increasing the weekly number of sessions rather than prolonging the duration of each session, since the effectiveness of HD to remove oxalate decreases over the course of a session as a result of decreasing plasma oxalate66. This strategy has been shown to effectively lower pre-dialysis plasma oxalate values71. Specific attention should be given to phosphate control in patients receiving intensive dialysis, as hypophosphataemia may further worsen bone disease and mineralization defects in patients with PH. We propose a shared decision-making approach to dialysis, taking into account whether patients and their families could tolerate daily dialysis sessions with or without nocturnal PD.
Rationale for transplantation
Liver transplantation remains the only cure for PH1. A substantial body of evidence demonstrates that liver transplantation can reverse hyperoxaluria and prevent the further development of oxalate-related disease in patients with PH1. The native liver should be removed at transplantation. Auxiliary liver transplantation is regarded as an obsolete procedure as it will not adequately reduce oxalate overproduction72. Three reports that advocate the use of auxiliary liver transplantation have not — in our opinion — shown convincing data to support their conclusion73,74,75. One study described a pyridoxine-responsive patient in whom plasma oxalate dropped from 34.8 µmol/l while on dialysis to 3.6–8.3 µmol/l after transplantation; however, both values are normal when corrected for eGFR, and there is no mention of oxalate urinary excretion74. A second study did not mention oxalate values at all75, whereas the third reported lowering of plasma oxalate levels (by up to 65 µmol/l 6 months after auxiliary liver transplantation) but not normalization, and also does not mention oxalate excretion73. For patients with PH and kidney failure who undergo kidney transplantation, combined liver and kidney transplantation (CLKT) results in better kidney graft survival than isolated kidney transplantation (87% versus 14% at 15 years, P < 0.0); adjusted HR for graft failure 0.14 (95% CI 0.05–0.41), although patient survival is similar76,77,78. Three other high-quality studies have reported 5-year kidney graft survival rates of 48–89% for CLKT and 14–45% for kidney transplantation76,79,80,81. Data from 267 patients with PH1 from the OxalEurope registry who had undergone transplantation confirmed that event-free survival was better after CLKT than after isolated kidney transplantation among patients who were insensitive to pyridoxine therapy (P < 0.001)82 but not among pyridoxine-sensitive patients (P = 0.411). This finding supports a strategy for isolated kidney transplantation in selected patients who respond to pyridoxine with normalization or near normalization of urinary oxalate excretion. The OxalEurope registry study reported comparable outcomes for simultaneous and sequentially performed liver–kidney transplantation (n = 159 and n = 37, respectively). Twelve patients underwent pre-emptive liver transplantation with poor outcomes82, and we therefore cannot recommend this approach.
Data on the impact of liver transplantation among patients with PH2 are scarce owing to the rarity of the disease and the assumed better outcome of these patients compared with patients with PH1. However, a 2019 study of 101 patients with PH2 found that 22 patients reached stage 5 CKD at a median age of 40 (34–48) years of age. Ten of these 22 patients underwent isolated kidney transplantation, for which 1-year and 5-year cumulative kidney allograft survival (censored for death) were only 43% and 29%, respectively2. One of these ten patients underwent two deceased-donor kidney transplantations within 2 years, and three of the patients received a second kidney transplant 5, 6 and 22 years after the first graft. Two of the three patients died 3 and 5 years after the repeat kidney transplantation2. Only one patient underwent CLKT; however, the transplanted liver demonstrated primary graft non-function, and the patient died 1 year later as a result of sepsis2. Adverse outcomes of isolated kidney transplantation were also described in a case report of a rare paediatric patient with advanced CKD due to PH2 (ref. 83). However, two cases of successful CLKT in patients with PH2 have been reported. The first was a 44-year-old man who underwent CLKT with normalization of urinary oxalate and glycerate excretion and good graft function of both organs84. The second was a 41-year-old man who had a failed isolated kidney transplant owing to oxalate nephropathy but underwent subsequent successful CLKT with normalization of urinary oxalate excretion after 9 months85. By contrast, a 2022 report described a 26-year-old man in whom liver transplantation after a kidney graft did not reduce oxalate excretion or prevent kidney graft loss due to oxalate nephropathy86.
Rationale for urological management
As evidence for stone management specifically in patients with PH is lacking, we recommend that clinicians follow the treatment algorithms for urolithiasis as outlined in the EAU guidelines87. Several studies have highlighted the superiority of percutaneous nephrolithotomy (PCNL) over external shock wave lithotripsy (ESWL) for stone removal in patients with PH1 (refs. 88,89,90). In a cohort study in paediatric patients with PH, ESWL resulted in a stone-free rate of just 20% (1 of 5 interventions) in patients with PH1 and 47% (8 of 17) in patients with PH2 (ref. 88). Patients with PH2 may respond better than those with PH1 to ESWL treatment, as these patients tend to have mixed oxalate–phosphate stones, although the numbers of patients studied are too small for a reliable comparison of the two approaches. In a study of 54 urological procedures performed in 14 patients with PH1, use of 23 primary ureteroscopy (URS) procedures in 11 patients achieved a stone-free rate of 57%; by contrast, ten primary PCNL procedures resulted in an initial stone-free rate of 70%, which increased to 90% after the second PCNL. PCNL was most successful and achieved the highest stone-free survival rate. ESWL was only performed in eight cases (five patients) of acute obstructive stones with a success rate (defined as a >50% reduction in stone burden) of 63%89. Furthermore, two small case series in children with PH1 described a decline in kidney function after ESWL, although this decline might reflect the normal course of PH91,92.
The finding that PCNL is typically the most successful approach for reducing the symptomatic need for multiple treatments under general anaesthesia needs to be balanced with the risks of stricture with ureteroscopy and the better availability and lower cost of ESWL.
Hyperhydration should be continued at all times in the peri-operative period of any urological or other surgical procedure, which also implies that any form of pre-operative liquid fasting should be avoided93, as even mild dehydration can lead to acute kidney injury in patients with PH. Otherwise, current anaesthesiological guidelines should be followed. Monitoring of fluid status peri-operatively is strongly recommended.
Patients with PH should undergo frequent imaging of the kidneys after surgery. Although ultrasonography is the most commonly used imaging modality for these patients94, a study in a non-PH cohort showed that non-enhanced CT might be more sensitive than ultrasound for the identification of renal calculi95. In that study, 77 of 101 calculi identified by CT scan were missed by ultrasonography95. It is unclear to what extent this finding can be applied to patients with PH, given the lifelong nature of the condition and ongoing risk of recurrent stones even after periods of being stone free. One study found that the number of stones did not correlate with kidney function over time or the risk of kidney failure among patients with PH; however, the risk of kidney failure was higher among patients with PH with nephrocalcinosis after adjustment for stone numbers94.
To determine the frequency of monitoring we recommend that the EAU guidelines for high-risk patients are followed, with the exception that the patients should not be discharged from follow-up. After 2 years of 6-monthly imaging, follow-up with imaging on at least a yearly basis should be considered for all patients with PH on medical treatment for stone disease.
Rationale for the management of infantile oxalosis
Infantile oxalosis, defined as stage 5 CKD due to PH before the age of 1 year is the most severe form of PH1 and is characterized by oxalate depositions causing multi-organ failure39. Registry data from 2022 showed that 96% of these patients had signs of systemic oxalosis16. Infantile oxalosis has only been reported in children with PH1. Although patients with PH2 are as likely as those with PH1 to present with nephrocalcinosis and urolithiasis in infancy, progression to stage 5 CKD does not usually occur before 15 years of age2.
More than 50% of children with PH1 diagnosed in infancy present with stage 5 CKD10,96,97,98. The most common clinical features include poor feeding and failure to thrive. Some children may exhibit seizures due to electrolyte disturbances in advanced CKD. Moreover, a large proportion of infants exhibit significant nephrocalcinosis and/or urolithiasis.
This group of patients is particularly challenging in terms of medical management, given issues relating to dialysis access, the need for tailored dietary prescription and tube feeding to meet nutritional targets, the frequent onset of fractures and the presence of electrolyte disturbances resulting from an intensified dialysis regimen. Therefore, these patients should be managed in highly specialized paediatric nephrology centres with expertise in dialysis and solid organ transplantation in small children, and with access to multidisciplinary care. To avoid unnecessary radiation exposure, bone X-rays should be performed only in case of bone symptoms, as proposed in general paediatric guidelines on bone impairment in kidney failure99.
New therapies
Until very recently, treatment of PH1 was supportive, burdensome to patients and only partly effective. Even good compliance with hyperhydration and citrate therapy cannot prevent the development of kidney failure in patients with PH1. New therapies, particularly those based on RNAi, have shown promise in reducing oxalate production in patients with PH1, at least in the short term. Emerging data that demonstrate clinical efficacy suggest that these drugs may indeed revolutionize the management of PH1 in the near future.
Indication and rationale for the uses of RNAi therapies
Two RNAi therapies are now available or under trial for patients with PH1 (Fig. 1d). Lumasiran (Oxlumo; Alnylam) has received marketing authorization by the EMA and FDA as an orphan drug for the treatment of PH1. Lumasiran is designed to silence the gene that encodes the enzyme glycolate oxidase, which catalyses the conversion of glycolate into glyoxylate. In the Illuminate A RCT of 39 patients with PH1, patients aged >6 years on lumasiran showed a mean reduction in urinary oxalate excretion of 65% compared with 11% in patients treated with placebo (P < 0.001). Of patients who received lumasiran, 84% had urinary oxalate levels below 1.5 times the upper level of reference values after 6 months of treatment, whereas no patient in the placebo group achieved a similar reduction. Fifty-two per cent of patients on lumasiran showed normalization of urinary oxalate excretion26. An extension study showed a sustained response after 12 months of follow-up100. In Illuminate B — an open label study of 18 children aged <6 years, including infants — lumasiran treatment was associated with a least-squares reduction in the spot urinary oxalate-to-creatinine ratio of 72% within 6 months of treatment, and 50% of patients achieved a urine oxalate-to-creatinine ratio within 1.5 times of the upper limit of normal101. In Illuminate C — an open label trial of 21 patients with eGFR <45 ml/min/1.73 m2 lumasiran treatment was associated with an average decrease in plasma oxalate level of 42% (95% CI 34–51%) after 6 months of treatment among patients on dialysis (n = 15) and 33% (95% CI −15 to −82%) in patients not receiving dialysis102. The adverse effects in all trials were minor (injection site reactions).
Nedosiran (Dicerna/Novo Nordisk) is another RNAi drug, which is designed to inhibit the production of l-lactate dehydrogenase A (LDHA), which is essential for the cytosolic conversion of glyoxylate into oxalate. In theory, this mechanism of action should make it effective for all types of PH. In the open label, phase I PHYOX 1 study of patients with PH1 or PH2, nedosiran was associated with an average 55% reduction in urine oxalate level and a lowering of urinary oxalate excretion to <1.5 the upper limit of normal in 67% of patients103. However, a sub-analysis of patients with PH2 showed no effect in these patients. Similarly, the PHYOX 2 RCT, which included patients with PH1 or PH2 and eGFR >30 ml/min/1.73 m2 reported a 59% reduction in urinary oxalate level with nedosiran treatment in patients with PH1, but no significant response in patients with PH2 (ref. 104); 81% of patients with PH1 achieved normalized or near normalized (<1.5 times the upper limit of normal) urinary oxalate excretion after 6 months of treatment. The lack of response in patients with PH2 may reflect the wide tissue distribution of GRHPR and the consequential systemic nature of PH2, which is difficult to target with a liver-specific therapeutic such as nedosiran. Only one case report has described the use of nedosiran in a patient with dialysis-dependent PH1. In this patient, nedosiran significantly reduced plasma oxalate levels105. No data are yet available on the effects of nedosiran in patients with PH3.
Impact of RNAi therapy on clinical disease and management
Lumasiran and nedosiran have the potential to markedly improve outcomes of patients with PH1. However, so far, insights into the impact of these agents on the clinical phenotype of PH1 — that is, on kidney stone recurrence, regression of nephrocalcinosis and, most importantly, the further deterioration of kidney function — can be gleaned only from case reports and short-term studies.
Six-month outcome data from the Illuminate C study show a trend towards a lower rate of kidney stone events among patients on lumasiran than among those in the placebo group, with no worsening of nephrocalcinosis102. This effect was maintained in the 12-month extension study, which also showed an improvement in nephrocalcinosis among those on lumasiran106. Kidney function remained stable in both the placebo and treatment groups in the extension study. A low titre of antidrug antibodies was found in one patient on lumasiran therapy. This finding must be followed up, as antibody formation could hamper the efficacy of the drug. Preliminary data from patients with lumasiran and preserved kidney function after 30–48 months of treatment with lumasiran support the notion that the drug positively impacts the clinical course of disease by preventing a decline in kidney function and the occurrence of new stones (E. Metry, unpublished work).
To what extent these drugs might replace the need for liver transplantation is unclear at present. One report of an adult patient who underwent an isolated kidney transplantation while receiving lumasiran therapy described an increase in serum creatinine level from 140 µmol/l to 240 µmol/l after transplantation. A kidney biopsy at the time revealed acute rejection and signs of oxalate nephropathy. The creatinine level dropped to 169 μmol/l after 10 weeks of anti-rejection therapy, and plasma oxalate levels dropped to 21 μmol/l107, suggesting that oxalate nephropathy had not been the reason for the temporary decline in kidney function108. Another case report described the use of lumasiran after kidney transplantation in a 5-year-old with a missed diagnosis of PH1. Graft function was maintained with a combination of intensive kidney replacement therapy and lumasiran, which suggests that this drug could potentially replace liver transplantation in selected patients with PH1 (ref. 109). A series of five patients with PH1 and isolated kidney transplantation also did not find any evidence of oxalate graft nephropathy in any of the patients treated with lumasiran110. Given the paucity of current knowledge, the decision not to perform liver transplantation in a patient with PH1 and stage 5 CKD on lumasiran should be taken carefully, with careful consideration of the specific aspects of each case and with extensive monitoring of the patient. For example, if a decision is made to perform isolated kidney transplantation and initiate lumasiran therapy, it is important that all possible precautions are taken to prevent the development of oxalate graft nephropathy from potentially stored oxalate, such as through prolonged intensive hyperhydration and alkalinization as soon as possible during the post-operative period, as lumasiran has no influence on previously systemically stored oxalate107. Indeed, patients undergoing CKLT can continue to release oxalate from bone for months or even years after transplantation111.
In summary, both RNAi therapies have been shown to be highly effective in lowering endogenous oxalate production in patients with PH1, and early clinical outcome data are encouraging. However, more follow-up data are warranted to fully appreciate the effect of these drugs on reducing stone disease and preventing kidney failure.
Stiripentol
Stiripentol is an LDHA-targeted oral commercial medication for the genetic epileptic encephalopathy Dravet syndrome. Of note, patients with Dravet syndrome typically have lower urinary oxalate excretion than healthy individuals. One report of a patient with PH1 and good kidney function described a significant reduction in urine oxalate level after 10 weeks of treatment112. In another report of an 18-month-old patient with PH1 characterized by a pyridoxine-responsive mutation, administration of stiripentol led to a significant reduction in urinary oxalate level to normal values113. However, other case reports — albeit in patients with advanced kidney failure — found no benefit of stiripentol therapy114,115. A trial is currently underway to determine the efficacy of stiripentol as monotherapy in patients aged >6 months with PH1–3 and an eGFR of >45 ml/min/1.73 m2 (ref. 116).
Management of PH in low-resource countries
The burden of PH varies between regions and is dependent on several factors, including its prevalence, its rate of early detection — which is determined by its recognition by health-care professionals and the availability of affordable diagnostic tools — and on access to therapeutic resources, including intensive dialysis regimens, transplantation and novel therapies117. High rates of consanguinity — which has been linked to socioeconomic status — may also result in a higher prevalence of rare inherited kidney diseases, such as PH1, in some regions12. Delayed diagnosis, the lack of availability of diagnostic tools and of therapeutic modalities or resources add to the challenge of diagnosing and managing PH118 and widen the health and life-expectancy gap between high-income and low-income regions. Low- and middle-income countries (LMICs) are therefore encouraged to act by promoting awareness of PH among physicians through a high index of clinical suspicion to enable early diagnosis and timely medical management, including testing of pyridoxine responsiveness. Screening of patients with dialysis and stage 5 CKD of unknown aetiology should also be encouraged, using available resources to diagnose cortical and medullary nephrocalcinosis that could otherwise be missed or inaccurately labelled as hyperechogenic or atrophic kidneys. Such an approach should avoid catastrophic diagnosis of PH after kidney transplantation, which is associated with a rate of early graft failure of >75% and serious comorbidities, and is relatively common in low-resource countries119.
The frequency of late diagnosis is reflected by the fact that the most common presentation of PH1 in LMICs is stage 5 CKD, especially in those aged <5 years96; 16–65% of patients with PH1 present with kidney failure96,120,121, and infantile oxalosis is particularly common in some regions96. No epidemiological data exist on PH2 and PH3 in LMICs.
The genetic landscape of PH1 in LMICs varies according to the geographical region96,120,121,122,123,124,125,126. Diagnosis is challenging, and diverse diagnostic tools are often unavailable12,96. However, ultrasound and X-ray are often available, inexpensive and provide valuable information on the extent of stone disease or nephrocalcinosis and on bone disease due to systemic oxalate and should be performed in all patients suspected to have PH. Proper measurement of 24 h urine oxalate and urine calcium are mandatory and should be repeated at least once, but preferably twice for diagnosis. Assessment of plasma oxalate is often not performed, but is extremely important in patients with CKD stage 4 or higher and concurrent nephrocalcinosis to establish or rule out a diagnosis of PH. Biochemical analysis to determine the presence of calcium oxalate monohydrate stones is recommended if available43. Facilities for genetic diagnosis of PH and PH type are lacking in most LMICs. The establishment of collaborative research programmes with institutions from high-resource countries might be a solution to address this problem and facilitate genetic diagnostics. In addition, advances in next generation sequencing technologies may lead to the availability of affordable genetic diagnosis and/or screening of PH1 in communities where it is prevalent in the future.
Conservative treatment is of paramount importance to prevent the formation of further stones and decline of renal function, particularly in regions with hot climates. Hydration, including the insertion of gastrostomy tube in infants, use of crystallization inhibitors and pyridoxine are recommended once the diagnosis is suspected, particularly in these settings127. Health systems in LMICs should focus on ensuring early access to such low-cost measures to avoid subsequent additional costs, morbidity including disability and to reduce mortality risk.
A key challenge in some LMICs is a lack of dialysis service and/or a lack of resources to finance intensive dialysis, which contributes to the poor outcome of patients with PH1 and stage 5 CKD. Access to organ transplantation, particularly liver transplantation, is also resource dependent and lacking in many LMICs. The limited possibilities for intensive dialysis and liver transplantation in LMICS makes access to the novel RNAi therapies even more important, given their relative ease of administration. Improved access to these medications might be a game changer in improving PH1 care and reducing global health-care disparities128. Implementation of differential pricing is a requirement for this to happen.
Unanswered questions and research agenda
Several topics require further research (Box 3), some of which have the potential to directly affect the management of patients with PH.
The impact of RNAi therapies on current management strategies
Although the two available RNAi therapies look promising, further research is needed to determine the extent to which they improve clinical outcomes. The clinical efficacy of these therapies in terms of reduction in stone disease, prevention of kidney failure and prevention of systemic oxalosis in patients with kidney failure at the start of therapy, as well as their cost-effectiveness, require careful study. One of the most urgent questions is whether and under what conditions RNAi therapy can safely replace liver transplantation in patients with PH1 and kidney failure and to what extent it may reduce the need for intensified dialysis regimens.
Pyridoxine responsiveness in PH1
More data are warranted to identify the factors that determine the responses to pyridoxine of patients with PH1 and point mutations other than p.Gly170Arg and p.Phe152Ile.
Other potential new therapies
New approaches for the reduction of oxalate and its substrate, such as CRISPR–Cas9-induced knockout of glycolate oxidase and LDHA, or the use of CHK-336, an oral LDHA inhibitor that is currently in phase I trials, may lead to new therapies for PH in the near future. Chaperone therapeutics are another promising approach to restore enzyme function and reduce oxalate levels. Some of these drugs, such as pyridoxal phosphate or dequalinium chloride (DECA), may provide benefits in patients with PH1 (ref. 129). DECA is FDA approved for the treatment of bacterial vaginosis; in a cell model of PH1 characterized by a pro11G170Arg mutation in AGXT, DECA promoted the peroxisomal retargeting of AGT and enhanced the effect of simultaneously administered pyridoxine.
Interpretation of plasma oxalate as biomarker for response to therapy
The management of patients with PH1 who present with stage 5 CKD can be challenging, as there are no good reference values for plasma oxalate level in stage 5 CKD and as systemic oxalate deposition can mask the clinical response of oxalate-lowering therapies. The in vivo measurement of endogenous oxalate and glycine production from glycolate by infusion of labelled glycolate and oxalate may be useful in this regard130. This method could potentially prevent unnecessary liver transplantation in patients with PH1 and stage 5 CKD who are fully sensitive to pyridoxine, as has been shown in two cases, although this possibility requires confirmation in further studies (S. Garrelfs, unpublished work).
The pathophysiology and management of PH2 and PH3
No specific therapies are currently available for the two rarest types of PH — PH2 and PH3. A key hurdle for the development of effective drugs is the fact that the pathophysiology of these diseases remains poorly understood. However, available evidence suggests that PH2 is less benign than previously thought and currently, the exact management strategy for patients with PH2 and stage 5 CKD5 remains unclear.
Conclusions
For a long time, PH has been an extremely challenging disease for physicians. Owing to its rarity, diagnostic hurdles and heterogeneity in phenotype, diagnosis was often established only in patients with advanced disease and kidney failure. New insights into the disease course for all three subtypes and into the outcomes of different transplantation strategies in pyridoxine-sensitive and non-sensitive patients with PH1, and the introduction of promising RNAi therapies for PH1, have changed the existing paradigms in management of PH. We intend these clinical practice recommendations to guide physicians in this new era for PH. Further developments in the near future will determine the extent to which RNAi therapies will improve the long-term outcomes for patients with PH and whether these developments may serve as a basis for the emergence of new therapies.
References
Cochat, P. et al. Primary hyperoxaluria type 1: indications for screening and guidance for diagnosis and treatment. Nephrol. Dial. Transpl. 27, 1729–1736 (2012).
Garrelfs, S. F. et al. Patients with primary hyperoxaluria type 2 have significant morbidity and require careful follow-up. Kidney Int. 96, 1389–1399 (2019).
Martin-Higueras, C. et al. A report from the European Hyperoxaluria Consortium (OxalEurope) registry on a large cohort of patients with primary hyperoxaluria type 3. Kidney Int. 100, 621–635 (2021).
Steering Committee on Quality Improvement Management. Classifying recommendations for clinical practice guidelines. Pediatrics 114, 874–877 (2004).
Linstone, H. A. & Turoff, M. The Delphi Method (Addison-Wesley, 1975).
Mandrile, G. et al. Genetic assessment in primary hyperoxaluria: why it matters. Pediatr. Nephrol. https://doi.org/10.1007/s00467-022-05613-2 (2022).
ACGS. Best Practice Guidelines https://www.acgs.uk.com/quality/best-practice-guidelines/ (2022)
Rumsby, G., Williams, E. & Coulter-Mackie, M. B. Evaluation of mutation screening as a first line test for the diagnosis of the primary hyperoxalurias. Kidney Int. 66, 959–963 (2004).
Hopp, K. et al. Phenotype-genotype correlations and estimated carrier frequencies of primary hyperoxaluria. J. Am. Soc. Nephrol. 26, 2559–2570 (2015).
Harambat, J. et al. Genotype-phenotype correlation in primary hyperoxaluria type 1: the p.Gly170Arg AGXT mutation is associated with a better outcome. Kidney Int. 77, 443–449 (2010).
Zhao, F. et al. Characteristics of the genotype and phenotype in Chinese primary hyperoxaluria type 1 populations. Urolithiasis 49, 17–25 (2021).
Talati, J. J. et al. Primary hyperoxaluria in populations of Pakistan origin: results from a literature review and two major registries. Urolithiasis 46, 187–195 (2018).
Belostotsky, R. et al. Mutations in DHDPSL are responsible for primary hyperoxaluria type III. Am. J. Hum. Genet. 87, 392–399 (2010).
Fang, X. et al. Nine novel HOGA1 gene mutations identified in primary hyperoxaluria type 3 and distinct clinical and biochemical characteristics in Chinese children. Pediatr. Nephrol. 34, 1785–1790 (2019).
Mandrile, G. et al. Data from a large European study indicate that the outcome of primary hyperoxaluria type 1 correlates with the AGXT mutation type. Kidney Int. 86, 1197–1204 (2014).
Deesker, L. J. et al. Improved outcome of infantile oxalosis over time in Europe: data from the OxalEurope Registry. Kidney Int. Rep. 7, 1608–1618 (2022).
Singh, P. et al. Clinical characterization of primary hyperoxaluria type 3 in comparison to types 1 and 2: a retrospective cohort study. Nephrol. Dial. Transplant. 37, 869–875 (2022).
van Woerden, C. S., Huidekoper, H. H., Groothoff, J. W., Wijburg, F. A. & Duran, M. Postponing urine acidification for 24 h does not change the oxalate concentration. Clin. Chim. Acta 384, 184–185 (2007).
Mazzachi, B. C., Teubner, J. K. & Ryall, R. L. Factors affecting measurement of urinary oxalate. Clin. Chem. 30, 1339–1343 (1984).
Ferraro, P. M. et al. Estimating 24-hour urinary excretion using spot urine measurements in kidney stone formers. Nephrol. Dial. Transplant. 37, 2171–2179 (2022).
Clifford-Mobley, O., Tims, C. & Rumsby, G. The comparability of oxalate excretion and oxalate:creatinine ratio in the investigation of primary hyperoxaluria:a review of data from a referral centre. Ann. Clin. Biochem. 52, 113–121 (2015).
Hoppe, B. et al. Influence of nutrition on urinary oxalate and calcium in preterm and term infants. Pediatr. Nephrol. 11, 687–690 (1997).
Siener, R., Hoppe, B., Löhr, P., Müller, S. C. & Latz, S. Metabolic profile and impact of diet in patients with primary hyperoxaluria. Int. Urol. Nephrol. 50, 1583–1589 (2018).
Witting, C. et al. Pathophysiology and treatment of enteric hyperoxaluria. Clin. J. Am. Soc. Nephrol. 16, 487–495 (2021).
Borghi, L. et al. Comparison of two diets for the prevention of recurrent stones in idiopathic hypercalciuria. N. Engl. J. Med. 346, 77–84 (2002).
Garrelfs, S. F. et al. Lumasiran, an RNAi therapeutic for primary hyperoxaluria type 1. N. Engl. J. Med. 384, 1216–1226 (2021).
Balchin, Z. E., Moss, P. A. & Fraser, C. G. Biological variation of urinary oxalate in different specimen types. Ann. Clin. Biochem. 28, 622–623 (1991).
Clifford-Mobley, O., Sjogren, A., Lindner, E. & Rumsby, G. Urine oxalate biological variation in patients with primary hyperoxaluria. Urolithiasis 44, 333–337 (2016).
Lumlertgul, N., Siribamrungwong, M., Jaber, B. L. & Susantitaphong, P. Secondary oxalate nephropathy: a systematic review. Kidney Int. Rep. 3, 1363–1372 (2018).
Marshall, D. J., Adaway, J. E. & Keevil, B. G. A combined liquid chromatography tandem mass spectrometry assay for the quantification of urinary oxalate and citrate in patients with nephrolithiasis. Ann. Clin. Biochem. 55, 461–468 (2018).
Frishberg, Y., Zeharia, A., Lyakhovetsky, R., Bargal, R. & Belostotsky, R. Mutations in HAO1 encoding glycolate oxidase cause isolated glycolic aciduria. J. Med. Genet. 51, 526–529 (2014).
McGregor, T. L. et al. Characterising a healthy adult with a rare HAO1 knockout to support a therapeutic strategy for primary hyperoxaluria. eLife 9, e54363 (2020).
Clifford-Mobley, O. et al. Glycolate oxidase deficiency in a patient with congenital hyperinsulinism and unexplained hyperoxaluria. Pediatr. Nephrol. 32, 2159–2163 (2017).
Clifford-Mobley, O., Hewitt, L. & Rumsby, G. Simultaneous analysis of urinary metabolites for preliminary identification of primary hyperoxaluria. Ann. Clin. Biochem. 53, 485–494 (2016).
Ventzke, A. et al. Systematic assessment of urinary hydroxy-oxo-glutarate for diagnosis and follow-up of primary hyperoxaluria type III. Pediatr. Nephrol. 32, 2263–2271 (2017).
Woodward, G., Pryke, R., Hoppe, B. & Rumsby, G. Rapid liquid chromatography tandem mass-spectrometry screening method for urinary metabolites of primary hyperoxaluria. Ann. Clin. Biochem. 56, 232–239 (2019).
Stokes, F. et al. Plasma oxalate: comparison of methodologies. Urolithiasis 48, 473–480 (2020).
Perinpam, M. et al. Plasma oxalate in relation to eGFR in patients with primary hyperoxaluria, enteric hyperoxaluria and urinary stone disease. Clin. Biochem. 50, 1014–1019 (2017).
Cochat, P. & Rumsby, G. Primary hyperoxaluria. N. Engl. J. Med. 369, 649–658 (2013).
Marangella, M. et al. Serum calcium oxalate saturation in patients on maintenance haemodialysis for primary hyperoxaluria or oxalosis-unrelated renal diseases. Clin. Sci. 81, 483–490 (1991).
Hoppe, B. et al. Plasma calcium oxalate supersaturation in children with primary hyperoxaluria and end-stage renal failure. Kidney Int. 56, 268–274 (1999).
Pfau, A. et al. Assessment of plasma oxalate concentration in patients with CKD. Kidney Int. Rep. 5, 2013–2020 (2020).
Daudon, M., Jungers, P. & Bazin, D. Peculiar morphology of stones in primary hyperoxaluria. N. Engl. J. Med. 359, 100–102 (2008).
Daudon, M. & Jungers, P. Clinical value of crystalluria and quantitative morphoconstitutional analysis of urinary calculi. Nephron Physiol. 98, p31–p36 (2004).
Servais, A. et al. Cystinuria: clinical practice recommendation. Kidney Int. 99, 48–58 (2021).
Daudon, M., Hennequin, C., Boujelben, G., Lacour, B. & Jungers, P. Serial crystalluria determination and the risk of recurrence in calcium stone formers. Kidney Int. 67, 1934–1943 (2005).
Jouvet, P. et al. Crystalluria: a clinically useful investigation in children with primary hyperoxaluria post-transplantation. Kidney Int. 53, 1412–1416 (1998).
Pak, C. Y., Sakhaee, K., Crowther, C. & Brinkley, L. Evidence justifying a high fluid intake in treatment of nephrolithiasis. Ann. Intern. Med. 93, 36–39 (1980).
Porowski, T. et al. Upper metastable limit osmolality of urine as a predictor of kidney stone formation in children. Urolithiasis 47, 155–163 (2019).
Lande, M. B., Varade, W., Erkan, E., Niederbracht, Y. & Schwartz, G. J. Role of urinary supersaturation in the evaluation of children with urolithiasis. Pediatr. Nephrol. 20, 491–494 (2005).
Skolarikos, A. et al. Metabolic evaluation and recurrence prevention for urinary stone patients: EAU guidelines. Eur. Urol. 67, 750–763 (2015).
Leumann, E., Hoppe, B. & Neuhaus, T. Management of primary hyperoxaluria: efficacy of oral citrate administration. Pediatr. Nephrol. 7, 207–211 (1993).
Fargue, S. et al. Effect of conservative treatment on the renal outcome of children with primary hyperoxaluria type 1. Kidney Int. 76, 767–773 (2009).
Sikora, P. et al. [13C2]oxalate absorption in children with idiopathic calcium oxalate urolithiasis or primary hyperoxaluria. Kidney Int. 73, 1181–1186 (2008).
Monico, C. G., Rossetti, S., Olson, J. B. & Milliner, D. S. Pyridoxine effect in type I primary hyperoxaluria is associated with the most common mutant allele. Kidney Int. 67, 1704–1709 (2005).
Monico, C. G., Olson, J. B. & Milliner, D. S. Implications of genotype and enzyme phenotype in pyridoxine response of patients with type I primary hyperoxaluria. Am. J. Nephrol. 25, 183–188 (2005).
Milliner, D. S., Eickholt, J. T., Bergstralh, E. J., Wilson, D. M. & Smith, L. H. Results of long-term treatment with orthophosphate and pyridoxine in patients with primary hyperoxaluria. N. Engl. J. Med. 331, 1553–1558 (1994).
Hoyer-Kuhn, H. et al. Vitamin B6 in primary hyperoxaluria I: first prospective trial after 40 years of practice. Clin. J. Am. Soc. Nephrol. 9, 468–477 (2014).
Hoppe, B., Latta, K., von Schnakenburg, C. & Kemper, M. J. Primary hyperoxaluria–the German experience. Am. J. Nephrol. 25, 276–281 (2005).
Hoppe, B., Beck, B. B. & Milliner, D. S. The primary hyperoxalurias. Kidney Int. 75, 1264–1271 (2009).
Leumann, E. & Hoppe, B. The primary hyperoxalurias. J. Am. Soc. Nephrol. 12, 1986–1993 (2001).
Singh, P. et al. Pyridoxine responsiveness in a type 1 primary hyperoxaluria patient with a rare (atypical) AGXT gene mutation. Kidney Int. Rep. 5, 955–958 (2020).
van Woerden, C. S. et al. Clinical implications of mutation analysis in primary hyperoxaluria type 1. Kidney Int. 66, 746–752 (2004).
Marangella, M., Petrarulo, M., Cosseddu, D., Vitale, C. & Linari, F. Oxalate balance studies in patients on hemodialysis for type I primary hyperoxaluria. Am. J. Kidney Dis. 19, 546–553 (1992).
Hoppe, B. et al. Oxalate elimination via hemodialysis or peritoneal dialysis in children with chronic renal failure. Pediatr. Nephrol. 10, 488–492 (1996).
Illies, F., Bonzel, K. E., Wingen, A. M., Latta, K. & Hoyer, P. F. Clearance and removal of oxalate in children on intensified dialysis for primary hyperoxaluria type 1. Kidney Int. 70, 1642–1648 (2006).
Franssen, C. F. Oxalate clearance by haemodialysis–a comparison of seven dialysers. Nephrol. Dial. Transpl. 20, 1916–1921 (2005).
Ogawa, Y. et al. Calcium oxalate saturation in dialysis patients with and without primary hyperoxaluria. Urol. Res. 34, 12–16 (2006).
Marangella, M. et al. Bony content of oxalate in patients with primary hyperoxaluria or oxalosis-unrelated renal failure. Kidney Int. 48, 182–187 (1995).
Díaz, C. et al. Long daily hemodialysis sessions correct systemic complications of oxalosis prior to combined liver-kidney transplantation: case report. Ther. Apher. Dial. 8, 52–55 (2004).
Tang, X. et al. Oxalate quantification in hemodialysate to assess dialysis adequacy for primary hyperoxaluria. Am. J. Nephrol. 39, 376–382 (2014).
Trotter, J. F. & Milliner, D. Auxiliary liver transplant is an ineffective treatment of primary hyperoxaluria. Am. J. Transpl. 14, 241 (2014).
Knotek, M. et al. Combined auxiliary split liver and kidney transplantation for type I primary hyperoxaluria and end-stage kidney disease. Nephrology 19, 814–815 (2014).
Onaca, N. et al. Cadaveric orthotopic auxiliary split liver transplantation and kidney transplantation: an alternative for type 1 primary hyperoxaluria. Transplantation 80, 421–424 (2005).
Elias, N. et al. Native portal vein embolization for persistent hyperoxaluria following kidney and auxiliary partial liver transplantation. Am. J. Transpl. 13, 2739–2742 (2013).
Metry, E. L. et al. Transplantation outcomes in patients with primary hyperoxaluria: a systematic review. Pediatr. Nephrol. 36, 2217–2226 (2021).
Compagnon, P. et al. Long-term results of combined liver-kidney transplantation for primary hyperoxaluria type 1: the French experience. Liver Transpl. 20, 1475–1485 (2014).
Monico, C. G. & Milliner, D. S. Combined liver-kidney and kidney-alone transplantation in primary hyperoxaluria. Liver Transpl. 7, 954–963 (2001).
Harambat, J. et al. Characteristics and outcomes of children with primary oxalosis requiring renal replacement therapy. Clin. J. Am. Soc. Nephrol. 7, 458–465 (2012).
Bergstralh, E. J. et al. Transplantation outcomes in primary hyperoxaluria. Am. J. Transpl. 10, 2493–2501 (2010).
Cibrik, D. M., Kaplan, B., Arndorfer, J. A. & Meier-Kriesche, H. U. Renal allograft survival in patients with oxalosis. Transplantation 74, 707–710 (2002).
Metry, E. L. et al. Long-Term transplantation outcomes in patients with primary hyperoxaluria type 1 included in the European Hyperoxaluria Consortium (OxalEurope) registry. Kidney Int. Rep. 7, 210–220 (2022).
Naderi, G., Latif, A., Tabassomi, F. & Esfahani, S. T. Failure of isolated kidney transplantation in a pediatric patient with primary hyperoxaluria type 2. Pediatr. Transpl. 18, E69–E73 (2014).
Dhondup, T., Lorenz, E. C., Milliner, D. S. & Lieske, J. C. Combined liver-kidney transplantation for primary hyperoxaluria type 2: a case report. Am. J. Transpl. 18, 253–257 (2018).
Del Bello, A., Cointault, O., Delas, A. & Kamar, N. Primary hyperoxaluria type 2 successfully treated with combined liver-kidney transplantation after failure of isolated kidney transplantation. Am. J. Transpl. 20, 1752–1753 (2020).
Jia, Z., Zhong, Q., Lin, T. & Song, T. Subsequent liver transplantation did not reverse recurrence of oxalate nephropathy after isolated kidney transplantation for primary type 2 hyperoxaluria. Asian J. Surg. 45, 483–485 (2022).
EAU Guidelines. Edn. presented at the EAU Annual Congress Amsterdam (2022).
Al-Abadi, E. & Hulton, S. A. Extracorporal shock wave lithotripsy in the management of stones in children with oxalosis–still the first choice? Pediatr. Nephrol. 28, 1085–1089 (2013).
Carrasco, A. Jr, Granberg, C. F., Gettman, M. T., Milliner, D. S. & Krambeck, A. E. Surgical management of stone disease in patients with primary hyperoxaluria. Urology 85, 522–526 (2015).
Kamoun, A. et al. Primary hyperoxaluria: Tunisian experience apropos of 24 pediatric cases [French]. Nephrologie 18, 59–64 (1997).
Kamoun, A. et al. Value of extracorporeal shockwave lithotripsy in primary hyperoxaluria type I [French]. Arch. Pediatr. 2, 747–749 (1995).
Boddy, S. A., Duffy, P. G., Barratt, T. M. & Whitfield, H. N. Hyperoxaluria and renal calculi in children: the role of extracorporeal shock wave lithotripsy. J. R. Soc. Med. 81, 604–605 (1988).
Frykholm, P. et al. Pre-operative fasting in children: a guideline from the European Society of Anaesthesiology and Intensive Care. Eur. J. Anaesthesiol. 39, 4–25 (2022).
Tang, X. et al. Nephrocalcinosis is a risk factor for kidney failure in primary hyperoxaluria. Kidney Int. 87, 623–631 (2015).
Vrtiska, T. J. Quantitation of stone burden: imaging advances. Urol. Res. 33, 398–402 (2005).
Soliman, N. A. et al. Clinical spectrum of primary hyperoxaluria type 1: experience of a tertiary center. Nephrol. Ther. 13, 176–182 (2017).
Jellouli, M. et al. Primary hyperoxaluria in infants. Saudi J. Kidney Dis. Transpl. 27, 526–532 (2016).
van der Hoeven, S. M., van Woerden, C. S. & Groothoff, J. W. Primary hyperoxaluria type 1, a too often missed diagnosis and potentially treatable cause of end-stage renal disease in adults: results of the Dutch cohort. Nephrol. Dial. Transpl. 27, 3855–3862 (2012).
Bakkaloglu, S. A. et al. Bone evaluation in paediatric chronic kidney disease: clinical practice points from the European Society for Paediatric Nephrology CKD-MBD and Dialysis working groups and CKD-MBD working group of the ERA-EDTA. Nephrol. Dial. Transpl. 36, 413–425 (2021).
Devresse, A. et al. 12-Month analysis of ILLUMINATE-A, a phase 3 study of lumasiran: sustained oxalate lowering and kidney stone event rates in primary hyperoxaluria type 1. Presented at Belgian society of Nephrology (2021).
Sas, D. J. et al. Phase 3 trial of lumasiran for primary hyperoxaluria type 1: a new RNAi therapeutic in infants and young children. Genet. Med. 24, 654–662 (2022).
Michael, M. et al. Lumasiran for advanced primary hyperoxaluria type 1: phase 3 ILLUMINATE-C trial. Am. J. Kidney Dis. https://doi.org/10.1053/j.ajkd.2022.05.012 (2022).
Hoppe, B. et al. Safety, pharmacodynamics, and exposure-response modeling results from a first-in-human phase 1 study of nedosiran (PHYOX1) in primary hyperoxaluria. Kidney Int. 101, 626–634 (2022).
Baum, M. A. et al. PHYOX2: a pivotal randomized study of nedosiran in primary hyperoxaluria type 1 or 2. Kidney Int. https://doi.org/10.1016/j.kint.2022.07.025 (2022).
Shee, K. et al. Nedosiran dramatically reduces serum oxalate in dialysis-dependent primary hyperoxaluria 1: a compassionate use case report. Urology 156, e147–e149 (2021).
Hulton, S. A. et al. Randomized clinical trial on the long-term efficacy and safety of lumasiran in patients with primary hyperoxaluria type 1. Kidney Int. Rep. 7, 494–506 (2022).
Joher, N. et al. Early post-transplant recurrence of oxalate nephropathy in a patient with primary hyperoxaluria type 1, despite pretransplant lumasiran therapy. Kidney Int. 101, 185–186 (2022).
Metry, E. L., Oosterveld, M. J. S., Groothoff, J. W. & Bacchetta, J. The appearance of oxalate crystals in a kidney biopsy is no proof of post-transplant oxalate nephropathy in primary hyperoxaluria type 1. Kidney Int. 102, 446 (2022).
Stone, H. K. et al. Primary hyperoxaluria diagnosed after kidney transplant: a review of the literature and case report of aggressive renal replacement therapy and lumasiran to prevent allograft loss. Am. J. Transpl. 21, 4061–4067 (2021).
Sellier-Leclerc, A. L. et al. Isolated kidney transplantation under lumasiran therapy in primary hyperoxaluria type 1: a report of 5 cases. Nephrol. Dial. Transpl. https://doi.org/10.1093/ndt/gfac295 (2022).
Duclaux-Loras, R. et al. Pediatric combined liver-kidney transplantation: a single-center experience of 18 cases. Pediatr. Nephrol. 31, 1517–1529 (2016).
Le Dudal, M. et al. Stiripentol protects against calcium oxalate nephrolithiasis and ethylene glycol poisoning. J. Clin. Invest. 129, 2571–2577 (2019).
Violier, P., Boyer, O., Berthaud, R. & Dorval, G. Treatment with stiripentol in a patient with primary hyperoxaluria type 1: lesson for the clinical nephrologist. J. Nephrol. 35, 1049–1051 (2022).
Martin-Higueras, C., Feldkotter, M. & Hoppe, B. Is stiripentol truly effective for treating primary hyperoxaluria? Clin. Kidney J. 14, 442–444 (2021).
Kempf, C. et al. Stiripentol fails to lower plasma oxalate in a dialysis-dependent PH1 patient. Pediatr. Nephrol. 35, 1787–1789 (2020).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03819647 (2021).
Soliman, N. A. & Mabrouk, S. Primary hyperoxaluria type 1 in developing countries: novel challenges in a new therapeutic era. Clin. Kidney J. 15, i33–i36 (2022).
Cochat, P. et al. Primary hyperoxaluria in infants: medical, ethical, and economic issues. J. Pediatr. 135, 746–750 (1999).
Cai, Z. et al. Primary hyperoxaluria diagnosed after kidney transplantation: a case report and literature review. BMC Nephrol. 22, 393 (2021).
Al Riyami, M. S., Al Ghaithi, B., Al Hashmi, N. & Al Kalbani, N. Primary hyperoxaluria type 1 in 18 children: genotyping and outcome. Int. J. Nephrol. 2015, 634175 (2015).
Almardini, R. I., Alfarah, M. G. & Salaita, G. M. The clinical pattern of primary hyperoxaluria in pediatric patient at Queen Rania Abdulla Children Hospital. Arab. J. Nephrol. Transpl. 7, 119–123 (2014).
Gargah, T. et al. Primary hyperoxaluria type 1 in Tunisian children. Saudi J. Kidney Dis. Transpl. 23, 385–390 (2012).
Boualla, L. et al. AGXT gene mutations and prevalence of primary hyperoxaluria type 1 in Moroccan population. Genet. Test. Mol. Biomark. 19, 623–628 (2015).
Murad, H. et al. Molecular analysis of the AGXT gene in Syrian patients suspected with primary hyperoxaluria type 1. BMC Med. Genomics 14, 146 (2021).
M’Dimegh, S. et al. Mutational analysis of agxt in Tunisian population with primary hyperoxaluria type 1. Ann. Hum. Genet. 81, 1–10 (2017).
Soliman, N. A. et al. Clinical and molecular characterization of primary hyperoxaluria in Egypt. Sci. Rep. 12, 15886 (2022).
Hoppe, B. An update on primary hyperoxaluria. Nat. Rev. Nephrol. 8, 467–475 (2012).
Devresse, A., Cochat, P., Godefroid, N. & Kanaan, N. Transplantation for primary hyperoxaluria type 1: designing new strategies in the era of promising therapeutic perspectives. Kidney Int. Rep. 5, 2136–2145 (2020).
Miyata, N. et al. Pharmacologic rescue of an enzyme-trafficking defect in primary hyperoxaluria 1. Proc. Natl Acad. Sci. USA 111, 14406–14411 (2014).
Garrelfs, S. et al. Endogenous oxalate production in primary hyperoxaluria type 1 patients. J. Am. Soc. Nephrol. 32, 3175–3186 (2021).
Acknowledgements
The authors would like to acknowledge all members of the external voting panel: G. Ariceta (University Hospital Vall d’Hebron, Barcelona, Spain); S. Bakkaloglu (Gazi University, Ankara, Turkey); B. Cellini (University of Perugia, Italy); P. Cochat (University of Lyon, France); L. Collard (CHU Liege, Belgium); E. Cornelissen (Radboud UMC, Nijmegen, Netherlands); A. Devresse (University Saint-Luc, Brussels, Belgium); F. Emma (Bambino Gesù Children’s Hospital-IRCCS, Rome, Italy); F. Erger (Cologne University, Germany); S. Fargue (University of Alabama at Birmingham, USA); C. Franssen (UMC Groningen, Netherlands); A. C. Gjerstad (Oslo University, Norway); V. Gillion (Catholic University Louvain-La Neuve, Belgium); D. Haffner (Hannover Medical School, Germany); J. Harambat (University of Bordeaux, France); W. Hayes (Great Ormond Street Hospital for Children, London, UK); S. Hulton (Birmingham Children’s Hospital NHS Foundation Trust, Birmingham, UK); F. Knauf (Charité University, Berlin, Germany); S. Lemoine (University of Lyon, France); E. Levtchenko (Catholic University Leuven, Belgium); G. Lipkin (University Hospital Birmingham, UK); D. Magen (Rambam Health Care Campus, Haifa, Israel); C. Martin Higueras (University de La Laguna, Tenerife, Spain); N. Mohebbi (Dialysis Centre Zurich, Switzerland); A. Nurmohamed (Amsterdam UMC, Amsterdam, Netherlands); J. Oh (University Hamburg/Eppendorf, Germany); L. Pape (Hannover Medical School, Germany); G. Reusz (Semmelweis University Budapest, Hungary); J. Roodnat (Erasmus MC Rotterdam, Netherlands); G. Schalk (University of Bonn, Germany); E. Salido (University de La Laguna, Tenerife, Spain); K. P. Schlingmann (University of Muenster, Germany); A. Servais (Necker Hospital, Descartes University, Paris, France); E. Simkova (Al Jalila Children’s Hospital Dubai, United Arab Emirates); R. Torra (University of Barcelona, Spain); A. Torres (University de la Laguna, Tenerife, Spain). The development of these clinical practice recommendations was supported by the European Reference Network for Rare Kidney Diseases (ERKNet), which is partly co-funded by the European Union within the framework of the Third Health Program ‘ERN-2016-Framework Partnership Agreement 2017–2021’ and by OxalEurope.
Author information
Authors and Affiliations
Contributions
All authors researched data for the article, contributed substantially to discussion of the content and reviewed and/or edited the manuscript before submission. J.W.G., E.M., L.D., S.G., R.A., B.B.B, O.B., B.K., G.M., G.R., N.A.S. and J.B wrote the article.
Corresponding author
Ethics declarations
Competing interests
J.W.F. has received study grants for stable isotope and registry studies from Alnylam, Dicerna and UniQure and commissions for advisory board work and presentations for Alnylam. All these payments were made to the Research unit; no personal payments were accepted. J.W.F is also chair of the Steering Committee of OxalEurope and vice-chair of the metabolic working group of ERKNet. S.G. received study grants from Alnylam. C.A. has received consulting fees from Alnylam. B.B.B. has received consultation fees from Alnylam Pharmaceuticals and is a member of the scientific advisory board of the German PH self-support group PH Selbsthilfe e.V. O.B. has received consultancy fees and travel grants from Alnylam and Biocodex. P.M.F. has received consultant fees and grant/other support from Allena Pharmaceuticals, Alnylam, Amgen, AstraZeneca, BioHealth Italia, Gilead, Otsuka Pharmaceuticals, Rocchetta, Vifor Fresenius, and royalties as an author for UpToDate. J.B. has received consulting fees from Alnylam, Dicerna, Amgen, Bayer, Biocordex and Amolyt, and honoraria for presentations from Alnylam, Alexion, Bayer and Kyowa Kirin. J.B., B.B.B and S.S.M. are members of the Steering Committee of OxalEurope. The other authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Nephrology thanks Efrat Ben-Shalom, Saeed Khan and David Sas for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Groothoff, J.W., Metry, E., Deesker, L. et al. Clinical practice recommendations for primary hyperoxaluria: an expert consensus statement from ERKNet and OxalEurope. Nat Rev Nephrol 19, 194–211 (2023). https://doi.org/10.1038/s41581-022-00661-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41581-022-00661-1
This article is cited by
-
Lumasiran: A Review in Primary Hyperoxaluria Type 1
Drugs (2024)
-
Native nephrectomy in advanced pediatric kidney disease: indications, timing, and surgical approaches
Pediatric Nephrology (2024)
-
Nephrocalcinosis can disappear in infants receiving early lumasiran therapy
Pediatric Nephrology (2024)
-
Diagnosis and management of primary hyperoxalurias: best practices
Pediatric Nephrology (2024)
-
Clinical characteristics, genetic profile and short-term outcomes of children with primary hyperoxaluria type 2: a nationwide experience
Pediatric Nephrology (2024)
